
Abstract
In 1981, Currarino et al described a triad of findings that consist of partial sacral dysgenesis, presacral mass (anterior meningocele, enteric cyst, or presacral teratoma) and anorectal malformation. Currarino syndrome exhibits variable expressivity and the clinical presentation tends to vary with the age of the subject such as spinal anomaly detected in the fetus, imperforate anus in the newborn, and intractable constipation or neurologic symptoms in the infant and older child. At any age, meningitis can be the presenting symptom and imaging is required for proper investigation. Meningitis, sepsis, urinary tract infections, and, rarely, malignant transformation of a teratoma are serious potential complications. This pictorial review describes the imaging findings, clinical history, surgical interventions, and genetic background in 5 children with this syndrome who presented in our hospital in the interval of 1 year.
In 1981, Currarino et al [1] described a triad that consists of partial sacral dysgenesis or hemisacrum, presacral mass (anterior meningocele, enteric cyst, or presacral teratoma) and anorectal malformation. This congenital syndrome is thought to be caused by malformation of the caudal notochord, which leads to aberrant secondary neurulation with incomplete separation of the ectodermal and endodermal layers in the developing embryo [2].
Most cases are related to an autosomal-dominant trait. In 1995, the underlying gene defect causing Currarino syndrome was localized in chromosome 7q36. Later, nearly all familial cases were found to be associated with a mutation of the gene HLXB9. Approximately 30% of sporadic cases also have HLXB9 mutation. No obvious genotype-phenotype correlation has been identified [3,4].
Currarino syndrome exhibits variable expressivity and may be diagnosed on prenatal ultrasound. It can present as an imperforate anus at birth, intractable constipation from anorectal stenosis or extrinsic compression from a presacral mass, or as acute meningitis [1,2,5–9].
This review describes the different clinical presentations, imaging findings, genetic profile, surgical management, and clinical outcome of 5 children with Currarino syndrome who attended our hospital in the interval of 1 year. Each patient presented at a different stage of life: a fetus (antenatal magnetic resonance [MR]), an infant, 2 siblings during their first years of life, and a teenager.
Imaging Cases
Case 1: The Fetus
The first patient is a 33 weeks gestational age fetus. A fetal MR imaging (MRI) was requested for a suspicious lumbar defect and possible presacral mass seen on prenatal ultrasound at 32 weeks. Fetal MR confirmed signs of low-lying cord, anterior sacral defect and a mass between the defect and the bladder (Figure 1, A-C). Absence of meconium in the rectum also raised the presence of an anorectal pathology.
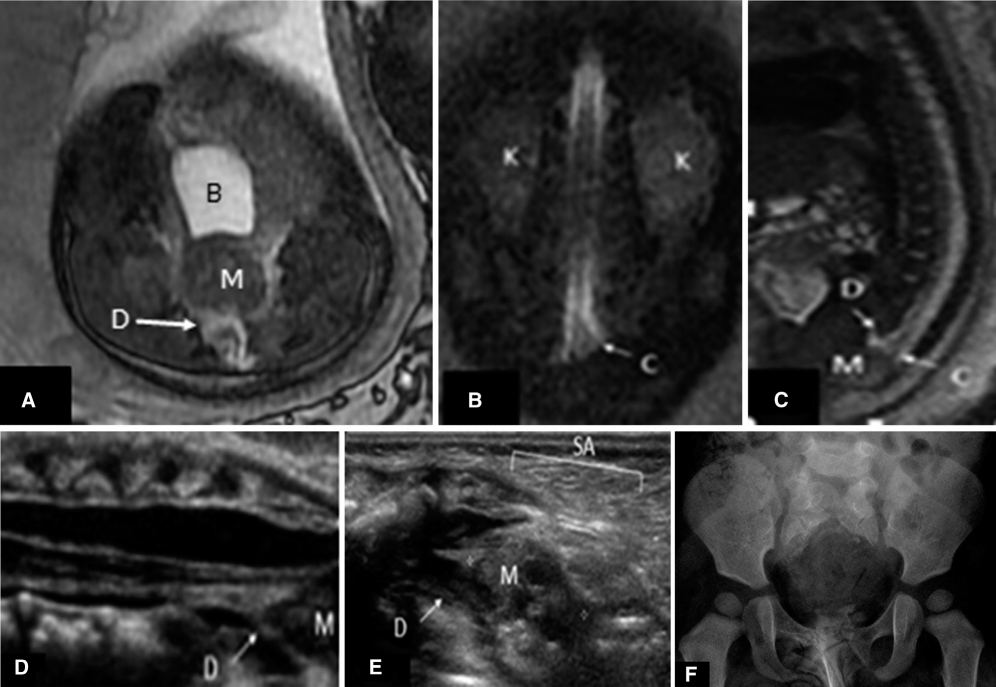
The fetus with prenatal ultrasound suggesting a sacral defect. Fetal magnetic resonance (A) axial steady-state free precession (SSFP), (B) coronal SSFP, (C) sagittal SSFP, (D, E) neonatal ultrasound, and (F) radiograph of the pelvis at 12 months. Fetal magnetic resonance imaging demonstrates an anterior spinal defect (D), sacral agenesis (SA), and a presacral mass (M) behind the bladder (B). The spinal cord extends below the level of the kidneys (K) with a broad-shaped conus (C) extending to the mass. Postnatal spinal ultrasound and radiography confirm these findings demonstrating fat signal within the mass suggestive of teratoma.
The infant was born by normal vaginal delivery without complication and postnatal ultrasound (Figure 1, D-E) and MR confirmed the anomalies adding the finding of a didelphys uterus. A colostomy was performed at 1 month of age, release of tethered cord at 2 months of age, and anorectal malformation was corrected by sagittal anorectoplasty at 5 months of age. The closure of the colostomy was performed at 13 months of age. A double STING (subtrigonal teflon injection) procedure was performed at 33 months of age due to recurrent urinary tract infection (UTI) related to bilateral grade 2 reflux. Presacral teratoma resection was performed at 3 years of age.
Genetic consultation showed a de novo mutation secondary to HLXB9 mutation. The possibility of the parents of having a second child with this syndrome is <1%. After 4 years and 8 months of follow-up, intermittent bladder catheterization is still necessary due to a neurogenic bladder and recurrent UTIs. The child is otherwise well.
Case 2: The Neonate
The second patient is a term baby, born by vaginal delivery at a community hospital after a normal pregnancy. At 9 days of life, he was brought to the emergency department for signs of dehydration. On examination, the infant had fever and bulging fontanel. Cranial and spinal ultrasound were performed and showed large ventricles with thick ependymal lining suggesting meningitis, low-lying cord and absence of normal sacrum with a presacral mass (Figure 2, A and B). MRI done on the following day confirmed these abnormalities (Figure 2C). A preoperative computed tomography scan of the pelvis was performed (Figure 2D).
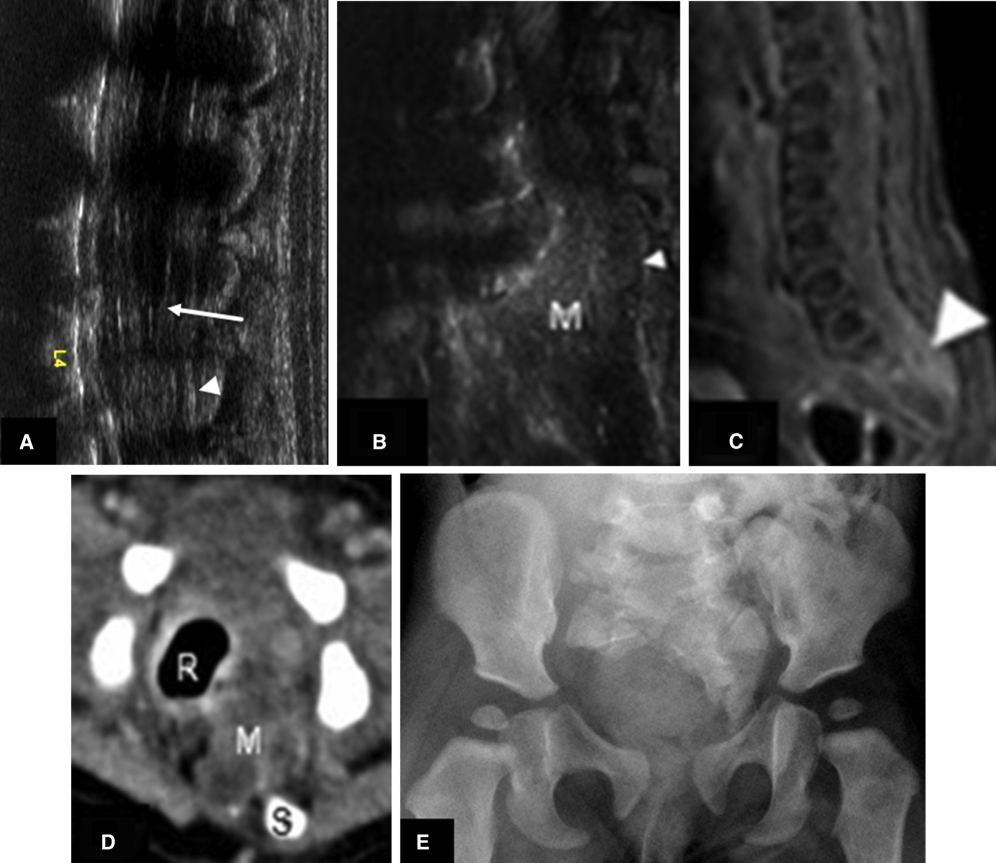
The neonate presenting with fever and dehydration. (A, B) Sagittal spine ultrasound, (C) spinal magnetic resonance imaging sagittal T1 postgadolinium, (D) axial computed tomography, and (E) frontal radiograph of the pelvis show a low-lying cord (arrow), signs of spinal meningitis (arrowhead), a presacral mass (M), and deficient sacrum (S). This figure is available in colour online at http://carjonline.org/.
Two days later, the infant was taken to the operating room for a posterior approach exploration of the rectum and partial excision of an infected presacral dermoid cyst. Closure of the rectal wall and sigmoid colostomy were performed.
At 5 months of age, tethered cord release was done. At 7 months of age the infant was taken to the operating room for posterior sagittal, closure of a rectal fistula and sinus, as well as anoplasty. One month later, the colostomy was closed.
Genetic consultation showed a de novo mutation of the HLXB9 gene and duplication of 9 base pairs (not previously reported). After 4 years and 11 months, this child is clinically well with signs of mild neurogenic bladder needing intermittent catheterization.
Cases 3 and 4: The Young Child
Patients 3 and 4 are siblings transferred from another centre due to a change of residence of the family. Both were diagnosed with Currarino syndrome by antenatal ultrasound. Their father was known for this condition, he underwent surgery of tight spinal cord and excision of presacral dermoid cyst at our Institution 20 years ago.
Both pregnancies and deliveries were normal without significant neonatal events. Early in life, the sister was diagnosed with vesicoureteral reflux as well as horseshoe kidney. Follow-up ultrasounds showed normal renal growth and the child remained asymptomatic. She has been assessed by neurosurgery, urology, orthopaedics, and general surgery to evaluate her development. She never underwent surgery for her syndrome because she is asymptomatic and on follow-up imaging, the mass remains unchanged. On MR, she has signs of low lying cord, a small complex presacral mass, and anorectal malformation (Figure 3, A and B).
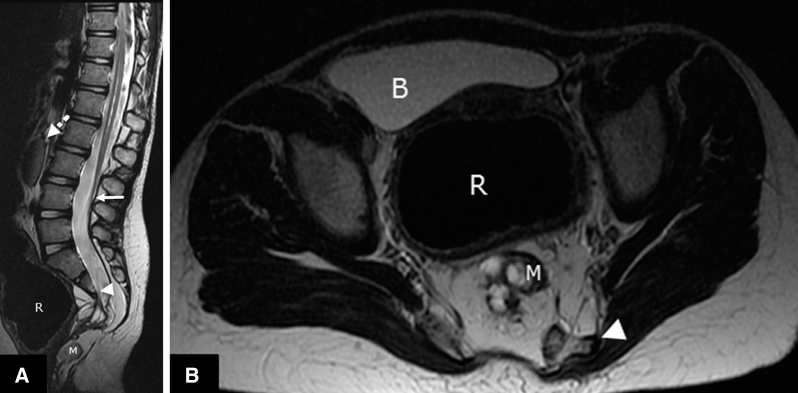
The young sister child, asymptomatic beside vesicoureteral reflux. (A) Sagittal and (B) axial T2 images show sacral deficiency (arrowhead), distention of the rectum (R) secondary to anorectal malformation and presacral mass (M). Low-lying cord (arrow) and horseshoe kidney (discontinuous arrow) are noted.
His brother was born with very mild distal glandular hypospadias, He was followed at another Hospital for frequent anal dilatations for anal stenosis during his first few years of life. At 2 years of age, he underwent partial excision of a presacral teratoma at that Hospital. He presented to our Hospital at the age of 5 years of age due to constipation and soiling accidents. He was then assessed by neurosurgery, urology and general surgery. On MRI (Figure 4), there are signs of low lying cord, nonexpansile syrinx, presacral mass and anorectal malformation Without specific further treatment, his clinical outcome after 3 years is very good, without GI or GU symptoms.
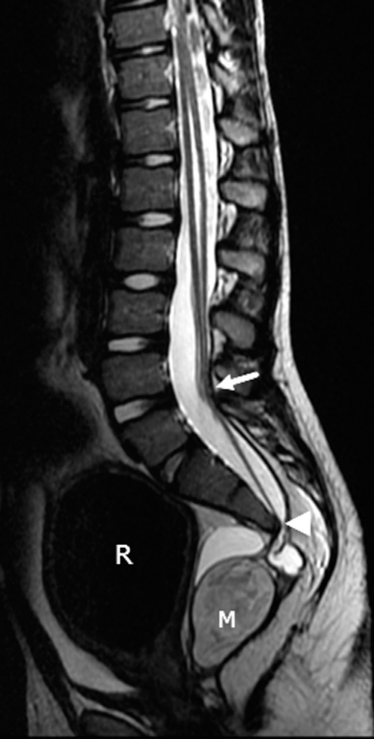
The young brother child with constipation and soiling accidents. Sagittal T2 image of the lumbosacral spine shows low lying cord (arrow), associated with sacral deficiency (arrowhead), distension of the rectum (R) secondary to anorectal malformation and a presacral solid-cystic mass (M).
Case 5: The Teenager
Patient 5 is a boy 17 years and 6 months of age transferred to our institution from a community hospital with a complicated history, which started 7 months before.
He initially presented with a persistent perianal abscess, which was drained under local anesthesia. He then developed meningitis and was hospitalized for 3 weeks. He was fine for 6 months when he started having fevers and recurrent pain in the perirectal area. Under general anesthesia, the patient underwent an attempted drainage at the community hospital but the procedure was aborted due to the palpation of a mass on digital rectal examination.
The patient was transferred to our hospital and an MRI showed a sacral defect with a complex solid-cystic mass (Figure 5, A and B). An abscess cavity was identified and presumed to be either an ischiorectal abscess or a noncommunicating infected meningocele. At that time, the patient did not have signs of sepsis or meningitis. Under ultrasound (Figure 5C), the abscess cavity was punctured, pus was drained, and multiple organisms including anaerobic and aerobic bacteria were identified.
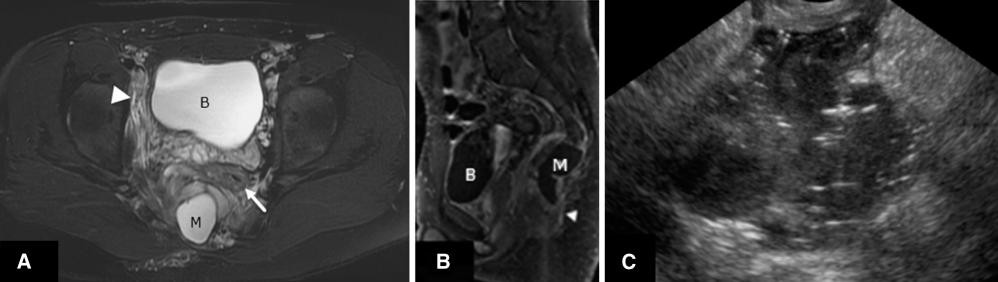
The teenager with persistent perianal abscess. (A) Axial T2 fat saturated, (B) sagittal T1-fat sat postcontrast, and (C) transrectal ultrasound images show sacral insufficiency, a presacral cystic mass (M) in close contact with the rectum (arrow), and associated inflammatory changes (arrowhead). Ultrasound guided transrectal drainage was performed and pus was obtained from an infected meningocele.
His familial history was remarkable with a father and 2 maternal aunts known for meningocele. After 1 year, he remained clinically asymptomatic and was transferred to an adult hospital for follow-up.
Discussion
In this article, through 5 different pediatric patients, the most frequent radiological findings of Currarino syndrome have been described. These findings include a 1) sacrococcygeal defect, which can range from an asymmetric sacral deformity such as a hemisacrum with scimitar or sickle shape [7,10–12] to a total sacral agenesis, a 2) presacral mass, and in this study 2 were sacral teratomas, 1 dermoid cyst, 1 anterior meningocele, and 1 remains unresected (patient 3). In the literature, anterior meningocele is the most frequent nature of the presacral masses. Teratomas, enteric cysts, dermoid or epidermoid cysts, lipomas, hamartomas, or rectal duplications are reported. Malignant transformation of a presacral teratoma has been described, but is rare [13]. 3) Anorectal malformations, including anal stenosis with or without a fistula, anal atresia or ectopia, imperforate anus, and cloacal anomalies are part of the syndrome [5–8].
Other associated manifestations include malformations of the urogenital system that comprise horseshoe kidneys, sigmoid kidneys, single pelvic kidneys, neurogenic bladders, multicystic kidneys, vesicouretral reflux, and partial or complete duplication of the vagina, clitoris, or uterus. Some of these anomalies have been seen in our population. Associated intraspinal anomalies include low-lying spinal cord, hydrofilum, intraspinal lipoma, and hydrocephalus [6,10,11]. There are incomplete forms of Currarino syndrome with absence of 1 or 2 characteristics, particularly in relatives of patients with Currarino syndrome [9,11,12]. Heterozygote patients can be asymptomatic and remain unrecognized. However, 80% of cases are diagnosed before 16 years of age [6,11].
Better knowledge of the different clinical signs and symptoms of Currarino syndrome at different stages of life enhances appropriate imaging and prompt diagnosis of this condition. The imaging approach includes a radiograph of the sacrum for detection of sacral defect in patients with unexplained severe constipation or anal anomalies. Pelvic and spinal MRI is required for the evaluation of a presacral mass and to detect associated intraspinal anomalies [5,8,11]. In newborn and infant before 4 months, spinal ultrasound is very useful and pelvic sonography is required for every patient with Currarino syndrome to explore for associated urogenital anomalies.
The treatment is often surgical to prevent eventual complications such as meningitis, perianal sepsis, urinary tract infections, and, rarely, malignancy transformation of a teratoma [8,9,11,13].
Conclusion
Currarino syndrome is a rare hereditary syndrome that includes anorectal malformation, sacrococcygeal defect and presacral mass. It usually manifests as persistent constipation in a child but its clinical presentation is variable.
Better knowledge of the different clinical signs and symptoms of Currarino syndrome at different stage of life enhances appropriate imaging and prompt diagnosis of this condition.
Early recognition of Currarino syndrome with adequate surgical treatment prevents eventual serious complications such as meningitis, sepsis, urinary tract infection, and, rarely, malignant transformation of a presacral mass.