
Abstract

The target paper of Johnson, Penke, and Spinath (2011) provides a broad up–to–date summary of the many reasons that limit the usefulness of the concept of heritability for understanding genetic contributions to personality differences. My comment focuses on one main reason why heritability is relatively silent about the roles of specific genes in creating personality differences: heritability refers to the outcome of personality development, not to its molecular basis, and therefore, the effects of nonshared genes can be transformed over development into effects that appear genetic in heritability estimates, and the shared genes can, over development, produce effects that appear to be nonshared environmental. Just as (non)shared environmental effects tell us little about (non)shared environments, (non)shared genetic effects tell us little about (non)shared genes. Copyright © 2011 John Wiley & Sons, Ltd.
Open Peer Commentary
Non–correspondence between Genes and Genetic Effects
Jens B. Asendorpf
Department of Psychology, Humboldt University Berlin, Berlin, Germany
In the context of personality research, heritability refers to the proportion of observed variance in a personality trait in a particular population that is attributed to trait–relevant genetic differences in this population. Because the attribution to genetic differences is based on assumptions about the genetic relatedness of relatives (MZ versus DZ twins, biological versus adopted siblings, etc.) that are ultimately based on the probability of sharing genes at the molecular–genetic level (more precisely: sharing alleles of genes), the concept of heritability seems to refer directly to the molecular–genetic level.
However, heritability is estimated on the basis of the phenotypic similarity of relatives and the extent to which they share genes overall. It thus refers to the similarity of trait–relevant overall gene expression, not to the similarity of particular trait–relevant specific genes. Inferences from the similarity in trait–relevant gene expression to the similarity in trait–relevant specific genes rest on the assumption of a 1–1 correspondence between genes and effects of these genes. Such a 1–1 correspondence is a tacit assumption underlying the reasoning of many geneticists and psychologists concerned with genetically informed explanations of personality differences.
For example, this tacit assumption drives gene association studies, both single allele and genome–wide; that is, the search for gene loci at which particular alleles are correlated with trait scores (trait–relevant genes). More specifically, it is assumed that for a trait with high heritability such as height, it is easier to find genes involved in the trait than for a trait with lower heritability (e.g. neuroticism), and the correlations are expected to be higher.
The failure to account for more than 5% of trait heritability by effects of specific genes even in large genome–wide association studies for very accurately measurable traits such as height led to the announcement of a ‘missing heritability problem’ (Maher, 2008), a bad choice of label because what is missing is not heritability—missing are trait–relevant alleles.
My argument here is that expectations to find trait–relevant alleles based on high heritability are misguided because the contribution of each gene to the trait may be too small to be detectable with reasonable sample sizes (one of the standard arguments for explaining ‘the missing heritability’) and because they rest on the assumption of 1–1 correspondences between genes and genetic effects, and this assumption is faulty.
Figure 1 illustrates the lack of correspondence between the molecular–genetic level of genes and the quantitative–genetic level of genetic effects. Assume that the two genomes G1 and G2 share particular alleles because they are genetically related and do not share other alleles because they are not MZ twins. A 1–1 correspondence view assumes that for any trait, the similarity of the trait–relevant genetic effects P1 and P2 is due only to the similarities between G1 and G2 (the solid lines in Figure 1). However, there may be cross–effects indicated by the dotted lines in Figure 1 such that alleles of G1 that are not shared with G2 influence the effects of the alleles shared with G2 and vice versa. Making the situation even more complicated, environmental circumstances may create different patterns of expression even in shared alleles so that they create differences rather than similarities in the trait. Moreover, the genome contains great redundancy so that even different alleles can contribute to trait similarity.
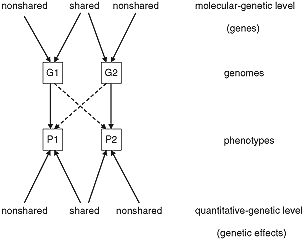
Non–correspondence between genes and genetic effects.
For an illustration, assume that the trait is attitude to death penalty, which shows a heritability close to 50% (Olson, Vernon, Harris & Jang, 2001), and G1 and G2 are the genomes of two DZ twins P1 and P2. Expectedly, multiple alleles influence this attitude, and G1 and G2 may share some of them but not all of them. The nonshared trait–relevant alleles of G1 not only influence P1's attitude but may also environmentally influence P2's attitude (e.g. because P1 is due to the additional genes a particularly strong advocate of death penalty, and she talks about it convincingly to P2, thus changing expression of some of P2's alleles that in turn affect P2's attitude). The cross–path in this case is caused by a genetic–environment effect that makes twins similar although it is based on alleles that they do not share.
Conversely, assume that G1 and G2 share trait–relevant alleles. These alleles may, nevertheless, have different genetic effects on P1 and P2 because of the differences in expression patterns.
Readers familiar with the critique by Turkheimer and Waldron (2000) of the concept of nonshared environment will recognize a clear analogy between my argument here and Turkheimer and Waldron's (and between Figure 1 here and Figure 2 in Turkheimer & Waldron, 2000): nonshared environmental conditions can have shared effects, shared environmental conditions can have nonshared effects. Both their and my line of reasoning distinguish between ‘objective’ similarity and similarity of effects, and in both lines of reasoning, the ‘cross–effects’ (dotted paths in Figure 1) are due to the fact that genes or environmental conditions serve as inputs to a cascade of developmental processes that lead to final effects where the originally shared may contribute to phenotypic differences and the originally nonshared may contribute to phenotypic similarities.
Heritability in the Era of Molecular Genetics: A Comment
S. Alexandra Burt
Department of Psychology, Michigan State University, East Lansing, MI, USA
The review by Johnson, Penke, and Spinath (2011) rightly highlights the limitations of heritability coefficients as indicators of molecular genetic associations. It is truly a must–read for all researchers interested in aetiology. I discuss two additional considerations: the influence of gene–environment correlations (rGE) on heritability estimates and the interpretation of shared environmental coefficients. It is concluded that rGE are more dynamic in their influence than previously thought and that the magnitude of shared environmental influences does matter, in that it specifies the likely bounds of identifiable shared environmental ‘main effects’.
The preceding review (Johnson, Penke, & Spinath, 2011) deftly describes the role of heritability coefficients in the modern genomic era. The authors rightly highlight the inherent limitations of the heritability coefficient as an indicator of molecular genetic associations, while also discussing the critically important role of twin and adoption studies in future aetiological research. The result is an incisive and important piece of scholarship. Despite my clear enthusiasm for this review, however, there are two interrelated points that warrant additional consideration.
My first point relates to the definitions of rGE, namely that gene–shared or common environmental correlations understate genetic influences, whereas gene–nonshared or unique environmental correlations overstate genetic influences. Although Johnson et al. (2011) are not the first to discuss rGE in this way (Purcell, 2002), I have always thought that this conceptualization of rGE overlooked the dynamic nature of rGE's influence, both in general and on genetic and environmental (i.e., ACE) estimates in particular. Specifically, rGE are likely to be fundamental to the formation of ‘reinforcement loops’, in which genetic predispositions guide the selection of particular environmental experiences, which then reinforce or otherwise shape the future expression of those genetic predispositions, which leads to additional environment selections and so forth. This role of rGE in progressively shaping outcomes is implicit in the vast majority of rGE examples (e.g. extroverted children seek out or elicit more social experiences, which exacerbate their tendency towards extroversion, which leads to more social experiences, etc.), but is rarely discussed explicitly. This is unfortunate, because this more dynamic conceptualization would alter the implied influence of rGE on genetic and environmental estimates. Specifically, to the extent that that exposure to a specific shared environmental experience serves to reinforce or further activate existing genetic predispositions, it would increase the genetic component of variance (because monozygotic or MZ twins, by virtue of their identical genetic predispositions, would emerge as more similar than would dizygotic or DZ twins). Similarly, to the extent that a nonshared environmental experience is serving to reinforce genetic predispositions in only one twin, it would increase the nonshared environmental component of variance (because MZ twins similarity would decrease).
As an example of the latter process, a recent paper found that men with lower levels of antisocial behaviour were more likely to marry and that once they did, their tendency to engage in antisocial acts decreased even more (Burt, Donnellan, Humbad, McGue, & Iacono, 2010). The act of marriage is an unusually strong and clear–cut candidate for both active and evocative rGE (individuals select into marriage and are also selected for marriage). MZ twins should thus be more or less equally prone to entering the state of marriage. Despite this, the influence of marriage on antisocial behaviour was observed even in those MZ twins discordant for marriage. In other words, the consequence of this rGE–driven event loaded at least partially on the nonshared environmental component of variance. In short, I would argue that rGE may be better viewed as intimately intertwined with GxE and, moreover, that their collective effects on ACE estimates effectively mirror those thought to result from ‘pure’ GxE.
Second, although the authors are quite right that the magnitude of estimated genetic influences has little relevance to the underlying biology of the trait in question, it is less clear to me whether this thesis would also apply to estimates of shared environmental influence. To the extent that heritability estimates are influenced by the psychometrics of the measure, this would certainly apply to estimates of shared environmental influence as well (it is worth noting, however, that psychometrics matter for virtually all statistical tests, and thus, heritability estimates should not be considered particularly biased in this regard). In other words, because estimates of genetic and environmental influence are obtained via statistical inference, they are susceptible to all of the normal vagaries of statistical findings.
But does this therefore mean that the magnitude of the estimate is necessarily meaningless? Johnson et al. (2011) argue that it does. For genetic effects, I am inclined to agree, although less because of the dependence of statistical inference on psychometric principles than because of the complicating role of gene–environment interplay in estimates of genetic influences. Namely, in addition to main effects of specific genes on the outcome, additive genetic estimates also contain GxE and active and evocative rGE (per the earlier discussion), and we cannot distinguish between these possibilities. By contrast, shared environmental influences are largely free of the effects of GxE and active and evocative rGE (as argued in Burt, 2009). They do contain passive rGE effects (although not all, as nicely spelled out by Johnson et al., 2011). However, the influence of passive rGE on shared environmental influences can be conclusively ruled out by examining adoptive sibling pairs (because they do not share segregating genes with their adoptive parents). In other words, unlike genetic and nonshared environmental estimates, the shared environmental component of variance is likely to primarily contain ‘main effects’ or those common risk and protective factors that influence individuals regardless of their genetic profile. 1 And, because main effects are typically easier to detect and are more replicable than are interactions, it follows that shared environmental influences should also be easier to identify than are genetic or nonshared environmental influences. This appears to be the case. For example, studies to date have already identified a quarter of the shared environmental variance in adolescent externalizing (Burt, 2009; Burt, Krueger, McGue, & Iacono, 2003).
Given all this, I would argue that the magnitude of the (unstandardized) variance accounted for by the shared environment actually does matter, at least in the sense that it defines the likely bounds of identifiable ‘main effects’ of the shared environment on the outcome (given the limits of statistical inference, of course). 2 This is not to say that shared environmental influences should be considered unbiased indicators of environmental influences; indeed, I suspect that these estimates vary considerably across environmental contexts (Burt, 2009). However, within a given context and/or a given population, I would argue that shared environmental effect estimates broadly illuminate the role of environmental ‘main effects’ on the outcome.
All that said, these are small considerations in an otherwise masterful piece of scholarship. Indeed, Johnson et al. (2011) will most certainly be on the required reading list in all of my future graduate behavioural genetics courses. All scientists interested in aetiology should pay very close attention to their review.
Where are the Genes? The Implications of a Network Perspective on Gene Hunting in Psychopathology
Angélique O. J. Cramer1, Kenneth S. Kendler2, and Denny Borsboom1
1Department of Psychology, University of Amsterdam, Amsterdam, The Netherlands
2Virginia Institute for Psychiatric and Behavioral Genetics, Richmond, VA, USA
The missing heritability problem is pervasive and Johnson, Penke and Spinath (2011) present a number of compelling reasons for its existence. In this comment, we present another reason for the apparent discrepancy between heritability estimates and gene–hunting results in psychopathological research: if syndromes are networks of causally related symptoms in which both symptoms and relations between them are driven by different sets of genetic polymorphisms, then gene hunting based on a phenotypic sumscore might be ill–advised because it will only capture genetic variance shared among those symptoms and their relations.
Depressed parents predispose their children to become depressed as well. This phenomenon is not so much attributable to a depressogenic environment (inadvertently created by the parents) as it is due to the fact that major depression is a moderately heritable syndrome, with heritability estimates ranging between 37% and 60% (Boomsma, Busjahn, & Peltonen, 2002; Kendler, Gatz, Gardner, & Pedersen, 2006; Sullivan, Neale, & Kendler, 2000). Combined with the high heritability of other mental disorders (Boomsma et al., 2002), it is surprising that despite many efforts, the genetic culprits have not been identified (see e.g. Sklar, 2002). For psychological traits in general, identified genetic polymorphisms typically account for less than 2% of the genetic variance (Levinson, 2006; Mitchell & Porteus, 2009).
The apparent discrepancy between high heritability and the inability to identify the responsible genetic polymorphisms has been termed the missing heritability problem and is pervasive in the realm of psychopathology (e.g. Manolio et al., 2009). In the present issue, Johnson et al. propose various plausible mechanisms that contribute to the missing heritability problem, ranging from methodological factors that result in inflated heritability estimates to problems with the specific research strategies employed in gene hunting. Pertaining to the latter, in this comment, we elaborate on a potential problem on which Johnson et al. (2011) did not reflect: what if the way we define a syndrome in current gene–hunting efforts is incorrect?
In psychopathological research, the most commonly used proxy for a phenotype is the typical operationalization of a syndrome, that is, a sumscore (i.e. the total number of symptoms of a disorder present) that can be further dichotomized, using, for example, criteria as specified by the Diagnostic and Statistical Manual of Mental Disorders Fourth Edition (DSM–IV) of the American Psychiatric Association, to reflect the absence/presence of a particular disorder. In its most rudimentary form, genetic association studies identify genes or genetic variants as predisposing to a mental disorder if they predict the dependent variable in the design, that is, the (dichotomized) sumscore (van der Sluis, Kan, & Dolan, 2010). So far, this strategy has not been very successful at identifying the important genetic polymorphisms in the onset of mental disorders. In our view, this may be partly due to the fact that one of its most important assumptions—that a sumscore forms a valid representation of a syndrome—is fundamentally flawed.
Current approaches to gene hunting rely on the assumption that relation between a phenotype—for example, major depression—and its observable attributes—for example, the ‘A criteria’ in DSM–IV is one of measurement: a psychological phenomenon causes its observable attributes (e.g. extraversion causes party–going behaviour: McCrae & Costa, 2008, p. 288). One of the far–reaching consequences of such a common–cause view is that correlations among the observable attributes themselves are deemed spurious; they only exist because they share a common cause: mental rotation skills and verbal intelligence are only correlated because they share a common cause, namely general intelligence. In terms of searching for genes that are implicated in the onset of a syndrome, this view translates into the following chain of events: genes, via a host of hypothesized endophenotypes, result in the expression of individual symptoms (because the relations between symptoms are spurious) and those, in turn, sum into a syndrome (because symptoms and syndrome have a measurement relationship). However, as we have argued extensively elsewhere (Cramer, Waldorp, van der Maas, & Borsboom, 2010), relations between symptoms might not only be non–spurious in nature but might also be the very essence of what constitutes a syndrome (similar arguments have been made for general intelligence: van der Maas et al., 2006). For example, consider the correlation between two symptoms of major depression: insomnia and fatigue. Under the assumption of a common cause, the correlation between these two symptoms is spurious; it only arises because insomnia and fatigue share a common cause, major depression. It is, however, more likely to assume that this correlation exists because there is a real, straightforward causal relation between these two symptoms: if you do not sleep, you will become tired. Similar arguments can be made for a host of other psychological phenomena—for example, consider feeling comfortable around people and party–going behaviour: do we need an overarching ‘extraversion’ trait to explain why these two observed behaviours tend to covary?—and as such, it is premature to dismiss direct relations between observed attributes as being mere spurious by–products of an overarching construct. What does this mean for gene–hunting efforts?
If constructs are indeed networks of causally related observables, individual differences are most likely to arise as differences in the strength of those relations: when Alice suffers from depressed mood, she fairly easily develops suicidal thoughts (i.e. strong relation between the observed symptoms ‘depressed mood’ and ‘suicidal ideation’), whereas Bob does not ever contemplate suicide while feeling depressed (i.e. relatively weak relation). Furthermore, it is likely that the strength of such relations stands at least partly under genetic control. Now, it is not likely that each relation is influenced by the same set of genes for the sheer number of relations (k2–k in a network containing k observables/symptoms) in any given network greatly diminishes this possibility and the relations probably differ in terms of the endophenotypes (and thus genes) involved (e.g. the more physiological homeostatic processes that are likely to govern relations between sleep and fatigue vis–à–vis the more cognitive processes that are probably involved in the relation between depressed mood and suicidal thoughts).
Hence, when trying to relate genetic polymorphisms to a sumscore, one only captures the genetic variance that is shared among those individual symptoms (including their relations); the different genetic polymorphisms that are responsible for individual differences in the strength of the relations between those symptoms are completely left unaccounted for. As such, the network approach may explain at least partly why current approaches cannot find the genetic culprits of mental disorders. By properly modeling their etiology, we increase our power to detect risk variants. It is, after all, the relations between symptoms that glue them together into a syndrome.
Acknowledgments
The work of Angélique Cramer and Denny Borsboom was supported by NWO innovational research grant no. 451–03–068.
Understanding Heritability by Explaining Heritability: Recent Developments in Behaviour Genetics Tell Us More
Marleen H. M. de Moor and Dorret I. Boomsma
Department of Biological Psychology, VU University Amsterdam, Amsterdam, The Netherlands
EMGO + Institute for Health and Care Research, VU University Medical Center Amsterdam, Amsterdam, The Netherlands
Neuroscience Campus Amsterdam, VU University Medical Center Amsterdam, Amsterdam, The Netherlands
mhm.de.moor@psy.vu.nl
Johnson, Penke, and Spinath (2011) extensively discuss the limitations of heritability estimates obtained from twin studies in understanding the role of genes on behavioural traits. They plead for more advanced modeling and a focus on gene–environment interplay. We review the results from advanced modeling and molecular genetic research and argue that gene–environment interplay is not likely to be the main factor in explaining heritability. It is anticipated that future developments in these areas will provide an even more complete picture on the genetic and environmental mechanisms underlying behavioural traits.
Johnson et al. (2011) extensively discuss the limitations of heritability estimates obtained from twin studies in understanding the role of genes and environment on behavioural traits. They make three important points about heritability. First, heritability depends on how the trait under interest is measured. For example, measurement error and low item endorsement frequencies tend to lower heritability. Second, when heritability is based on the classical twin design, it can be biased if the assumptions of this design are violated. These assumptions include the absence of assortative mating, gene–environment interaction and correlation. Third, heritability tells us little about the underlying biology of a trait, because biology is only one of the factors that influence the magnitude of heritability estimates. They plead for more advanced modeling and a focus on gene–environment interaction and correlation (‘interplay’).
In this commentary, we introduce two types of advanced modeling that we feel remain underexposed: extended twin family designs and causal modeling. We further review some recent molecular genetic studies and theoretical developments that illustrate the progress in explaining the heritabilities of complex traits. We argue that the results from these approaches suggest that gene–environment interplay is not likely to be the main factor in explaining heritability.
Extended twin family designs
Extended twin family designs test assumptions underlying the classical twin design and can address more complex questions about the influence of genetic and environmental factors than the classical twin design (Eaves et al., 1978; Keller et al., 2009). Twin–sibling studies on personality (e.g. neuroticism, extraversion, sensation seeking) and related traits converge on the finding that the heritabilities of these traits represent both additive and non–additive genetic influences (Keller et al., 2005; Stoel et al., 2006; Distel et al., 2009b). This has been confirmed by studies adding data from parents and other types of first–degree and second–degree relatives (Eaves et al., 1998; Eaves et al., 1999; Rettew et al., 2008; Distel et al., 2009a). These studies further show that the spouse correlation, an indicator for assortative mating, for personality traits is generally very low (0–0.2) and has an almost negligibly small impact on the estimates of heritability and the proportion of variance explained by environmental influences. There is typically no evidence for cultural transmission, a shared sibling or a shared twin environment. These findings are consistent with studies in adult twins reared apart or together (Bouchard et al., 1990), although in adolescent adoptees, some evidence for shared environment has been found (Buchanan et al., 2009). The absence of cultural transmission renders passive gene–environment correlation unlikely, although gene by environment interaction may still be present. Overall, the findings from extended twin family designs suggest that the initial heritability findings obtained with the classical twin design are less biased than Johnson et al. (2011) suggest, at least for personality and related behavioural traits.
Causal modeling
Twin family designs can be informative in testing for causal mechanisms among behavioural traits. Available methods include direction–of–causation modeling (Heath et al., 1993; Duffy & Martin, 1994), the children–of–twins design (D'Onofrio et al., 2003) and the co–twin control method (Kendler et al., 1993). These methods have been applied to test for causal effects of parenting, life events and lifestyle behaviours on mental health (Kendler et al., 1993; Kendler et al., 1999; Gillespie et al., 2003; Stubbe et al., 2006). A combination of the standard bivariate genetic model and the co–twin control design, has been applied to cross–sectional and longitudinal data on exercise behaviour and anxiety/depression, showing that the association can be best explained by common genetic factors rather than a causal effect (De Moor et al., 2008). These methods are also suitable to test causal hypotheses between personality and related behaviours. They show that twin (family) data are informative beyond establishing the heritability of a trait. They allow for genetic influences on behavioural traits to be mediated by other traits and hence give a more complete picture on the aetiology of behavioural traits. Future developments in this area are expected that may even better exploit the longitudinal nature of many twin (family) data and that incorporate data on measured genetic variants.
Molecular genetic studies
Recent molecular genetic studies show the progress in finding the genetic variants for complex traits. This holds not only for height, the example used by Johnson, Penke, and Spinath (2011), but also for psychiatric diseases and personality. The largest genome–wide association (GWA) study on height in 180 000 subjects identified 180 loci, explaining about 10% of the variance (Lango Allen & et al., 2010). These loci are enriched for genes in biological pathways involved in skeletal growth defects. Almost simultaneously with the Lango Allen paper, a study was published that showed that the proportion of variance captured by analysing all measured single–nucleotide polymorphisms (SNPs) on a common micro–array explained about 45% of the variance in height (Yang et al., 2010). Thus, common SNPs explain a large proportion of the heritability for human height.
One example to illustrate progress for psychiatric disorders is CACNA1C, which was identified as a gene for bipolar disorder, major depression and schizophrenia (Ferreira et al., 2008; Green et al., 2010) and which has also been shown to affect brain structure and activity (e.g. Wessa et al., 2010; Franke et al., 2010). Another example is the PCLO gene in relation to major depression (Sullivan et al., 2009; Hek et al., 2010) and bipolar disorder (Choi et al., 2010). For personality, the largest GWA study identified two loci for Openness to Experience and Conscientiousness in over 17 000 subjects (De Moor et al., 2010). The variants in these studies explain only very small portions of the heritabilities. This cannot simply be ascribed to the factors that Johnson, Penke, and Spinath (2011) mention, such as bias in heritability as a result of gene–environment interplay. Other reasons include imperfect genomic coverage of common variation by SNP chips, the high penalty that needs to be paid for multiple testing, small effect sizes of common variants and the possible influence of other types of variants (see also Manolio et al., 2009). The application of new technologies (e.g. sequencing) and methods (e.g. Yang et al., 2010) is expected to give more insight into the biological underpinnings of heritability estimates.
Conclusions
We discussed how extended twin family designs, causal modeling and molecular genetic studies have yielded knowledge that helps us better understand the nature of heritability. It is anticipated that future developments in these areas will provide an even more complete picture on the genetic mechanisms underlying personality and related behavioural traits.
Acknowledgments
Marleen de Moor (VENI–016–115–035) is financially supported by the Netherlands Organization for Scientific Research (NWO). Dorret Boomsma is financially supported by the European Research Council (ERC–230374).
Introducing Neur–X, a Style of Being: The Transformation of a Concept
Elena L. Grigorenko1,2,3
1Child Study Center, Department of Psychology and Department of Epidemiology & Public Health, Yale University, New Heaven, CT, USA
2Columbia University, New York, NY, USA
3Moscow State University, Moscow, Russia
Discussing psychology's attempts to incorporate genetic findings into its inquiry into the aetiology of complex behaviour traits, Johnson, Penke, and Spinath (2011) deliver an engaging and well–informed précis of the powers and limitations of the concept of heritability. Here, another dimension is added to this discussion—the blurring of behaviour traits, even when viewed through different concepts (e.g. disorder, style, trait). This overlap, in part, generates the perception of ‘ubiquitous’ heritability. Understanding this overlap might clarify the now rather convoluted view of the connections between the genome and behaviour.
We are loping sequences of chemical conversions, acting ourselves converted. We are twists of genes acting ourselves twisted; we are wicks of burning neuroses acting ourselves wicked. And nothing to be done about it. And nothing to be done about it.Gregory Maguire, Son of a Witch, 2005, p. 128
Psychology is replete with concepts that are related by content and form. Perhaps it cannot be otherwise, because psychological theories and, therefore, psychological concepts are developed in proximity, both epistemologically and temporally. Quests to understand psychological phenomena are often launched simultaneously within different theoretical and empirical paradigms. This results in somewhat redundant conceptualizations that, in turn, make psychology a continuous science, despite apparent contradictory interpretations of the same phenomenon within different theories. Such redundancy contributes to the observation of Johnson et al. (2011) that ‘all behavioural traits are moderately heritable’ (p. 5).
This commentary illustrates this point by using the constructs of neurosis (or psychoneurosis or neurotic disorder), neurotic styles and neuroticism. Etymologically, these words are related to the Greek word νεῦρον (neuron, ‘nerve’). Phenomenologically, they all refer to a particular behavioural presentation—style—that is recognizable no matter which of these three terms is used. And the heritable (or temperamental, Johnson et al., 2011, p. 4) nature of this style has long been recognized in all its ‘incarnations’ in psychological theory.
The concept of neurosis, where the ending—osis signifies a diseased or abnormal condition, was introduced by Scottish doctor William Cullen (Cullen, 1769) to refer to disorders of sense and motion, whose physiology was evasive and, because no other organs seemed to be jeopardized, could be attributed to disturbances in the nervous system. The concept, mostly unnoticed for about a century, attracted much attention through the work of Sigmund Freud and his disciples, becoming one of the cornerstones of psychoanalysis. The place of neurosis within psychodynamic literature is impossible to underestimate—it is simply essential to it. What is especially important, however, is its transformation into the psychiatric diagnoses we know and use today. Although present in earlier versions of the DSM, neurosis was eliminated from the DSM–III (Bayer & Spitzer, 1985). Yet, its connectedness to current diagnostic categories is ever present in various types of anxiety (Frances, Pincus, Manning, & Widiger, 1990) and depression (Torgersen, 1986) disorders. Thus, a single concept was first elevated to superior heights as an all–encompassing descriptor of a type of behaviour and its particular underlying etiological mechanism, then shredded into numerous related concepts.
The concept of neurosis was fundamentally transformed from a categorically discrete disorder to a continuous normative (i.e. observed in all people) behavioural trend, or ‘neurotic’ style, by Karen Horney (1950). Horney viewed child development as fueled by a conflict between nature and culture. For Horney, nature is in the genetically endowed real self; when this real self is abandoned under cultural pressures, neurosis arises (Rendόn, 2008). Each child has a trajectory of self–realization, enacted as the genetic predisposition is translated into a unified self. This translation can unfold in multiple modes, but in each, the child's relationships with others are crucially important. Horney delineates three main styles of relating to others, specifically, the following: (i) the ‘toward’ style, assuming the desire to receive and give affection; (ii) the ‘against’ style, assuming the desire to assert and fight for one's interests; and (iii) the ‘away’ style, assuming the desire to withdraw from people and keep to oneself. These three styles provide a child with alternative modes of dealing with external stressors, which may challenge the process of self–realization and cause neurosis.
As a result of the disorder–to–normative–behaviour–style–transformation of neurosis that occurred as Freud's ideas were interpreted and further developed by Horney, neurosis has been further transformed within trait theories of personality. Capitalizing on the descriptions generated in volumes of studies of neurosis, Eysenck (1963) particularly focused on the notion that neurosis arises from some inherent biological properties of the nervous system, whose quintessential characteristics are emotional instability and high basal arousal. On the basis of this notion, he conceptualized neuroticism as an indicator of central nervous system excitation, with high levels of excitation being a source of vulnerability for illnesses. Neuroticism is typically captured by items depicting self–centredness, irritability, anger, depression, anxiety, worry, hostility, high levels of self–criticism and criticism of others, feelings of inadequacy and vulnerability (McCrae & Costa, 2003; Watson, Clark, & Harkness, 1994); these items can be directly mapped onto Freud's descriptions of neurotic patients and Horney's accounts of neurotic styles. Neuroticism, either as a heterogeneous multifaceted trait or as a component of a higher–order dimension of negative emotionality, is embraced by most, if not all, theories of personality.
The idea that Neur (neurosis, neurotic style, neuroticism)–X has a biological foundation has always been present. Even Freud (1986) referred to neurosis as heritable, although it was an ingenious vision rather than an empirically supported assertion. Horney, although more engaged with the family (or caregiver) role in the formation of neurotic styles (1937), never doubted and even re–enforced the importance of the inherited potential for neurosis in her later writings (1950). Both visions, however, have been confirmed in various quantitative–genetic and molecular–genetic research studies of both neuroses (with whatever diagnostic labels attached) and the trait of neuroticism. Notably, although the heritability estimates of neuroticism are 50–60%, it has been difficult to reliably pinpoint other sources of variance in the individual differences on this trait. The estimates of shared environmental influences have been reported as negligible, attributing the remaining variance to nonshared environmental effects; yet, these effects remain unidentified. Although these findings indicate that Neur–X has a strong heritable component, the texture of this component is far from understood.
In sum, whether conceived as a disorder, style or trait, Neur–X presents as a recognizable, identifiable and persistent cluster of behaviours—a behaviour style. There is a convincing amount of evidence indicating that Neur–X is genetically grounded and emerges from a complex constellation of ‘twists of genes’. These twists, however, are far from deterministic in their impact, and, although forming a predisposition, can be either twisted further or untwisted by non–genetic forces. The realization of this predisposition is complex but uninterrupted within the lifespan of a single person. Thus, when by virtue of probabilistic processes this predisposition is in place, a preferential type of behaviour unfolds, with all of the due developmental fluctuations and deviations from its course. Yet, the course is there; otherwise, it would have not been captured by only that many theorists and in only that many empirical studies. This course is called the Neur–X of development: a holistic style of being from beginning to end. Neur–X's prominence in many complex behaviours guarantees, at least in part, the ubiquitous heritability–ness of correlated behaviour traits. Understanding Neur–X's psychological structure might clarify the now rather convoluted view of the connections between the genome and behaviour.
Author note
The work on this commentary was supported, in part, by R01 DA010726 from the National Institutes of Health, USA. I am thankful to Mei Tan for her editorial assistance.
Sociogenomic Theory as an Answer to the Heritability Problem
Joshua J. Jackson, Patrick L. Hill, and Brent W. Roberts
Department of Psychology, University of Illinois, Urbana, IL, USA
We present typical misinterpretations of heritability statistics that Johnson, Penke, and Spinath (2011) failed to thoroughly discuss. These misinterpretations stem from difficulties in incorporating the environment into genetic models. We present the sociogenomic model as a solution, which was initially introduced to personality psychology in response to similar concerns that prompted Johnson et al.'s review (Roberts & Jackson, 2008). Specifically, the sociogenomic model is better able to incorporate the environment in genetic models through switching the focus from genes to the genome.
We were pleased to see a target article updating the current thinking on heritability and genetics, in part, for personal reasons. As much of our research focuses on personality traits, a typical focal point of behaviour genetics research, we are often called on to interpret behaviour genetics findings for constituencies outside of psychology. Often, we are asked for clarification concerning the meaning of heritability and the mystery of the missing heritability in the current generation of molecular genetics studies (Manolio et al., 2009). Johnson et al. (2011) provide an excellent overview of current thinking on heritability, but we believe that they did not go far enough in the exposition. Specifically, we hoped that their paper would more thoroughly address common misperceptions of heritability estimates and offer a reason of why these misperceptions persist. We suggest that using a sociogenomic model (Roberts & Jackson, 2008) can help rectify these misperceptions better.
Heritability ≠ immutability
Many scholars outside of the field of behaviour genetics and personality psychology tend to equate the fact that personality traits are heritable with the idea that there is some component within traits that is unchanging (e.g. genetic polymorphisms). Therefore, heritability is used as a rough indicator of the changeability of a construct (i.e. 1—heritability = changeability). Most portrayals of the findings from behaviour genetics are more nuanced than this, of course. Nonetheless, a nuanced approach still fails to go far enough to retract this misperception. This can be seen in the current article where the authors tie heritability to test–retest reliability (p. 9). This connection is made without an acknowledgment of the difference between reliability and meaningful change. It should be noted that high levels of heritability do not equal stability nor is it even necessary for stability. For example, according to Rutter (2006), two of the least heritable qualities are the belief in god and attitudes towards racial integration, yet these constructs are quite stable over time (Alwin & Krosnick, 1991; Wink & Dillon, 2002).
Heritability ≠ similarity
We were very pleased to see the concrete admission that ‘even quite substantial heritability does little to create actual similarity among family members’. That said, we do not believe that the authors, nor past behaviour geneticists, have come clean on the real numbers. Specifically, in a rather illuminating chapter, Loehlin (2005) reports the meta–analytic estimates of the correlations between parent personality and the personality of their biological offspring. The average correlation across all traits from over 90 studies was a rather unimpressive .13. Now, we do believe that small effect sizes can be quite important (e.g. Roberts et al., 2007). But, a correlation of .13 means that any similarity between children and their parents is best thought of as random. Contrast the .13 correlation with the average correlation between MZ twins personality traits, which is .50. Because the MZ results tend to be overemphasized in behaviour genetics research, it is easy to see why many people would conclude that parent–child correlations should be close behind that. In fact, an oversimplified conclusion would be that it should be half .50 or about .25. In fact, it is half that again.
The relatively low correlation of .13 is more than marginally important. First, it shows that personality traits do not ‘breed true’ to the extent that is often inferred from behaviour genetics research. In Behavioral Genetics (BG) terms, there is a significant portion of the heritable variation in personality that results from dominance heritability (Loehlin, 2005). This fact should partially inform the missing heritability problem in genome–wide association studies. Moreover, it means that a decent portion of our personalities are ‘genetic’ in origin but unpredictable from our parents’ personality—an idea that takes some time to get used to. Even more perplexing is the fact that .13 also includes the effect of the environment parents create. This relationship is half of what we would expect if all of the heritability from parents to kids was ‘under genetic control’, which implies that parents do little or nothing to make the children ‘chips of the old block’. Unfortunately, findings such as these are often overlooked because they do not correspond with what heritability statistics imply (e.g. high levels of similarity).
Heritability ≠ genetic substrate
Of course, heritability does not equal ‘genetic control’, but the misperceptions surrounding immutability and similarity (and the misperceptions outlined in Johnson et al., 2011) stem from the perception that heritability is isomorphic with an unchanging biological substrate (i.e. devoid of environmental influence, a genetic destiny). Although the authors point out this is not the case, we believe that many individuals have difficulties coming to grips with this idea, partially due to a tendency not to meaningfully incorporate the environment in typical behaviour genetics research. Although newer work is starting to include objective measures of the environment in behaviour genetic models (Johnson, 2007), most research and thinking in behaviour and molecular genetics has provided lip service at best to the role of the environment. Moreover, even incorporating the environment into current BG models does not solve this misconception because a heritability statistic is still estimated and will thus still be interpreted as a genetic substrate devoid of environmental influence. The authors do well to speak against this simplistic interpretation, explaining that heritability estimates include gene–environment correlations and interactions, but it is still difficult for lay individuals to wrap their heads around the complexities—and if they do, what to do with that information.
Sociogenomics
We hoped that the authors should have provided a theory that helps address why misconceptions about heritability and genetics arise (especially for those who are neither behavioural geneticists nor biologically savvy). We believe that sociogenomic theory (Roberts & Jackson, 2008; Robinson et al., 2005) can be used to both better understand how genetic factors influence personality and provide a generative way to inform theoretical models of personality.
Sociogenomic theory offers a way to combat the confusion that surrounds heritability estimates by switching the focus from genes to what genes produce at the genome level. Doing this allows a better integration of the environment into genetic models. Our genes cannot be altered by environmental influence, and thus, on the surface, it may be difficult to think about how unchanging genes interplay with the environment—other than in the deterministic way argued against in Johnson et al. (2011). The role of genes become clearer when the focus of genetic effects are at the level of the genome. The genome, in contrast to one's DNA, is dynamic and depends on the environment for activation and maintenance (Robinson, 2004; Robinson et al., 2005). Both variation in genes and the environment can influence gene products, with the effect being that nature and nurture should not be thought of as two distinct processes but rather as two sides of the same coin. We feel that this simple premise better allows one to think about the complexities of examining the influence of genetic factors and, by extension, clarifies why one cannot make any strong inference about underlying biological processes, stability or similarity from a single heritability estimate.
Conclusion
Difficulties in interpreting heritability statistics are frequent, and the utility of simple heritability statistics has run its course, as such, the field must move on to incorporate more advanced conceptualizations of the genetics of personality. We feel that the sociogenomic model offers a better way to understand how genetic and environmental factors combine to influence personality so as to overcome misinterpretations that stem from behaviour genetic models. Sociogenomics also serves as way to structure future research in the direction of gene–environment transactions, the area that both Johnson et al. (2011) and we believe is necessary to understand the role of genetics in personality.
Bringing the Person into Research on Gene–Environment Interplay
Sara R. Jaffee and Thomas S. Price
Institute of Psychiatry, King's College London, London, England, UK
sara.jaffee@kcl.ac.uk; thomas.price@kcl.ac.uk
Quantitative genetic studies have firmly established that virtually all complex traits are heritable. The question is how genes act to establish individual differences. Researchers are now beginning to investigate the complex, reciprocal transactions between genes and environments, and these studies are starting to point to the mechanisms that underlie health and behaviour. These studies highlight the importance of individual differences in personality and perceptions in bridging genes and environments. However, research on gene–environment interplay is in its infancy, and researchers should be cautious of overstating its implications.
What does it mean to show that a trait is heritable? This is the central question that Johnson, Penke, and Spinath (2011) address. As they rightly perceive, heritability is a heuristic measure. It provides a simple reckoning of the relative magnitude of genetic and non–genetic influences on trait values in a population. Since the 1980s, behavioural geneticists have demonstrated the ubiquity of genetic influences on trait after trait. The list of phenotypes that are heritable includes not only attitudes and beliefs, personality traits, cognitive abilities, and behaviours but also our environments. Genes influence the quality of our relationships with parents, peers, and partners; our tendency to experience stressful life events; and the degree to which our homes are chaotic and disorganized rather than organized and calm (Hanscombe, Haworth, Davis, Jaffee, & Plomin, 2010; Jaffee & Price, 2007; Kendler & Baker, 2007; Plomin & Bergeman, 1991). This body of research has provided an important corrective to the view held by many social science researchers in the 1960s and the 1970s that individual differences arose from differences in parenting or access to opportunities rather than biological differences that were seen to be resistant to change.
Heritability is a slippery concept to grasp. A heritability estimate is specific to one population. Gene–environment interactions and correlations, secular trends, genetic drift, selection and so on mean that any heritability estimate is just a snapshot of a particular set of circumstances at a particular point in time. Heritability estimates therefore have neither relevance for any investigation of differences between populations nor mean differences between groups. The idea that heritable traits are immutable persists: it is wrong.
Research in genetics has also misconstrued the meaning of heritability. Contrary to some claims, molecular genetic investigations are not necessarily more likely to succeed for highly heritable traits (Visscher, Hill, & Wray, 2008). For example, the BRCA1/BRCA2 mutations that predispose to breast cancer were discovered even though the disease is not especially heritable. The BRCA1/BRCA2 example also illustrates the disconnect between heritability, which is a feature of a population and individual genetic risk. A woman who carries one of these mutations is estimated to have a 60% chance of developing breast cancer (National Cancer Institute, 2011). In the general population, however, the BRCA1 and BRCA2 mutations are rare and so account for a relatively small proportion of breast cancer cases in the population and explain only a fraction of the trait's heritability.
How does the field progress from here? For some time now, there has been a consensus—articulated by Johnson et al. (2011)—that the important questions are no longer about nature versus nurture but rather about nature and nurture—about the ways in which genes get ‘outside the skin’ to influence our environments, the ways in which environments get ‘under the skin’ to influence gene expression and epigenetic processes and the ways in which gene variants condition our responses to specific environments. These questions about gene–environment interplay can be situated in a broader framework for the study of person–environment interactions, including life course theories of personality (Elder, 1998) and developmental theories about the effect of the child on his or her environment (R. Q. Bell, 1968). In general, these theories emphasize complex, reciprocal transactions over time between persons and their environments, and this complexity is borne out in what we are starting to learn from molecular and quantitative behavioural genetics research about the interplay of genes and environments.
For example, epidemiological studies have established that social isolation is a robust risk factor for cardiovascular disease. Why? The answer may lie in the ability of the social environment to turn off and on genes that regulate inflammatory processes implicated in heart disease. In one study of healthy older adults, those who perceived themselves as feeling more lonely and distant from others differentially expressed genes involved in inflammation, immune response to viral infection and production of antibodies (Cole et al., 2007). Our social environments may also colour our perceptions of social interactions, with implications for gene function. In another study of children with asthma, those from low socioeconomic status groups were more likely than children from higher socioeconomic status groups to view socially ambiguous stimuli as threatening. In turn, these cognitive appraisals influenced whether genes involved in inflammation—a key component of asthma—were turned off or on (Chen et al., 2009). These studies demonstrate the effects of social context on gene function in genes that are likely to be implicated in health and disease. Being healthy or not being healthy will, in turn, change our environments even more by potentially influencing our relationships with significant others and constraining (or enhancing) our opportunities for work and recreation.
These studies of gene–environment interplay highlight the role of the person and the person's perceptions and interpretations of his or her environment. The extent to which genes are turned off or on is influenced by a person's subjective sense of isolation rather than an objective count of social contacts (Cole et al., 2007) and by a child's subjective perception of threat rather than objective measures of educational attainment or occupational status (Chen et al., 2009). Here, personality psychology has much to offer the study of gene–environment interplay through its knowledge of how situations affect behavioural and cognitive response tendencies and how stable individual differences in such response tendencies influence how we will perceive our environments.
The interplay between genes and environments is complex and unfolds over time. Even the expression of Mendelian disorders such as phenylketonuria depends on factors other than the risk genotype. What we currently know, however, barely scratches the surface, and the interplay of genes and environments may not be the answer to every question. Recent discussions of ‘missing heritability’ have been illuminating in this regard. It is clear that the main effects of variants discovered by genome–wide association studies account for relatively little trait variance (Manolio et al., 2009). The reason may simply be that true associations have intractably small rather than intractably complex effects (Yang et al., 2010).
As Johnson et al. (2011) so clearly articulate, the way forward will involve more than a simple decomposition of variance into genetic and environmental components. The real rewards lie in knowing the underlying biological and environmental mechanisms.
The Concept of ‘Reactive Heritability’: How Heritable Personality Variation may Arise from a Universal Human Nature
Aaron W. Lukaszewski
Haas School of Business, University of California, Berkeley, CA, USA
Johnson, Penke, and Spinath (2011) provide many productive insights regarding the study of gene–environment transactions and go further than most others in questioning the basic premise that specific genotype–personality linkages exist at all. The current commentary elaborates upon these issues in relation to the concept of ‘reactive heritability’, which occurs when a personality trait is facultatively calibrated over development in response to other heritable phenotypic features. Importantly, this concept may help resolve perceived conflicts between the existence of heritable personality variation and the hypothesis of a universal human nature.
Since the discovery that all psychological and behavioural traits exhibit substantial heritability, behavioural geneticists have assumed that the next step in their research should be to identify specific genotypes that influence the endophenotypes of personality in reliable ways—which has made the apparent lack of such genotypes across molecular genetic studies (including genome–wide association studies) understandably surprising to many scientists. In their excellent target article, Johnson et al. (2011) clearly outline a variety of conceptual and methodological tools that may help lead to fruitful discoveries regarding the complex gene–environment transactions that could potentially underlie personality development. Notably, Johnson et al. (2011) also go further than most previous theorists in seriously questioning the basic premise that consistent patterns of gene–personality linkages will, for a given heritable trait, ever be found to exist at all. The current commentary elaborates upon these issues in relation to an important concept that has been heretofore neglected by behavioural and evolutionary geneticists: reactive heritability.
Reactive heritability: Heritable personality variation from a universal human nature
More than two decades ago, Tooby and Cosmides (1990; see also Lukaszewski & Roney, in press) theorized that specific gene polymorphisms that directly organize personality in reliable ways may be few and far between. Instead, they emphasized the possibility that personality variation may be facultatively calibrated over development via evolved conditional rules of the form ‘given condition x, pursue behavioural strategy y’. Theoretically, such mechanisms of facultative calibration should often outcompete genetically fixed strategies over evolutionary time by orchestrating a relatively precise functional match between strategy and circumstance, assuming that particular cues did, in fact, predict the reproductive payoffs of alternative trait levels better than the identities of specific genotypes (Penke, in press).
Importantly, the cues to which personality traits are calibrated may be other phenotypic features. For instance, because physical strength and physical attractiveness are two characteristics that would likely have predicted the reproductive payoffs of extraverted (versus introverted) behavioural strategies across human ancestors, Lukaszewski and Roney (in press) theorized that extraversion levels should be facultatively calibrated over development to variations in these phenotypic features. In support of this, they reported that approximately a third of the variance in extraversion scores could be jointly predicted from physical strength and attractiveness. Crucially, in this example, because physical strength (Silventoinen, Magnusson, Tynelius, Kaprio, & Rasmussen, 2008) and physical attractiveness (Rowe, Clapp, & Wallis, 1987) are both highly heritable phenotypic features, extraversion should also exhibit heritability even if there are no specific gene polymorphisms that directly influence the endophenotypes of this behavioural trait—a phenomenon Tooby and Cosmides (1990) referred to as ‘reactive heritability’.
The concept of reactive heritability thus provides a mechanism through which heritable personality variation may arise from a species–typical psychological architecture and thereby illustrates the fundamental lack of conflict between the existence of genetic variance in adaptively patterned personality variation and the hypothesis of a universal human nature. This is especially important because it has become increasingly common in recent years for theorists to argue that adaptationist approaches to understanding individual differences—which focus on species–typical mechanisms of facultative calibration—are essentially impotent in the explanation of heritable personality variation and that evolutionary genetic models are therefore required (e.g. Nettle, 2006; Penke, Denissen, & Miller, 2007). If reactive heritability is discovered to be a relatively widespread phenomenon, such claims will require serious re–evaluation.
Implications of reactive heritability for understanding gene–environment transactions
The concept of reactive heritability has a variety of potential implications for scientists moving forward in attempting to understand the gene–environment transactions underlying personality development.
First, the discovery that a personality trait is reactively heritable implies that any specific genes that predict personality variation (which, it bears repeating, have thus far proven elusive) will be those that underlie variation in the other phenotypic features on the basis of which the personality trait of interest is calibrated. For instance, Lukaszewski and Roney (in press) replicated a few previous findings that a specific polymorphism in the androgen receptor gene sequence was predictive of men's extraversion, but they also found that this association was almost entirely mediated through the gene's effect on men's physical strength. This suggests that the apparent genotype–personality linkage was in fact attributable to the effects of facultative calibration, rather than direct hormone–mediated effects of the gene polymorphism on the neural endophenotypes of extraversion.
By the same line of reasoning, if there are no specific genotypes that underlie variation in the phenotypic features upon which a personality trait is reactively heritable, then this will also be true for the personality trait itself. For instance, genetic variance in physical attractiveness is likely maintained over evolutionary time as a function of stochastic interactions among the following: (i) effects of chance mutation occurring across the entire genome; (ii) massive pockets of noisy genetic polymorphisms maintained in the context of parasite–host coevolution; and (iii) unpredictable exposure to environmental factors such as pathogens and nutrition (Lukaszewski & Roney, in press; Tomkins, Radwan, Kotiaho, & Tregenza, 2004). The upshot of this state of affairs is that different genotypes will predict physical attractiveness across different individuals—and that this will always shift in evolutionary perpetuity across both time and space. Thus, to the extent that the heritable variance in a personality trait arises via facultative calibration to physical attractiveness, it does not make sense to hunt for the specific genotypes that reliably predict the personality trait across individuals.
Finally, and most importantly, the study of reactive heritability requires the field of behavioural genetics to broaden its tool kit beyond twin studies and molecular genetics (although these will remain indispensible). This is because reactive heritability places the causal structure of species–typical psychological mechanisms at centre stage in the explanation of genetic variance in personality. Because the study of such mechanisms is the specialization of more traditional cognitive, developmental and social psychologists, these scientists may find themselves playing a surprisingly large and important role in explaining the gene–environment transactions that have in recent decades been seen as the exclusive province of behavioural genetics.
The Genetic Architecture of Behavioural Traits
Kevin J. Mitchell
Smurfit Institute of Genetics and Institute of Neuroscience, Trinity College Dublin, Dublin 2, Ireland
Johnson, Penke, and Spinath (2011) raise interesting issues regarding how to move beyond the now well–established demonstration that many behavioural traits are heritable. Progress towards identifying the genes involved crucially depends on having the right model of the genetic architecture of the traits. New evidence indicates that such traits are dominated by effects of rare variants, which collectively can explain the population heritability. Future progress will depend on identifying such variants and elucidating their effects at the neurobiological level, providing the context to examine additional factors that contribute to the phenotype.
Although the moderate heritability of most behavioural traits is well established, efforts to identify the causal genetic variants have met with limited success thus far. Johnson et al. (2011) consider several possibilities for why this might be and suggest future approaches to advance the field. The major outstanding questions are as follows: how can the causal genetic variants be identified? What will their identification tell us about underlying brain mechanisms? What is the genetic architecture of particular traits in individuals and across populations? What are the limits of genetic effects? What other factors contribute to the individual's behavioural phenotype?
The demonstration of heritability for any particular trait leaves open the crucial question of what the genetic architecture of the trait is. Does it involve the combined effects of multiple variants in each individual, each with small effect alone? Or is the point at which an individual lies upon the phenotypic continuum largely determined by one or a small number of mutations with large effect? And are the genetic variants involved common across the population or rare, possibly even unique to individuals or their close relatives? The answers to these questions will determine the appropriate approach to search for the responsible variants.
For some reason, there is a common misconception regarding the contribution of rare variants to overall heritability, which the authors repeat here. This derives from the correct observation that if a particular variant is very rare, then it will make little contribution to the overall heritability of a trait across a population. Although true, this does not imply that such rare variants, collectively, could not contribute to or even completely explain the heritability of a trait. The mistake is in thinking that heritability is a measure of effects across the population. It is a measure of effects within families, that is averaged across the population. It does not matter whether the genetic variants involved are the same or different in different families. Indeed, de novo mutations will make a disproportionate contribution to heritability estimates derived from twin studies because they will always be shared between pairs of monozygotic twins and never between pairs of dizygotic twins.
There is in fact mounting empirical evidence that phenotypic variance in behavioural traits, like that in many others, is mainly determined by rare variants (McClellan & King, 2010; Mitchell, 2011). First, genome–wide association studies for behavioural traits, including personality (Verweij et al., 2010), general intelligence (Davis et al., 2010), mathematical ability (Docherty et al., 2010), body mass index (a measure of eating behaviour rather than metabolism; Speliotes et al., 2010) and susceptibility to psychiatric disorders (Purcell et al., 2009; Weiss et al., 2009) have all yielded very meagre returns, in terms of the total variance explained. Because of the massive scale of some of these studies, involving hundreds of thousands of individuals in some cases, these negative results positively indicate that common variants do not make a consistent and major contribution to the overall phenotypic variance. In contrast, there is a growing number of rare variants now being identified that have large effects on the behavioural phenotypes of individuals who carry them (Bevilacqua et al., 2010; Mitchell, 2011; Vissers et al., 2010; Walters et al., 2010). These have so far mainly been associated with extreme phenotypes, but there is no reason to think the same model will not apply across the phenotypic range. These findings suggest a far more optimistic outlook for finding specific variants affecting behavioural traits, especially with rapid advances in sequencing technologies (Durbin et al., 2010).
The identification of such mutations will provide entry points to elucidate the neurobiological mechanisms that ultimately impinge on the trait in question. The authors rightly highlight the potential complexity of such mechanisms. This is especially true for a phenotype that is defined at the psychological level, as it may be affected by many different underlying mechanisms. These could include variants affecting any number of neurodevelopmental or neurophysiological processes, ultimately impinging on the structure and function of local circuits and long–range networks.
The next key step will thus be to find ways to better define the phenotypes in question at the neurobiological level (e.g. through neuroimaging), so as to tease out the effects of different mutations and infer the normal functions of the disrupted genes. These may not be obviously related to each other. For example, genes that cause dyslexia when mutated are not genes ‘for reading’. At least some of them are genes for controlling cell migration in the cerebral cortex—when this process is defective, it can indirectly impair white–matter connectivity and communication between areas required for the acquisition of reading expertise (Gabel et al., 2010). We should similarly not expect to find genes ‘for’ other cognitive functions or behavioural traits.
Behavioural genetic studies have also demonstrated the limits of genetic effects on behavioural traits, illustrated by the fact that heritability estimates are far less than one. This observation does not imply, however, that the remaining variance is due to differences in the environment. For most behavioural traits, the shared environment component of variance is very small. The most straightforward interpretation of this is that these traits are not strongly, or at least not consistently, affected by environmental variables. The counter–argument that nonshared environmental variables (such as idiosyncratic experiences) can have an impact whereas shared environmental variables cannot is nonsensical. This is like claiming that something can affect my phenotype only if my identical twin is not also exposed to it.
It seems far more likely that the nonshared ‘environment’ component of variance is largely driven by measurement error and by intrinsic developmental variation. The developmental programme of brain development is complex, intrinsically noisy and variable, and will generate diverse outcomes even from identical starting genotypes. This will always be a significant source of differences between individuals, even identical twins, in the structure and function of neural circuits and networks and is thus likely to impose intrinsic limits on genetic effects on behaviour (Mitchell, 2007). The corollary is that many behavioural traits may be more innate than heritability estimates would seem to imply.
Of course, how those dispositional traits are expressed in terms of actual behaviour will be highly dependent on the environment and the experience of the individual. These important factors may be profitably explored through what might be termed‘genetic sociology’ approaches of the kinds described by the authors in the final section of their article. But the surest route to understanding variation in the traits themselves will be from the identification of genetic variants to the elucidation of neurobiological mechanisms.
Heritability and its Discontents
Marcus R. Munafò1 and Jonathan Flint2
1School of Experimental Psychology, University of Bristol, England, UK.
2Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, England, UK.
In an era of genome–wide association studies (GWAS), where in excess of half a million genetic markers are used to isolate genetic loci associated with a multiplicity of traits, what hope is there for the venerable heritability (h2) statistic? Genetically informative twin and adoption samples can continue to be used to explore the effects of cultural transmission, assortative mating and gene–environment interplay on developmental processes. However, there is growing evidence that the genetic architecture of complex behavioural traits consists of the combined effect of many hundreds, if not thousands, of small effect loci—a model sometimes referred to as quasi–infinitesimal. If this is correct, then we will need much larger sample sizes than currently considered—perhaps consisting of hundreds of thousands of individuals. Although this may seem daunting at present, advances in genotyping technology suggest that we will soon see case–control samples of tens of thousands subject to genome sequencing.
In an era of GWAS, where in excess of half a million genetic markers are used to isolate genetic loci associated with a multiplicity of traits, what hope is there for the venerable heritability (h2) statistic? If the statistic appears to tell us little beyond excluding the possibility of an extreme behaviourist position, its interpretation continues to confound and confuse. Modern incarnations of twin analyses, informed by path analysis and an initial (but subsequently subdued) enthusiasm for deconstructing causal pathways from gene to phenotype, have considerably expanded the information content of h2. Most of what we know of the extent and nature of gene–environment correlation, and gene × environment interaction comes from twin studies, partitioning out the relative contributions to heritability. Nevertheless, as Johnson et al. (2011) describe, understanding exactly what is meant by interaction and correlation in this context is not straightforward (Kendler, 2001; Neale, Roysamb, & Jacobson, 2006).
Heritability estimates tell us how important (or, more rarely, how unimportant) genetic variation is as a contributor to phenotypic variation; they tell us how genes act, through and with the environment, and they tell us whether phenotypes are genetically related. This may be at odds with our views of how diseases are classified: anxiety and depression, for example, turn out to be closely related (Kendler, Gardner, Gatz, & Pedersen, 2007). But, as Johnson et al. (2011) make clear, heritability tells us nothing about the genetic variants out of which it emerges. Given the confusions and inconsistencies in the burgeoning literature dealing with the molecular genetic analysis of behaviour, this is perhaps just as well. Heritability is no guide to the likely success or otherwise of a genetic assault on the molecular basis of behaviour. Phenotypes with high heritabilities (such as autism and schizophrenia) can prove to be far harder to dissect than those with a much lower heritability (such as type 2 diabetes). This is because the single most important factor governing the success of a molecular genetic study is the effect size of the individual loci that contribute to variation. Heritability is the sum of those effects and can be achieved equally well by four loci each contributing 10% of the variation or 4000 loci contributing 0.01%. Those working with heritability are able to remain ignorant of the radically different underlying genetic architecture of those two scenarios, because it matters not at all to the conclusions they reach. For molecular geneticists, however, the first scenario spells success, the second disaster.
Over the last two decades, most attempts to find genes have worked on the assumption that existing knowledge of the neurobiology of behaviour, together with hypotheses about how genes might influence behaviour, would be sufficient to identify some culprits. In using sequence polymorphisms in candidate genes, the question was straightforward: is the frequency of one or other allele significantly higher in people with this psychological characteristic or disease? Nevertheless, it is fair to say that candidate gene association studies have largely failed to fulfil their early promise. Few robust associations between variation in the ‘usual suspects’ of serotonergic and dopaminergic candidates and behavioural traits have been reported (Munafo, 2009). Even if they were to be observed, it is not clear what these associations would tell us—if our choice of candidates is predicated on existing knowledge of the neurobiology of the trait of interest, then candidate gene associations, however robust, can only be confirmatory.
This leads us to GWAS, which continue to grow in both popularity and scale (of sample size and marker density) since their advent around 2007 (Wellcome–Trust–Case–Control–Consortium, 2007). GWAS carry out association at every gene in the genome, rather than just testing one candidate gene, and are therefore agnostic in nature; they require no prior knowledge of neurobiology. As such, they have the potential to lead to genuinely new understanding. One clear example of this is the recent interest in the cluster of nicotinic acetylcholine receptor genes on chromosome 15, and in particular the alpha–5 subunit gene CHRNA5 (Berrettini, et al., 2008). It was previously generally believed that the alpha–4 and beta–2 subunits were of primary importance in nicotine dependence. The identification of a clear signal on chromosome 15 led to renewed interest in the alpha–5 subunit, and recent preclinical evidence confirms that it plays an important role on modulating nicotine self–administration (Fowler, Lu, Johnson, Marks, & Kenny, 2011). GWAS are therefore already beginning to bear fruit.
Despite unarguable successes, GWAS face a problem. They test association at common genetic variants (conventionally defined as alleles with frequencies greater than 5%) and typically identify a handful of significant, replicated associations, each of vanishingly small effect (odds ratios of less than 1.2 are typical; Manolio, et al., 2009). The problem is that when the effects of all confirmed loci are added up, they account for only a small proportion of the total genetic variance (Manolio, et al., 2009). As Johnson et al. (2011) note, this may in part be due to a failure to appreciate what other factors may contribute to a high h2 estimate, such as gene × environment interactions and gene–environment correlations. Some claim that common genetic variation, the sort that GWAS test, is only part of the story (Dickson, Wang, Krantz, Hakonarson, & Goldstein, 2010)—the DISC1 gene has been reported to play a role in schizophrenia, but the contribution of common variation in this gene may be negligible (Mathieson, Munafo, & Flint, 2011). Rare mutations may confer large effects but be difficult to detect except in very large or phenotypically enriched samples.
Beyond this, the importance of epigenetic mechanisms (whereby gene expression is modified by environmental influences) is unknown but likely to be considerable. Johnson et al. (2011) suggest that genetically informative twin and adoption samples, which would previously have been used to calculate h2, can instead be used to explore the effects of cultural transmission, assortative mating and gene–environment interplay on developmental processes. This is a sensible way to use existing samples, because there is no longer much value in simply calculating h2. Some caution is necessary, however. The history of candidate gene studies tells us that over–exuberance in the pursuit of genetic markers for complex behavioural traits resulted in a large literature with an impenetrable signal–to–noise ratio (Munafo, 2009). The recent gene × environment literature compounded this problem by, among other reasons, opening a Pandora's box of subgroup analyses, flexibility in the choice of statistical models to use and variable phenotype (and even genotype) definition (Munafo & Flint, 2009). Genetically informative designs that make use of twin, family and adoption samples will therefore only generate robust new findings if they incorporate the lessons of the past.
How, then, can we ensure success in the future? It would be reasonable to start by accepting the standards adopted by GWAS, such as large sample sizes, independent replication (ideally in larger samples than the discovery sample) and stringent statistical criteria for declaring association (a p–value of 10−8 is regarded as reflecting ‘genomewide significance’). After all, GWAS have been successful for many phenotypes and, all other things being equal, their current standards will serve to increase the likelihood that a reported effect is genuine. Of course, things are not always equal—for example, larger samples can often only be achieved at the expense of phenotype precision. The chromosome 15 signal accounts for ∼1% of phenotypic variance in self–reported cigarettes per day but as much as ∼5% of phenotypic variance in objective measures of nicotine exposure (Keskitalo, et al., 2009). This is simply because two people who smoke 10 cigarettes per day, for example, may do so in very different ways (such as inhaling more or less deeply), but biochemical measures of exposure are expensive and time–consuming to collect compared with self–report measures.
There are, however, other ways of thinking about how to detect genetic effects. For some, the concept of ‘genomewide significance’ is misguided (Wellcome–Trust–Case–Control–Consortium, 2007). Instead, appropriate thresholds can be set by the prior probability of association at each locus rather than correcting for the number of tests performed. We now have enough results to know what that prior probability is: effect sizes expressed as odds ratios are typically less than 1.2 (or the equivalent for continuously distributed phenotypes such as personality). The probability of detecting that effect size can then be estimated from the sample size used in any study, allowing a more nuanced interpretation of the reported p–value (the posterior probability). Simply put, for example, a notionally significant result (i.e. a corrected p–value < 0.05) is a false positive if the study is underpowered. Conversely, we may be able to obtain useful information from much higher p–values than would be conventionally accepted. A good example is Peter Visscher's analysis of the schizophrenia GWAS results. Although very few individual loci achieve conventional genome–wide significance, there is predictive information even in markers with p–values up to .5. This was shown by using results from one GWAS to predict the results in another, independent sample (Purcell, et al., 2009). Wray and Visscher point out that even with 10 000 case subjects and 10 000 controls, power to detect a variant with a relative risk of 1.05 and a frequency of 0.2 at a low threshold of 1 × 10−6 is only 0.2% (Wray & Visscher, 2010). But they then argue that power in the published schizophrenia GWAS (containing only a few thousand subjects) is such that 72% of the time variants of this effect size will feature in the top half of the list of all results. They showed that these single–nucleotide polymorphisms sets were predictive of case–control status, (Purcell, et al., 2009) and, most importantly, they argued that genotyped single–nucleotide polymorphisms accounted for about a third of the variance in liability. In other words, the genetic architecture of schizophrenia consists of the combined effect of many hundreds, if not thousands, of small effect loci—a model sometimes referred to as quasi–infinitesimal. Small effects dominate and are consistent with available data on the genetic architecture of complex traits (Wray, Purcell, & Visscher, 2011).
If this insight is correct, then the successful genetic dissection of behavioural and cognitive phenotypes will need much larger sample sizes than currently considered—perhaps consisting of hundreds of thousands of individuals. Whereas current genotyping costs make it difficult to see how such studies would be funded, molecular technologies continue to improve and costs fall. The first population–scale genome sequencing has recently been achieved (Durbin, et al., 2010), and it is not inconceivable that within the next five years, case–control samples of tens of thousands will routinely be subject to genome sequencing, with even large samples to follow soon after. At that point, we may, at last, begin to understand the molecular basis of heritability.
Acknowledgements
MRM is a member of the UK Centre for Tobacco Control Studies, a UKCRC Public Health Research Centre of Excellence. Funding from the Economic and Social Research Council, the British Heart Foundation, Cancer Research UK, the Department of Health and the Medical Research Council, under the auspices of the UK Clinical Research Collaboration, is gratefully acknowledged. JF is supported by the Wellcome Trust.
Size Matters! Heritability is not Dichotomous
Rainer Riemann, Christian Kandler, and Wiebke Bleidorn
Department of Psychology, Bielefeld University, Bielefeld, Germany
Johnson, Penke, and Spinath (2011) thoughtfully examine the meaning of quantitative behavioural genetics and especially heritability estimates with respect to psychometric effects on heritability estimates and the information that heritability estimates provide on genetic processes underlying complex behavioural traits in humans. We critically examine some of the authors’ conclusions but agree with their central tenet that quantitative behavioural genetic research should further proceed to the study of gene–environment transactions.
In the target, Johnson et al. (2011) stress the limitations of the heritability index as an indicator of the aetiology of psychological traits and the likelihood of discovering genetic associations with behavioural traits. On the one hand, we applaud Johnson and colleagues for thoughtfully reconsidering the meaning of quantitative behavioural genetics and especially heritability estimates. This is very important because all too much myths are associated with the concept of heritability. On the other hand, we critically comment on some of their conclusions basically because they do not properly take into account what can be expected from a heritability coefficient or quantitative behavioural genetics in general.
By stating ‘everything is heritable’, Johnson et al. (2011) point out their conviction that there are genetic effects on any systematically manifested behaviour and the differences in the size of this influence are not worth to be considered. Indeed, most studies confirm a moderate heritability of personality and ability traits and falsify, for example, Cattell (1950) distinction between ‘environment mold’ and ‘constitutional traits’ and the idea (e.g. Strelau, 1987) that among other criteria, temperament traits can be distinguished from personality traits by the size of their respective heritability. Finding moderate genetic effects on many personality traits is remarkable indeed, possibly reflecting that most personality traits are jointly affected by several biological systems (e.g. McCrae, Jang, Livesley, Riemann, & Angleitner, 2001) and the almost exclusive reliance on the self–report method that is itself genetically influenced (e.g. Kandler, Riemann, Spinath, & Angleitner, 2010).
To dismiss differences in heritability estimates between personality measures is—at best—not helpful. Although it is comfortable for a divided scientific community to conclude that genes are about as much important for explaining individual differences as environmental factors are, this position ignores important and theoretically meaningful distinctions. Constructs (such as the dimensions of the Five–Factor Model or general mental ability) measured by multiple methods with reference to ‘longer’ time intervals yield heritability estimates well above .5 (Kandler, Riemann, Spinath, & Angleitner, 2010; Riemann, Angleitner, & Strelau, 1997), whereas the heritability of (reliable or latent) state or situation specific measures is substantially smaller than .50 (Riemann, Angleitner, Borkenau, & Eid, 1998; Borkenau, Riemann, Spinath, & Angleitner, 2000; Borkenau, Riemann, Spinath, Angleitner, 2006). These differences are predictable from trait theory; however, this does not make them trivial. There a numerous other examples (some of them are presented by Johnson and colleagues) that demonstrate that heritability estimates differ substantially and predictably between trait measures (Borkenau et al., 2000). The study of these differences between social settings (environments), age groups, sexes, or (sub–)cultures is the means to study gene–environment transactions, a goal we share with Johnson and colleagues.
It would really be surprising, if heritability estimates, which in humans exclusively rely on correlational methods, would not be affected by the well–known psychometric principles. Heritability is the ratio of genetic variance to total variance. Thus (all else being equal), a reduction of measurement error results in a higher heritability estimate for this measure, if there is genetic influence on that particular measure. Correction for attenuation or the calculation of heritability estimates for latent variables is required to compare heritability estimates. With respect to the aggregation principle, we should keep in mind that the aetiology of the covariation among measures has a crucial effect on the heritability of the aggregate. Single behavioural measures may show little genetic influence (such as state measures or measures of behaviour in specific situations), and yet, their aggregate may show substantial heritability, if the covariation among measures is mainly due to genetic factors. To the contrary, aggregates of robust personality measures with a well–established genetic aetiology (e.g. domain scores on the NEO Personality Inventory Revised) may be less heritable than their component measures (Riemann & Kandler, 2010). What is true of aggregation analogously holds for the temporal stability of measures.
Neither aggregation nor reliability nor temporal stability ‘contribute’ to heritability. Behaviour registration (mechanical or observational) or the registration of important life events (e.g. number of arrests) can be accomplished almost error free and yet results in zero or very small heritability estimates. Heritability of temporally stable measures tends to be higher than for temporally instable ones because of explicit aggregation over time (like suggested by Epstein, 1979) or by raters’ implicit averaging of behaviours or experiences. This is not an artefact but tells us as much about the underlying biology as a heritability coefficient can tell us: effects of genetic variance show up more clearly in broad, temporally stable, reliable, multi–source measures of individual differences.
Some of the arguments put forward by Johnson et al. (2011) against heritability estimates are quite meaningless, if we have in mind what heritability is. It is a population statistic that is not defined for an individual, it considers variance not constants and is based on variance ratios not the absolute size of variance components. Thus, of course, heritability studies cannot answer all the important questions put forward by Johnson and colleagues. Just like a correlation does not inform us about the underlying processes that link two variables, heritability estimates do not inform us about the genetic processes underlying individual differences. If we want to study these processes in detail, heritability estimates serve as the best guide to select measures to be used in molecular genetic studies in the absence of additional knowledge (Visscher, Hill, & Wray, 2008). Modern quantitative behavioural genetic studies, which of course go beyond estimating the heritability of just another trait, are based on a theoretically sound measurement models, models of genetic as well as environmental influence on a trait.
Johnson et al. (2011) give numerous examples of possible distortions of heritability estimates. This list of manifold genetic processes that might play a role in the aetiology of complex behavioural traits is impressive and important. Some of these processes (such as gene–environment correlation and interaction) can and should be controlled in quantitative behavioural genetic studies by collecting measures of the environment. Since, for example, epigenetic effects have not been described for behavioural traits in humans, there is no way to estimate whether and to what degree they distort heritability estimates(see Hill et al. for dominance and epistasis effects).
In sum, quantitative behavioural genetic studies are important. They shed light on questions on top–down or bottom–up genetic influence, contribution of genes to stability and change, on gene–environment covariance and interaction, on environmental effects of traits controlled for genetic effects and many more. And of course the answers to these questions imply differential heritability.
Equifinality and Endophenotypes Enrich Personality Research
Susan L. Trumbetta1 and Irving I. Gottesman2
1Department of Psychology, Vassar College, Poughkeepsie, NY, USA
2Departments of Psychiatry and Psychology, University of Minnesota—Twin Cities, Minneapolis, MN, USA
Heritability estimates for complex physical or mental phenotypes often reflect a composite of genetically influenced processes. The concept of equifinality is critical to understanding how genetic and epigenetic factors may lead to phenotypic personality. The endophenotype strategy should help identify and parse some of the aetiological heterogeneity we see under conditions of equifinality as it identifies genetically influenced mechanisms that give rise to observable phenotypes. The endophenotype strategy also should help illuminate the nature of yet–uncharacterized genetic correlations between related behavioural phenotypes.
Heritability estimates are complex, and their complexities rest, in part, upon underlying complexities of the genome, gene expression, human development from conception through the lifespan, environmental factors (some correlated with genotype), and their diverse interplay, as well as upon the critical processes of genotypic, environmental, and phenotypic measurement. Johnson, Penke, and Spinath's paper (2011) thoughtfully illuminates these intricacies as they also sound some necessary cautionary notes about the limitations of evolutionary inference from simple heritability estimates and also about the failure so far of genome–wide association studies to account for very much of the genetic variance expected from heritability estimates. In some ways, they are preaching to a choir that already appreciates their admonitions, but even the choir appreciates occasional inspirational preaching to keep up their morale. Increasing phenotypic complexity for physical (Wang & Snieder, 2010) and for mental variables continues to reveal itself, as a recent study moved beyond a simple gene by environment interaction to explore a more complex three–way interaction of the brain–derived neurotrophic factor val–66–met polymorphism with the 5–HTTLPR length polymorphism and with obstetric complications, childhood events and long–term difficulties as they separately and in combination influenced the neuropsychological phenotype of adolescents’ effortful control (Nederhof et al., 2010). Given the sheer size and complexity of the genome, plus the myriad trajectories of environmental and developmental processes that may co–act and interact with genotype, and the multiple possible levels of gene by gene and gene by environment interactions, as well as other non–linear dynamics (cf. the burgeoning field of epigenomics, C. G. Bell & Beck, 2010) and the ongoing emergence of de novo mutations, it is no wonder that all of our known polymorphisms still leave large proportions of known genetic variance unspecified. This ‘dark matter’ of the genome remains a frontier to be explored (Manolio et al., 2009; Wang & Snieder, 2010) because it is still quite early in the game.
Complex phenotypes, by definition, may arise as a result of multiple processes, and there may be more than one path to any given phenotypic outcome. This principle of equifinality (von Bertalanffy, 1967), in which multiple developmental and mechanistic processes may ultimately lead to the same phenotypic outcome, may help ultimately in mapping the genome's dark matter. People of the same adult height, for example, may arrive there from different birth lengths and through different patterns of growth spurts, as well as by diverse patterns of nutrition, parenting, and exercise across the lifespan, all associated with different combinations of genetic polymorphisms activated at different times. Each bone contributing to linear height has its own complex story to tell. For complex behavioural phenotypes, one also can imagine a similar variety of developmental and mechanistic pathways. The ability to separate and characterize these pathways will be essential to understanding the full range of genetic effects on a phenotype that shows equifinality.
How is this related to heritability? When any phenotype shows equifinality, then the heritability estimate for that phenotype is essentially a composite of diverse genetic effects. At a genetic level, this composite may include the effects of a few rare genes of large effect and any number of genes of small effect across any number of loci, whether inherited from a long parental line or the result of rare de novo mutations (Visscher et al., 2008). This genetic diversity occurs against a background of similarly diverse environments and developmental processes. A first step in detecting the so–called missing heritability may be to identify and separate some of the underlying, heritable traits and processes that give rise to the same phenotype. These heritable traits and processes, typically inaccessible to the unaided eye, may be revealed through neuroscans, genome–wide association studies, assays and other means. The increasing use of this endophenotype strategy (Gottesman & Shields, 1972; Gottesman & Gould, 2003) may aid in the detection of the genome's ‘dark matter’, insofar as endophenotypes represent traits and processes more proximal to immediate genetic effects than are more distal, obvious personality phenotypes. With luck, they may even be less complex than the complex phenotype under study. The endophenotype approach may help parse the composite of genetic effects on behavioural phenotypes by examining genetic effects on more specific aetiological pathways involving various neurotransmitter systems, both facilitating and suppressing. In combination with twin–family studies that also control for selection into relevant environments, and with Mendelian randomization studies that may characterize the effects of different modifiable environments on the phenotypes of individuals possessing polymorphisms of known effect, the use of endophenotypes should illuminate the many pathways to a single behavioural phenotype as well as the nature of yet–uncharacterized genetic correlations between behavioural phenotypes.
(Mis)understanding Heritability
Peter M. Visscher1 and Matthew C. Keller2,3
1Queensland Statistical Genetics laboratory, Queensland Institute for Medical Research, Brisbane, Queensland, Australia
2Department of Psychology and Neuroscience, University of Colorado at Boulder, Boulder, CO, USA
3Institute for Behavioral Genetics, University of Colorado at Boulder, Boulder, CO, USA
Johnson, Penke and Spinath (2011) review the concept of heritability in the light of previous findings from genetically informative designs and of modern findings from molecular genetics. Several of their points about the interpretation of heritability and the search for genetic variants underlying trait variation appear to us to obscure rather than clarify the state of behavioural genetics. In this response, we review points of disagreement with the hope of simplifying and clarifying fundamental concepts in the field.
We applaud Johnson et al. (2011) for revisiting the scope and limits of statistical inference from empirical data regarding the genetics of human behaviour, in particular in the light of the modern tools of molecular genetics. Our comments centre on points of agreement and subsequently on point of disagreement and what we consider to be misleading or erroneous statements.
The points of agreement are many; we touch on a few that we wish to highlight. Variation between people in most if not all aspects of human behaviour is in part due to genetic factors, and the scientific question is not if there is genetic variation but how much of the variation in the population is related to genetic differences between individuals. Increasingly, even this question is becoming passé, and scientists have moved towards trying to understand this variation in terms of individual gene variants and their interaction with other genetic variants and the environment.
We agree with the authors that there are potential biases and inaccuracies involved in estimating heritability so that heritability estimates should not be over–interpreted (Keller, Medland, & Duncan, 2010). The information to estimate heritability for human behavioural traits from twin studies comes essentially from twice the difference between the observed phenotypic correlation of MZ twins and that of DZ twins, and there may be non–genetic factors that cause the difference in MZ–DZ correlations to be positive. We also fully endorse the comments about the difficulties in inferring the types of evolutionary selection from apparent levels of additive versus non–additive genetic variance, and we applaud the authors’ attempts to add proper nuance into the interpretation of heritability; see for example their discussion of the ‘Flynn effect’. The Flynn effect is a classic example of a false paradox because high heritability says nothing about the cause of differences between groups (e.g. mean differences between people today and people 20 years ago or mean differences between different populations of humans; Visscher, Hill, & Wray, 2008).
There were several statements by Johnson et al. (2011) about heritability and attempts to understand the molecular basis of that heritability that we found obfuscating when they could have been elucidating. We review these in the hope of clarifying what otherwise might confuse readers not versed in quantitative genetics:
As Johnson et al. (2011) point out, animal and plant breeders focus on narrow sense heritability because additive variation is the raw material for selection. It is not additive genetic variation that is transmissible to the next generation (cf. Johnson, Penke & Spinath, 2011, p. 11) but rather gene variants and their additive effects. Response to selection can be expressed in the so–called ‘Breeder's Equation’, R = h2S (Falconer & Mackay, 1996). S is the difference between the mean of the population and the mean of the individuals that reproduce and is not the same as the truncation point for selection (cf. Johnson, Penke & Spinath, 2011, p. 12). We found confusing the authors’ use of the Breeder's Equation to argue that ‘…quite substantial heritability does little to create actual similarity among family members’ (Johnson, Penke & Spinath, 2011, p. 12). The difference in phenotypic scores between relatives (e.g. MZ twins) is a function of their correlation only. If the MZ correlation is rMZ = .80, then the SD of their pairwise differences is √(2(1 − rMZ)) SD units or 0.63. The SD of differences scores between unrelated individuals is √(2(1 − 0)) or 1.41, for a ratio of ∼.45. (In all cases, the mean of the difference scores is 0, so it is not sensible to compare the mean differences, cf. Johnson, Penke & Spinath, 2011, p. 12). So given high familial correlations, relatives are indeed much more similar to one another than they are to randomly chosen individuals, as should be expected. Johnson et al. (2011) state that the heritability of a rare Mendelian disease is essentially zero because the causal alleles are rare. However, the authors appear to confuse the estimate of heritability from a sample in which nobody is affected with the true heritability in the entire population, where affected individuals do exist. In fact, the broad sense heritability of a Mendelian disease, measured on an all–or–none scale, is 1.0 because there is a perfect relationship between genotype and phenotype. The narrow sense heritability, on the other hand, is ∼1 for a rare dominant disease and ∼0 for a rare recessive disease. Johnson et al. (2011) state (p. 17) that epistasis causes the appearance of additive genetic variance. However, the variance generated by non–additive effects at the molecular level is partly additive at the population level. The simpler example is a single locus model with dominance. The additive genetic variance is 2p(1 − p)[a + (1 − 2p))d]2, with 2a the difference between the two homozygotes and d the deviation of the heterozygote from the mean of the two homozygotes (Falconer & Mackay, 1996). So long as p, the allele frequency, is not .5, dominance genetic effects contribute to the additive genetic variation. In their section on attempts to find the genes underlying trait heritability (p. 20–24), Johnson et al. (2011) claim that results of genome–wide association studies have ‘mystified’ geneticists and seem to suggest that the complexity of the system is a reason for the missing heritability. In fact, there is little need for mystification or appeals to ‘complexity’ (e.g. epigenetics, copy number variants or regulatory effects) to make sense of current findings in genetics. Heritability studies show that variation in additive genetic effects exists, regardless of how complicated the pathway between genes and the phenotypes is, and so in principal the alleles behind those effects should be detectable. Indeed, there is now overwhelming evidence across a range of traits and diseases that associated single–nucleotide polymorphisms (SNPs) can be reliably detected, although Johnson et al. (2011) rightly point out that the sum of the variance explained by these reliably associated SNPs has been small (<10%) to date. Johnson et al. (2011) are right that association studies cannot detect signals from very rare causal variants, but recent advances have begun to shine light on the importance of common variants. Yang et al. (2010) estimated that over half the genetic variation in height is explained by considering all SNPs simultaneously, that is due to correlations (linkage disequilibrium) between the unknown causal variants and the common SNPs on the commercial genotyping arrays. Because rare alleles (with frequencies <.01) tend to have very low correlations with common variants, this evidence strongly suggests that a substantial amount of the heritability of height is due to alleles with frequencies over .01. Similar findings are forthcoming for several other complex phenotypes. Thus, alleles responsible for much of the heritability in complex traits have not previously been detected because their individual effects are too small to pass stringent significance tests, and larger sample sizes are likely to uncover more associated SNPs. It can be debated whether the quest to find as many SNPs that explain as much trait variation as possible is the best way to make progress in understanding the genetic basis of traits. However, it is neither scientifically useful nor didactically instructive to hand wave towards ‘complexity’ when trying to explain the current state of genome–wide association studies findings.
In conclusion, we appreciate the review by Johnson et al. (2011) and agree with many of their general points regarding the interpretation of heritability but disagreed on a number of technical points, in particular those relating to understanding the genetic basis of complex trait variation in human populations.
Footnotes
1
Of note, these same risk and protective factors may also interact or correlate with genetic risk. For example, parent–child conflict accounts for 23% of the shared environmental influences on adolescent externalizing, even though it also shares genetic and non–shared environmental influences with delinquency (Burt et al., 2003). Such findings suggest that conflict contributes to externalizing both via main effects and via GxE or rGE (although we can only be sure of the former, given that traditional twin models do not discriminate between GxE and rGE).
2
Although originally dismissed as universally small and non–significant, recent meta–analytic work has indicated that shared environmental influences on common forms of psychopathology are moderate in magnitude prior to adulthood (accounting for 10–30% of the phenotypic variance; see Burt, 2009). It is less clear whether this pattern of effects persists to personality per se, however.