
Abstract
OBJECTIVES: The study goals were (1) to determine if the degree and pattern of semicircular canal dysmorphology and the presence or absence of a cochlea in patients with congenital sensorineural hearing loss predict audiologic outcome, severity, or the frequencies involved and (2) to review the recent advances in molecular genetics of the semicircular canals and correlate this information with audiologic and anatomic patterns seen in our series of patients
DESIGN AND SETTING: We conducted a retrospective study at a tertiary care center with a large otologic and cochlear implant service.
PATIENTS AND METHODS: The study population consisted of 16 patients with congenital sensorineural hearing loss in 28 congenitally malformed inner ears consisting of semicircular canal dysplasia or aplasia, with or without cochlear malformation. History, physical examination, computed tomography scans, and serial audiograms were reviewed. Factors analyzed included other phenotypic dysmorphology characteristic of syndromes, audiometric configuration, severity and type of hearing loss, and the presence of associated inner ear anomalies other than the vestibular system. An extensive review of the literature regarding molecular genetic factors in semicircular canal anomalies, with or without cochlear abnormalities, was performed.
RESULTS: Sixteen patients (31 ears) were identified with profound sensorineural hearing loss and semicircular canal abnormalities. Only 3 patients had known syndromes, although 4 patients had other congenital anomalies. Most radiographic detectable abnormalities were bilateral. Audiograms of the patients demonstrated pure tone averages between 90 and 100 dB in the affected ears with few exceptions. No correlation was found between type and severity of malformation of either the cochlea or semicircular canals with the severity of hearing loss. There was no stepwise progression of hearing loss increasing malformation severity. Seven of the 16 patients received cochlear implants. Of these 7, 3 patients had cochlear hypoplasia and 1 patient had a common cavity deformity. Audiologic follow-up on all 7 patients revealed improvement in both speech assessment threshold and pure tone average. Presence or absence of the cochlea was not a factor in outcome after cochlear implantation.
CONCLUSION: We have assembled the largest series of patients with semicircular canal dysmorphology, with or without various cochlear abnormalities. Our study failed to correlate the type and severity of semicircular canal malformation with any specific audiologic outcome. The variation in hearing loss severity and pattern even in patients with similar bony radiographic findings must be explained by other non-radiologically detectable defects, likely abnormalities in membranous labyrinthine development. New molecular genetic discoveries have linked specific genes to the development of certain inner ear structures in mice studies. The independent development of the individual semicircular canals in relation to the cochlea and vestibule and the variability in hearing loss suggest a more complex embryologic process than merely an arrest in development as previously thought. As genetic studies are extended into humans, we will likely be able to stratify these patients by molecular defect and severity of hearing loss. (Otolaryngol Head Neck Surg 2003;129:637-46.)
The adult inner ear is divided into an auditory apparatus, consisting of the organ of Corti in the cochlea, and a vestibular apparatus, consisting of the saccule, utricle, and semicircular canals (SCCs). These structures are composed of a membranous labyrinth encased in a bony labyrinth, situated in the petrous portion of the temporal bone. The inner ear develops from a thickening of ectoderm on the lateral surface of the neural tube termed the otic placode. During week 4 of human gestation, the otic placode invaginates into underlying mesenchyme, forming the otic vesicle (otocyst). The mesenchyme surrounding the otocyst is the precursor of the cartilaginous capsule of the otocyst termed the otic capsule.
The membranous labyrinth is composed of the pars inferior, which gives rise to the membranous cochlea and saccule, and the pars superior, the phylogenetically older structure, which gives rise to the SCC, utricle, and endolymphatic duct. The vestibular structures begin development during week 6 of human gestation and attain adult configuration by 8 weeks. SCC embryogenesis first appears as diverticular outpouchings in the dorsal portion of the otocyst during week 6 of gestation. The lateral wall of these outpouchings delaminates from the underlying mesenchyme and grows toward the medial wall, forming a fusion plate. This fusion plate eventually disappears to form the characteristic closed tubular form of the SCC. 2 The lateral SCC (LSCC), also called the horizontal canal, is the last to mature and is more susceptible to anomalous development. The membranous cochlea differentiates from the ventral portion of the otocyst at week 7 of gestation. The number of turns of the cochlea increases with progressive development. The adult configuration of 2 ½ to 2 turns form by week 8 of gestation. The endolymphatic sac forms from the dorsal portion of the otocyst at week 6 of gestation and is the only inner ear structure to continue to grow after birth. The hair cells and auditory sensory network are largely complete by week 26 to 28 of gestation.
The bony labyrinth encloses the membranous labyrinth and forms in 3 stages. The initial stage occurs between weeks 4 and 6 of gestation and consists of mesenchymal condensation surrounding the membranous labyrinth. The second stage involves the formation of the bony vestibule, which encloses the utricle, saccule, and cochlear duct. The perilymph-filled scala tympani and scala vestibuli surrounding the endolymph-filled cochlear duct form at this time. The third stage involves ossification of the otic capsule, which begins at week 15 of gestation. Ossification begins in 14 centers and is complete by week 23 of gestation, resulting in the formation of the petrous portion of the temporal bone.
Jackler et al 2 published a classification system in 1987 to standardize the description of inner ear abnormalities. They noted that the hierarchy of radiographically detectable malformations corresponded to the embryologic development of inner ear structures, indicating that the spectrum of malformations can be explained by an arrest in the normal development of the inner ear at different times. Their classification system included malformations of both the osseous and membranous labyrinths, based on cochlear involvement. The theory behind their classification system focused on arrest in development at various stages of embryogenesis. For example, an arrest in development at the primitive otic placode would result in complete labyrinthine aplasia—the Michel deformity. Failure of the otocyst to differentiate would result in the common cavity deformity, first described by Edward Cock. Arrested development of the cochlear bud would result in cochlear aplasia with a normal vestibule and SCC. Arrest in progressive cochlear development would result in varying degrees of cochlear hypoplasia. The classic Mondini deformity is formation of the basal turn of the cochlea with a sac instead of the apical turns. Other variations in malformations may result from aberrant rather than arrested development. This was an attractive hypothesis to explain the myriad of malformations that were seen in radiographic studies. 2 However, Parnes and Chernoff 3 challenged the hypothesis in 1990 when they were the first to report 2 cases of bilateral SCC aplasia with concomitant normal or near-normal cochlear development. This report conflicted with the traditional hypothesis that malformations of the inner ear resulted from arrest of development since the SCC develops during week 6 of gestation and the cochlea develops during week 7 of gestation.
This is a retrospective study of 16 patients with 28 congenitally malformed labyrinths. In light of recent molecular genetic discoveries linking specific genes to development of certain inner ear structures in mice, we also present a discussion of these studies. The discovery of homologous genes that, when mutated, cause cochlear and vestibular aberrancies in mice may have many applications in humans. As genetic studies are extended into humans, we will likely be able to stratify patients by molecular defect.
PATIENTS AND METHODS
Patients from 16 different families were compiled through the Department of Otolaryngology at a tertiary care university hospital with a large otologic and cochlear implant (CI) practice. All patients were identified by temporal bone computed tomography (CT), which were obtained for the evaluation of SNHL and possible cochlear implantation. The criterion for inclusion in this study was unilateral or bilateral SCC dysplasia or aplasia identified by CT, regardless of the status of cochlear development. We did not exclude or specifically target patients with known syndromes or other phenotypic abnormalities. The audiograms of each patient were then reviewed to determine if (1) the severity of dysmorphology correlated with the severity of hearing loss, (2) the unilateral deformities correlated with normal hearing on the contralateral side, and (3) certain frequencies or audiologic patterns were more commonly affected with certain anatomic patterns. Audiograms from initial presentation were analyzed and compared with post-CI audiogram, if applicable, to determine whether the severity of radiologic abnormality implied poorer outcome after cochlear implantation.
CT findings were divided into ossicles, vestibule, cochlea, vestibular aqueduct, SCC (lateral, posterior, superior), external auditory canal (EAC), and oval window. Radiologic interpretations were based on the following definitions: small cochlea (<7 mm in diameter measured vertically [normal, 8 to 10 mm]), enlarged vestibule (>5 mm in vertical dimension), and enlarged vestibular aqueduct (>2 mm in diameter in its intraosseous portion). Mondini malformation was defined as formation of the basal turn of the cochlea with a sac instead of the apical turns.
RESULTS
Sixteen patients (28 ears) were identified (Table 1). A majority of the cases were nonsyndromic. Three patients had defined syndromes: 2 children with CHARGE (Coloboma, Heart defects, Atresia choanae, Retardation of growth and development, Genitourinary problems, and Ear abnormalities) association and 1 with Goldenhar syndrome. Three patients had other nonotologic congenital anomalies: specifically tetraology of Fallot, esophageal atresia, and cleft lip/palate. Only 2 patients had abnormalities of the middle ear, consisting of a lateralized incus and fusion of the malleus and incus. This was not surprising considering the distinctly separate embryologic origins of the middle and inner ear.
Twelve patients had bilateral deformities, with anatomic patterns on each side usually identical. Those with unilateral deformities (patients 10, 12, 15, and 16) demonstrated normal hearing on the unaffected side and profound SNHL on the radio-graphically abnormal side.
Six of 16 patients had complete aplasia of the SCC (12 ears). Of the 12 ears, 7 had normal cochleas and vestibules, 1 had bilateral cochlear hypoplasia, 1 had an ipsilateral Mondini malformation, and 1 had bilateral Mondini malformations. Two patients had common cavities (3 ears) with associated malformation of the SCC. Five patients had classic Mondini malformations (9 ears). Of these 5 patients, 1 had SCC aplasia, 3 had SCC dysplasia, and 1 had preserved SCC with only a dilated crus of the superior canal noted. Three of the 16 had isolated lateral SCC dysplasia (patients 6, 15, and 16). One of the 16 had isolated superior SCC dysplasia (patient 11). Three of the 16 had complete aplasia of the posterior SCC (patients 1, 2, and 10), with some development of the lateral and superior SCC. Of these 3, patient 10 had Goldenhar syndrome with normal lateral and superior SCC bilaterally and a normal posterior SCC on the contralateral side. Patients 1 and 2 had dysplastic but present lateral and superior SCC. The vestibular aquaduct was normal bilaterally in all cases.
Computed tomography findings of patients with congenital sensorineural hearing loss and semicircular canal abnormalities
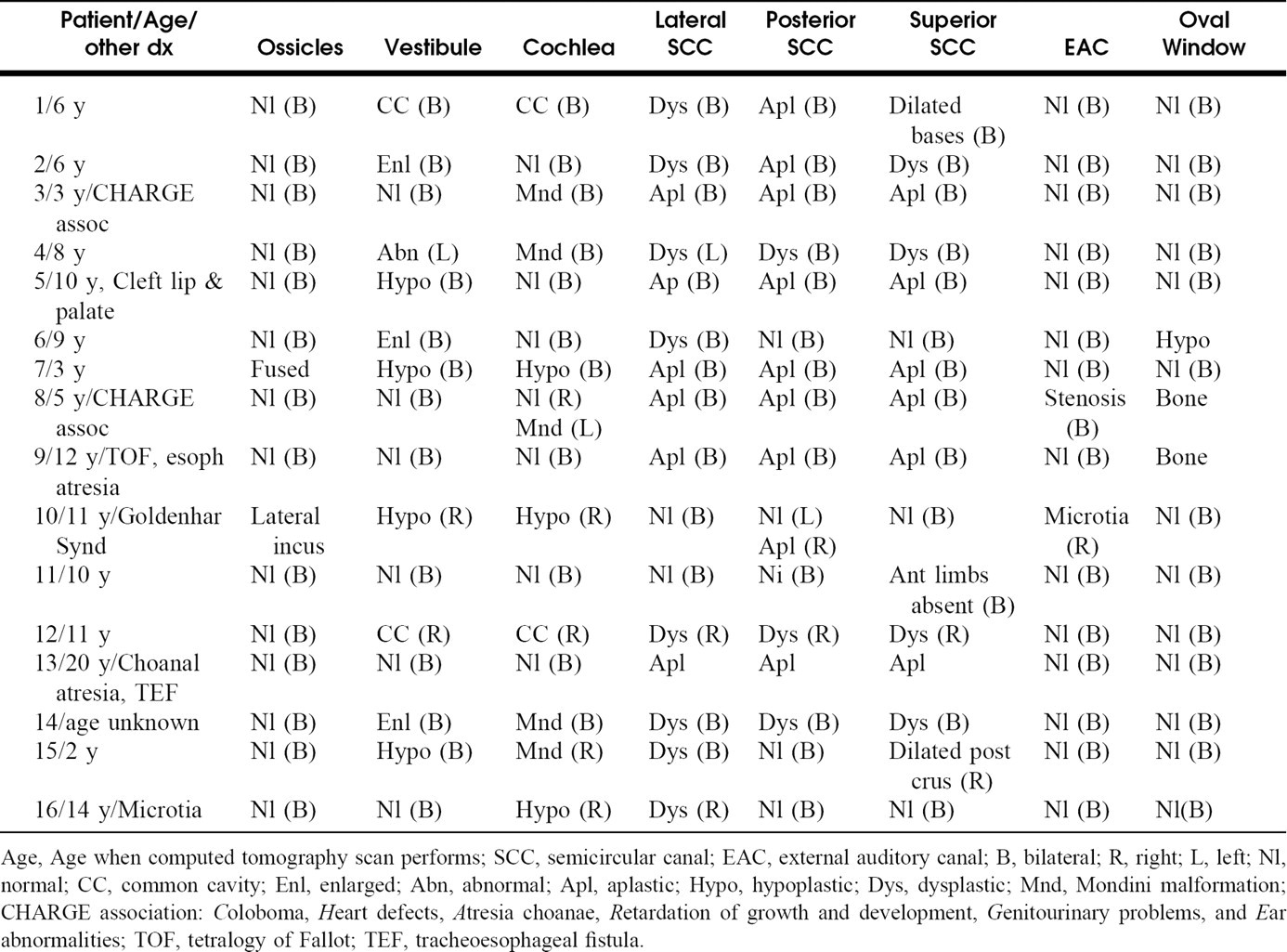
Age, Age when computed tomography scan performs; SCC, semicircular canal; EAC, external auditory canal; B, bilateral; R, right; L, left; Nl, normal; CC, common cavity; Enl, enlarged; Abn, abnormal; Apl, aplastic; Hypo, hypoplastic; Dys, dysplastic; Mnd, Mondini malformation; CHARGE association: Coloboma, Heart defects, Atresia choanae, Retardation of growth and development, Genitourinary problems, and Ear abnormalities; TOF, tetralogy of Fallot; TEF, tracheoesophageal fistula.
Audiograms of the patients demonstrated pure tone averages (PTAs) between 90 and 120 dB in the affected ears with a few exceptions (Table 2). There was no PTA better than 55 dB at the time of initial presentation. The data demonstrate no specific patterns of audiologic findings that would correlate with the severity of cochlear radiographic findings. Patients with deformities that preserved the cochlear architecture did not consistently have better hearing than patients with cochlear malformations. For example, patient 12 with a common cavity had the best PTA in this series of patients.
Seven of the 16 patients received a CI (Table 2). Age at implantation ranged from 1.8 to 6 years. Implanted cochleas ranged from normal (3 patients) to hypoplastic (3 patients) to a common cavity (1 patient). There were no intraoperative or postoperative complications.
Audiologic follow-up was available for 0 to 12 months after implantation. Improvements in speech assessment thresholds (SAT) and PTAs were independent of cochlear maldevelopment (Table 2). The common cavity patient had a good result with an SAT of 30 dB and a PTA of 35 dB. The patients with cochlear hypoplasia/Mondini malformation had postoperative SATs of 10 to 50, whereas the patients with normal cochleas had SATs ranging from 5 to 25 dB. Patient 3 has CHARGE association and at 1-year follow-up, his auditory brainstem response (ABR) indicated responses but his performance remains poor with PTAs of 60 to 80 dB. Although with only 7 patients with CI it is not possible to prove statistical significance, all of the implanted patients achieved improvement in both SAT and PTA.
Radiographic findings with associated audiologic data in patients with sensorineural hearing loss and semicircular canal abnormalities
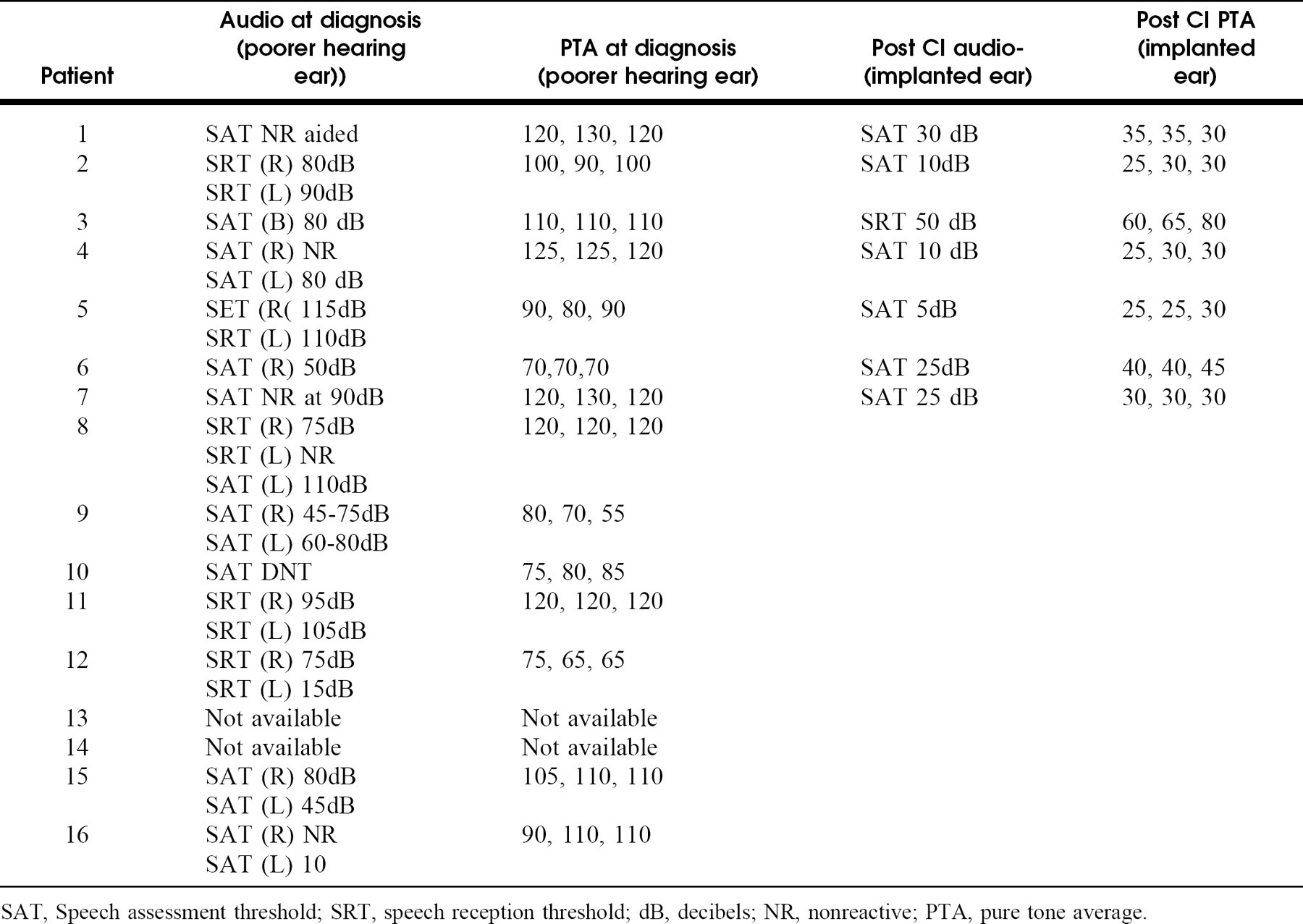
SAT, Speech assessment threshold; SRT, speech reception threshold; dB, decibels; NR, nonreactive; PTA, pure tone average.
DISCUSSION
Of the patients with congenital SNHL, 30% demonstrate abnormalities on radiographic studies, emphasizing the significance of both the membranous and osseous components of the inner ear with regard to functional hearing. 4 As a general rule, it was believed that the more severe the radiographic abnormality, the poorer was the hearing. However, this was not the case in our study. There was variation in hearing loss frequencies even among patients with very similar osseous anomalies. Our analysis failed to find any correlation between severity of hearing loss and frequencies involved with radiographic abnormality. The hearing loss associated with SCC dysplasias is most likely due to anomalous membranous labyrinth development, which is not radiologically detectable.
The hierarchy of development or maldevelopment of the SCC is most likely not simply an arrest in embryogenesis, as has been previously suggested. The high degree of variability in radiologic findings or associated cochlear malformations argues against a single classification system based on embryogenic arrest. With the advent of new molecular genetic information, a much more complex story evolves regarding the etiology and pathogenesis of SCC anomalies.
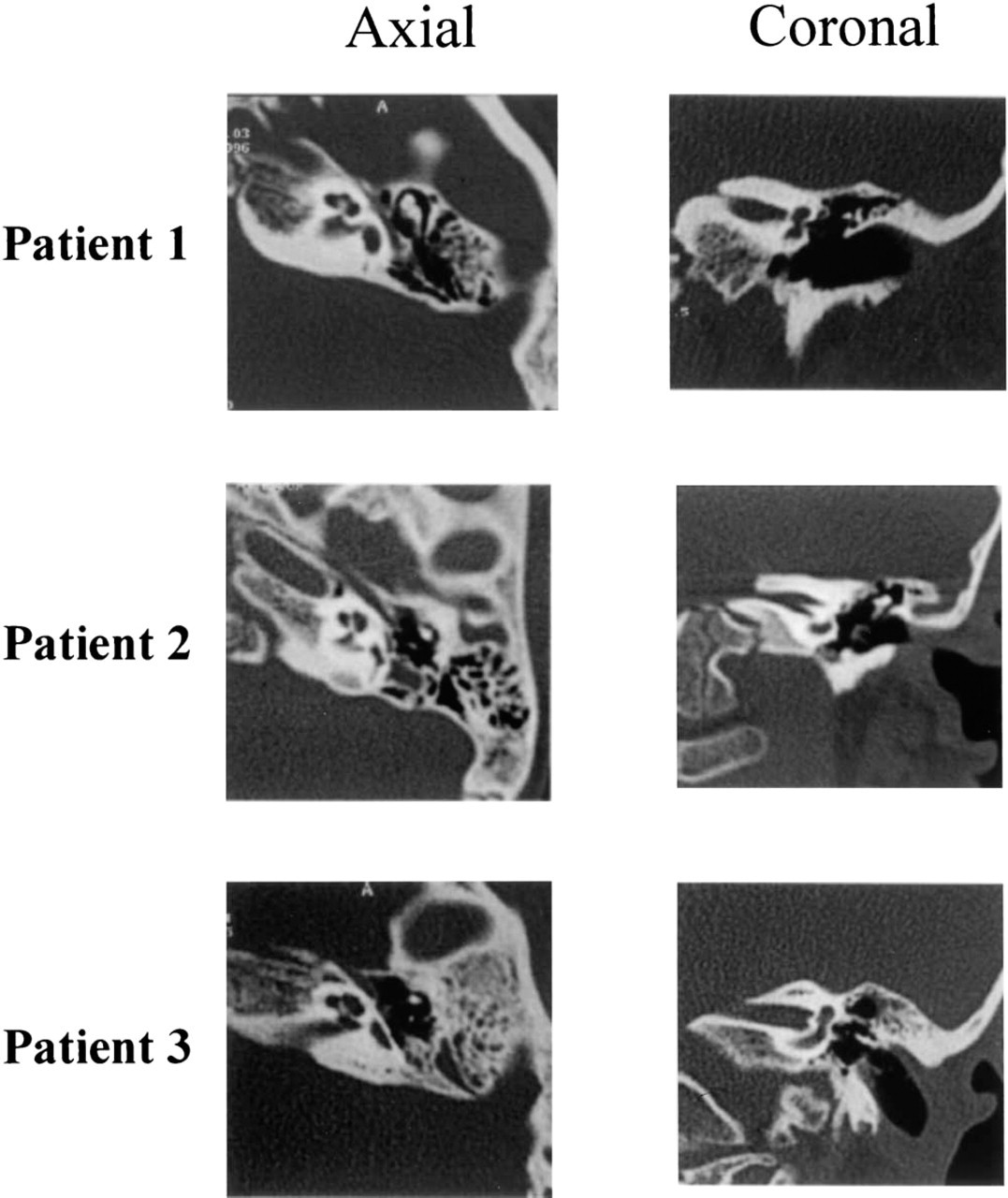
Axial and coronal CT scans of these 3 patients with SNHL demonstrate complete aplasia of the SCCs with normal development of the cochlea and vestibule.
Molecular genetic research is progressing at an astonishing rate, fueled by innovative technologies such as knockout mice, which allow researchers to examine the effect of a single gene inactivation. Disruption of specific genes can result in gross structural abnormalities, as seen in patients reviewed here, or in disruption of cellular functions within a normal bony labyrinth and cochlea. 5 This discussion concentrates on the former type of mutations.
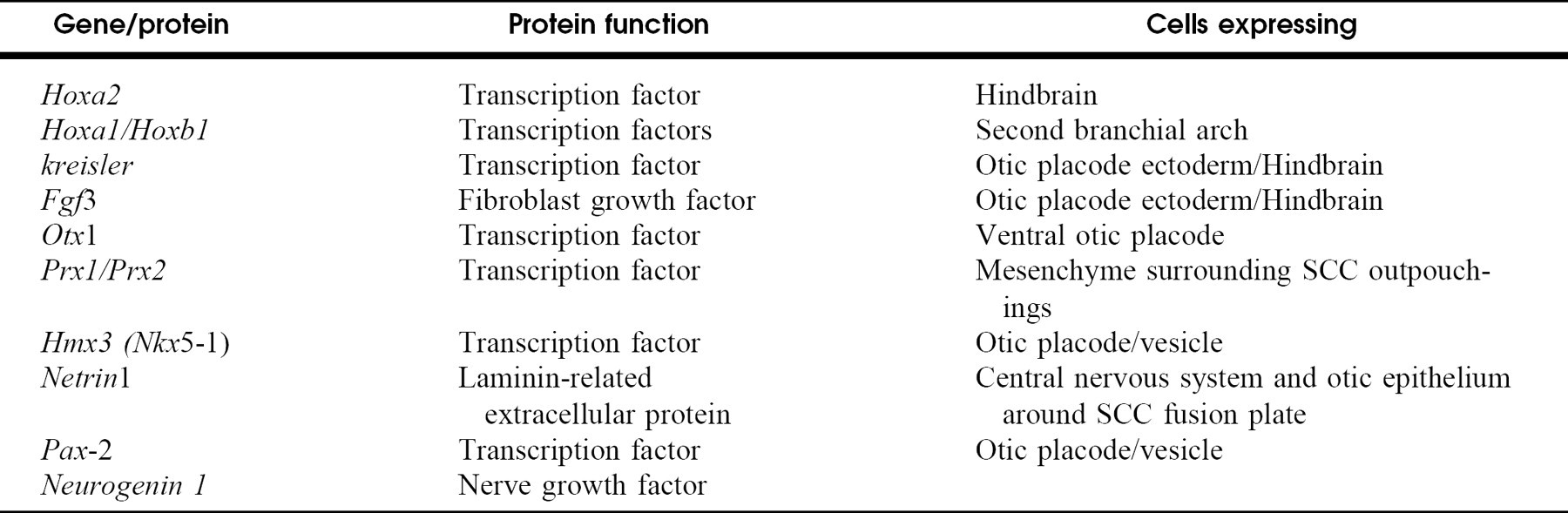
Genes with mutations leading to gross anatomic alteration of the inner ear of mice generally encode transcription factors (Table 3). Mutation in these genes results in a variety of inner ear defects with variable penetrance (Fig 2). Transcription factors are proteins that modulate the expression of other genes. During embryonic development, transcription factors serve critical roles as switches in the elaboration of general body plan, organ commitment, and differentiation of specific cell types. Most of the genes discussed display variable penetrance of abnormalities, even in knockout mice. Transcription factors ultimately act in an all-or-none fashion: either the regulated gene is turned on or not, and many transcription factor genes are redundant, with more than one (particularly in the same family) functioning to activate the same genes. For example, Hoxa1 knockouts have variable inner ear dysmorphogenesis, while Hoxa1/Hoxb1 double mutants have uniform hypomorphic development of the inner ear. 6 Mutations in the homoebox genes Prx1 and Prx2 also act synergistically in the development of inner ear abnormalities. If activation depends on the local concentration of related transcription factors and one gene is mutated, than by a simple twist of fate some cells will turn on the downstream genes, and some will not. This results in variable deficiencies from one individual to another, even when the same mutation is expressed in identical genetic backgrounds.
Genes with mutations leading to gross structural abnormalities of the inner ear
Mutations in Hoxa2, Hoxa1/Hoxb1, Kreisler, and Fgf3 result in aplasia/dysplasia of the entire inner ear 5,7 (Fig 2). Patients 1, 7, 12, and 14 have abnormalities that span all areas of the inner ear and may harbor mutations in the human homologues of these genes. Interestingly, patients 1 and 7 underwent successful implantation, proving that some cochlear nerve fibers had formed in the inner ear and could support auditory signaling.
Two transcription factors, Nkx5-1 and Pax2, are expressed in a complementary pattern in the otic placode and vesicle in mice. 2 Homozygous mutation in Nkx5-1, also called Hmx3, results in aplasia of most of the vestibular system with apparently normal hearing, whereas homozy-gous mutations of Pax2 result in cochlear aplasia. 9 Nkx5-1 mutant mice show no defects in hearing ability nor display any morphologic or histologic abnormalities of the cochlea. 10 This suggests that the cochlea and vestibule develop via independent mechanisms and do not interchange critical information during their developmental course. Another gene mutation that can affect all 3 SCCs is netrin-1. 10,11 netrin-1 is expressed at high levels in otic epithelium, particularly around cells of the SCC fusion plate. When mutated, no posterior or lateral SCCs form and the superior SCC is diminished. It is believe that netrin-1 is involved in stimulating mesenchyme to push together the medial and lateral walls of the fusion plate. netrin-1 is not expressed in hair cells, and all sensory areas, including the cochlear ganglions, develop normally in netrin-1 mutant mice.
Malformation of all 3 SCCs was the most common pattern seen in our patients. Patients 2, 5, 9, and 13 had abnormalities of all 3 SCCs with normal cochleas, whereas patients 3, 4, and 14 also had similar SCC abnormalities along with Mondini malformations of the cochlea. The 2 patients in our series with CHARGE association both had aplasia of the semicircular canals. In a series of 15 patients with semicircular canal aplasia, Satar et al 12 found 9 patients with CHARGE association. Patient 8 had 3 abnormal SCCs, with 1 normal cochlea and 1 Mondini malformation. The human homologues of Nkx5-1 or netrin-1 may be mutated in this patient population. All of our patients were originally selected because of hearing loss, so that if the human homologue of Nkx5-1 were involved in these changes, mutations would have a different phenotype in mice and humans. It may be possible to define a human population with an aplastic vestibular system but normal hearing, by obtaining temporal bone CT scans oforchildren with a developmental delay in walking (>18 months). This vestibular phenotype is seen in children with Usher's syndrome type 1, who lack a functional vestibular system, but have normal temporal bone CT scans. 13,14
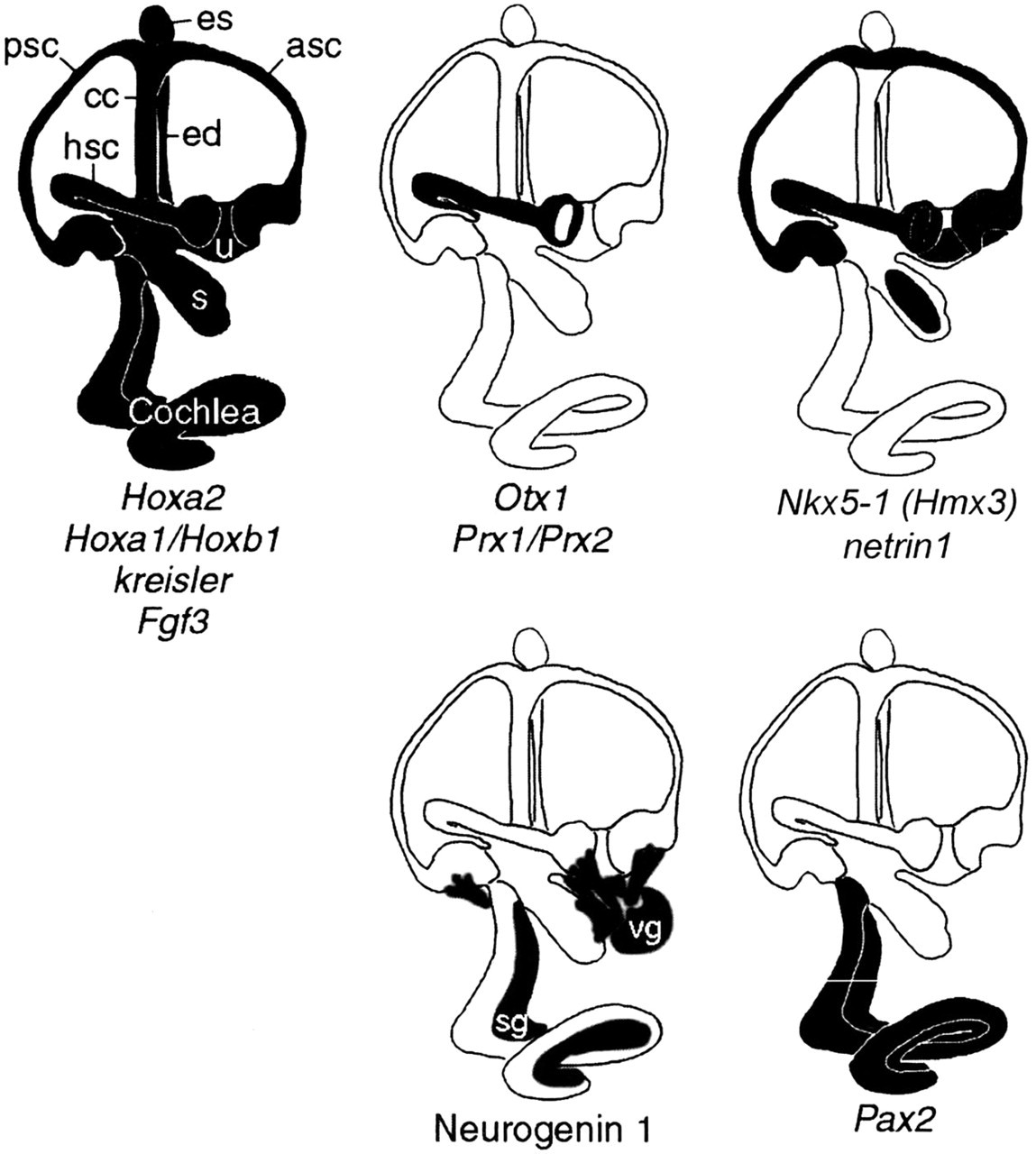
Mutations that alter the morphology of the inner ear. Phenotypes can vary between individual mice within the same strain, between strains of mice, and for different mutation. Shaded areas indicate malformations that result from mutations in these genes. (Reproduced with permission from Fedete 1999. 6 ) (Asc, ascending semicircular canal; cc, common crus; ed, endolyphatic duct; es, endolymphatic sac; hsc, horizontal semicircular canal; psc, posterior semicircular canal; s, saccule; u = utricle.) The most severe phenotypes are shown.
In our series, the most common isolated SCC malformation is seen in the lateral SCC (patients 6, 15, and 16). In mice, genetic pertubations also most often affect this canal. 5 Mutations in otx1 lead to isolated malformations of the lateral SCC and lateral ampulla. 15 Changes in the prx1/prx2 gene pair lead to aplasia of the lateral SCC and diminished superior and posterior SCCs, 16 and Nkx5-1 mutations preferentially affect the lateral SCC. 2,10 The maxim “embryology recapitulates phylogeny” is a recurring theme in developmental biology. This maxim emphasizes the relationship between a structure's phylogenetic age and its resistance to developmental abnormalities. 16 The lateral SCC is phylogenetically the most recent, as demonstrated by jaw-less vertebrates that exhibit a 2-canal inner ear composed of the anterior (superior) and posterior SCC. 6 This suggests that Nkx5-1, otx1, and prx1/prx2 are responsible for lateral SCC development and their human homologues may be mutated in patients exhibiting this abnormality.
Our series of patients exhibit a wide range of inner ear structural abnormalities. Parallel phenotypes are seen in mice and humans, and as murine phenotypes are translated into human genetics, it will be possible to direct genetic testing of patients with inner ear abnormalities. This will lead to effective genetic counseling for these families. So far, we have had reasonable results with cochlear implantation in these patients, but it is likely that some mutations that result in cochlear nerve abnormalities will not support cochlear implants. For example, mutations in the mouse transcription factor neurogenin-1 results in the failure of development of all inner ear neural elements, including the spiral and vestibular ganglia. 17 Humans with a similar phenotype would undoubtedly fail to respond to cochlear implantation. Specific genetic testing will allow the eventual identification of patients who will and will not benefit from cochlear implantation.