
Abstract
Striatal medium-sized spiny neurons (MSNs) are highly vulnerable to ischemia. A brief ischemic insult, produced by oxygen and glucose deprivation (OGD), can induce ischemic long-term potentiation (i-LTP) of corticostriatal excitatory postsynaptic response. Since nitric oxide (NO) is involved in the pathophysiology of brain ischemia and the dopamine D1/D5-receptors (D1-like-R) are expressed in striatal NOS-positive interneurons, we hypothesized a relation between NOS-positive interneurons and striatal i-LTP, involving D1R activation and NO production. We investigated the mechanisms involved in i-LTP induced by OGD in corticostriatal slices and found that the D1-like-R antagonist SCH-23390 prevented i-LTP in all recorded MSNs. Immunofluorescence analysis confirmed the induction of i-LTP in both substance P-positive, (putative D1R-expressing) and adenosine A2A-receptor-positive (putative D2R-expressing) MSNs. Furthermore, i-LTP was dependent on a NOS/cGMP pathway since pharmacological blockade of NOS, guanylate-cyclase, or PKG prevented i-LTP. However, these compounds failed to prevent i-LTP in the presence of a NO donor or cGMP analog, respectively. Interestingly, the D1-like-R antagonism failed to prevent i-LTP when intracellular cGMP was pharmacologically increased. We propose that NO, produced by striatal NOS-positive interneurons via the stimulation of D1-like-R located on these cells, is critical for i-LTP induction in the entire population of MSNs involving a cGMP-dependent pathway.
INTRODUCTION
An important feature of ischemic brain damage is the selective vulnerability of specific neuronal populations. Striatal neurons are particularly vulnerable to ischemia1–3 and medium-sized spiny neurons (MSNs), representing the large majority of the entire striatal neuronal population, are rapidly lost during ischemia and excitotoxic injury. In vitro studies have shown that in the striatum a brief oxygen and glucose deprivation (OGD) insult induces a pathological form of synaptic plasticity, named ischemic long-term potentiation (i-LTP).4,5 This aberrant form of synaptic plasticity has been considered the electrophysiological correlate of molecular apoptotic cell death. 6 In fact, neurons located in the core of a focal cerebral ischemia are often largely and irreversibly compromised, mainly by excitotoxic processes that can enhance glutamate-mediated neurotransmission. However, the function of neurons within the ischemic penumbra, an area of injured tissue that surrounds the central core of the focal cerebral ischemia, might be rescued. Thus, i-LTP could facilitate neuronal death but, at the same time, it might also help functional recovery and the induction of novel connections between neurons. Whether i-LTP represents the consequence of the enzymatic cascades triggered by the ischemic injury or a potential protective and/or reparative form of plasticity leading to a dynamic recovery after stroke is still a matter of debate. 7
Nitric oxide (NO) is involved in the pathophysiology of brain ischemia8–11 as well as in the formation of activity-dependent synaptic plasticity.12,13 Accordingly, inhibition of nitric oxide synthase (NOS) attenuated anoxic LTP in the hippocampus. 14 The NOS family consists of three isoforms: neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS).15,16 Since nNOS and eNOS have been suggested to play a role in activity dependent and i-LTP in the hippocampus,17,18 we hypothesized that the blockade of the striatal NO production would also affect striatal i-LTP.
In the striatum ischemia causes a large increase of dopamine (DA) levels 19 that may become neurotoxic, either directly or by interacting with the glutamatergic system.20,21 The role of D1-like-R/cAMP/PKA intracellular pathway appeared to be critical in MSN i-LTP induction. 22 Ischemia induces long-lasting increase of the amplitude of postsynaptic potentials (EPSPs), however, pharmacological blockade or genetic inactivation of the D1-like-R/cAMP/PKA pathway, rather than D2-like receptor pathway, prevented this increase. Since the selective expression of D1-like-R in a subpopulation of MSNs23–25 is still matter of debate, the mechanism by which D1-like-R stimulation mediates i-LTP induction in the entire MSN population is far from being clear.
Within the striatum, D1-like-Rs are also expressed by NOS positive GABAergic interneurons, cells representing less than 5% of the total striatal neuronal population and virtually projecting to all MSNs. These neurons express both DA D1/D5 receptor mRNA and protein26–28 and a relation between D1-like-Rs and the release of NO by NOS striatal interneurons has been demonstrated. 29 In fact, administration of D1-like-Rs agonists increased striatal NO efflux in an in vivo animal model. 29 NO also contributes to the induction of DA-dependent physiological synaptic plasticity in MSNs. 30 Moreover, NO modulates activity-dependent LTP in the hippocampus. 18 Interestingly, transient ischemia increases the expression of nNOS 31 suggesting that an ischemic event may lead to NO production, thus triggering the induction of both physiological and pathological forms of synaptic plasticity.
It has been suggested that the biochemical pathways activated by the ischemic insult might mimic the molecular key steps required for the induction of activity-dependent synaptic plasticity, finally causing, via the modulation of nuclear transcription factors, long-term changes of excitatory synaptic transmission in various neuronal subtypes. 7 According to this hypothesis, activity-dependent LTP and i-LTP share some downstream biochemical mechanisms such as an increase of intracellular calcium and they can be mutually occlusive.4,5
Nevertheless, the involvement of NO in corticostriatal i-LTP has not been demonstrated yet. Therefore, we aimed at investigating the involvement of the NO/cGMP intracellular pathway in MSN i-LTP and we explored the mechanism by which D1-like-R stimulation mediates i-LTP formation in both MSNs of the direct and indirect pathway.
METHODS
Electrophysiology
All the experiments were conducted in conformity with the European Communities Council Directive of November 1986 (86/609/ECC), in accordance with a protocol approved by the Animal Care and Use Committee at the University of Perugia and with the ARRIVE guidelines. Two- to three-month-old male Wistar rats (Harlan) and 5- to 6-week-old male C57BL/6J-Swiss Webster mice were used. Preparation and maintenance of corticostriatal slices (270 to 300 μm) have been described previously. 4 A single slice was transferred to a recording chamber and submerged in a continuously flowing Kreb's solution (34°C; 2.5 to 3 mL/min) bubbled with a 95% O2–5% CO2 gas mixture. The composition of the Kreb's solution was (in mmol/L): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3. For intracellular recording sharp microelectrodes were filled with 2 mmol/L KCl and an Axoclamp 2B amplifier (Molecular Devices) was used. Only neurons electrophysiologically identified as spiny neurons were considered for experiments. 5 They presented highly hyperpolarized resting membrane potentials (–80 to −90 mV), tonic firing activity during a long-duration depolarizing pulse and current-voltage relationship showing current rectification (Figures 1A and 1B). For patch-clamp recordings, neurons were visualized using differential interference contrast (Nomarski) and infrared microscopy (Olympus). Whole-cell voltage-clamp recordings (Vhold −50 mV) were performed with borosilicate glass pipettes (4 to 7 MΩ; access resistance 5 to 30 MΩ) filled with a standard internal solution containing (in mmol/L): 125 K+-gluconate, 0.1 CaCl2, 2 MgCl2, 0.1 EGTA, 10 HEPES, adjusted to pH 7.3 with KOH. Signals were amplified with a Multiclamp 700B amplifier (Molecular Devices), recorded and stored on PC using pClamp 10 (Molecular Devices). In all patch-clamp experiments 50 μmol/L picrotoxin was added to the external medium to block GABAA receptors. The recording electrodes were placed within the dorsolateral striatum. Glutamatergic EPSPs and excitatory postsynaptic currents (EPSCs) were evoked every 10 seconds by means of a bipolar electrode connected to a stimulation unit (Grass Telefactor). The stimulating electrode was located in the white matter between the cortex and the striatum to activate glutamatergic fibers. In vitro ischemia was delivered by switching the standard solution to a solution in which sucrose replaced glucose, gassed with 95% N2 and 5% CO2 (OGD). 4 Ischemic LTP was induced by 2 to 3 minutes OGD episodes. External magnesium ions were omitted trough all the experiments to maximize the contribution of NMDA receptors during all i-LTP experiments.
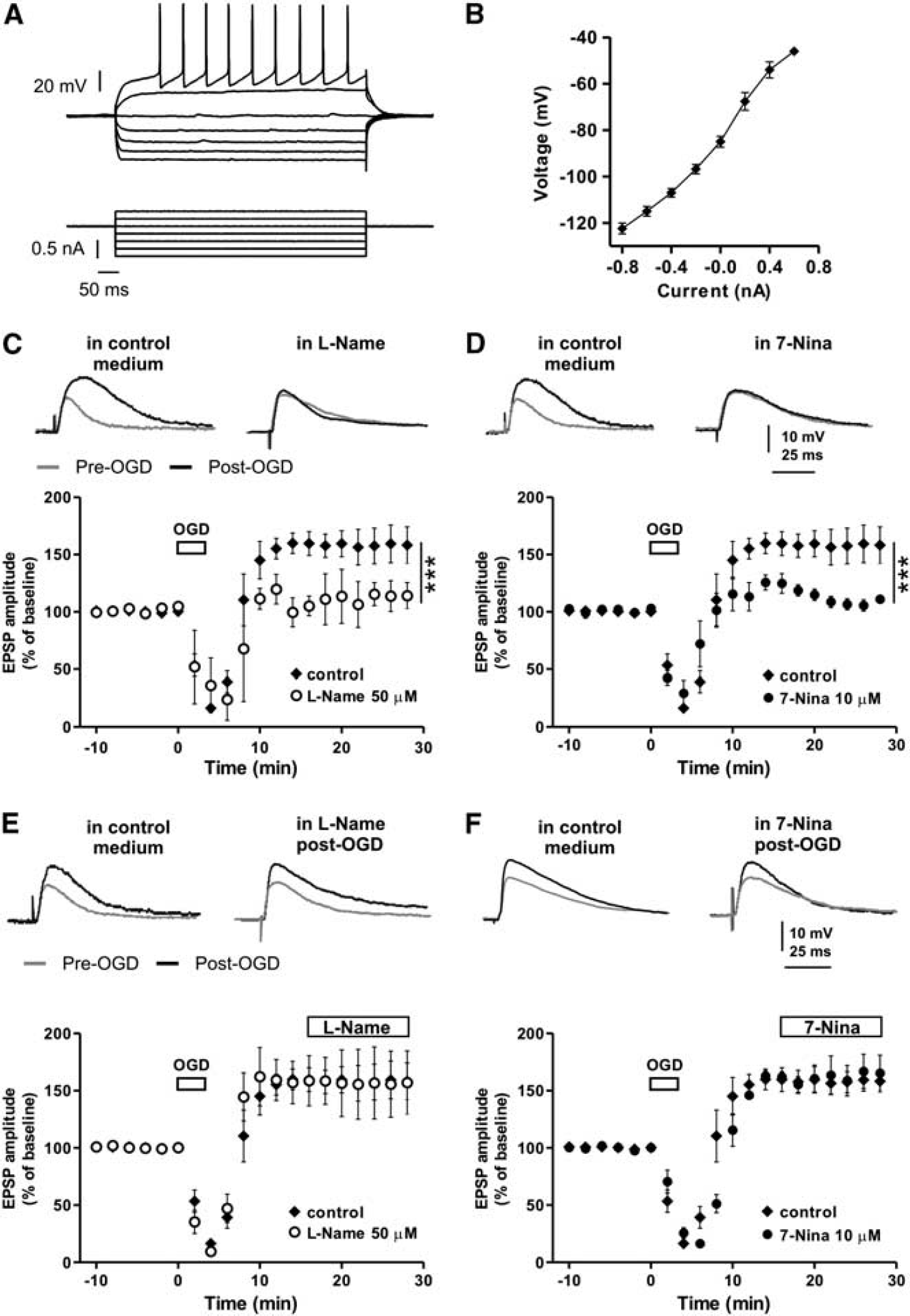
Inhibition of NOS affects the induction but not the maintenance of striatal i-LTP. (
Tissue Processing and Double Immunofluorescence
Corticostriatal brain sections were postfixed overnight at 4°C with 4% paraformaldehyde in saline solution and then cryoprotected in phosphate buffer (PB 0.1 M) with sodium azide 0.02% for 48 hours at 4 °C. The sections filled with biocytin (during electrophysiological recordings) were incubated with streptavidin-Cy3 (Sigma) diluted 1:600 in PB-TX-100 0.3% for 2 hours at room temperature (RT) to verify the presence of cells filled with biocytin. Sections (from each group) were preincubated with a primary antibody goat anti-SP (Immunological Science) or rabbit polyclonal anti-adenosine A2A receptor (A2AR) (Alexis) to label the medium spiny projection neurons that are component respectively of the ‘direct’ or ‘indirect’ basal ganglia pathway.32–34 The primary antisera were used at a concentration of 1:400 for SP and 1:250 for A2A in 0.1 M PB containing Triton X-100, 0.3% and sodium azide, 0.02% for 24 hours at RT and 48 hours at 4°C. Sections were then rinsed three times for 15 minutes at RT, and subsequently incubated with a cocktail of anti-goat Cy2 and anti-rabbit Cy5-conjugated secondary antibody (Jackson Immunoresearch) for 2 hours at RT. All the secondary antibody were used at 1:200 concentration. Tissue was mounted on gelatin-coated slides, coverslipped with GELMOUNT and all the images were acquired with a Confocal Laser Scanning Microscope (CLSM) Zeiss LSM700 with 40 × oil magnification. To acquire the inserts showing the branched dendrites a 100 × oil zoom 6 magnification was used.
6-Hydroxydopamine-Induced Lesion
Procedures for obtaining rats with 6-hydroxydopamine (6-OHDA)-induced striatal DA denervation have been previously given in detail.35,36 In brief, deeply anesthetized rats were unilaterally injected with 6-OHDA (12 μg/4 μL of saline containing 0.1% ascorbic acid) into the medial forebrain bundle. Sham-operated rats were injected only with vehicle at the same stereotaxic coordinates. Fifteen days after surgery, rats were tested with 0.05 mg/Kg subcutaneous administration apomorphine, and turns contralateral to the lesion were counted for 40 minutes. Rats with more than 200 turns were assigned to the group of the DA-denervated animals. Sham-operated animals did not show turning behavior. Two months after surgery, rats were used for electrophysiological experiments. The severity of the lesion was confirmed afterward by striatal and nigral immunohistochemistry tyrosine hydroxylase.
Drugs
Drugs were applied by dissolving them to the desired final concentration in the Krebs' solution and by switching the perfusion from control solution to drug-containing solution. Picrotoxin, SCH23390, 7-nitroindazole (7-Nina), NG-nitro-L-arginine methyl ester (L-Name), 1H-[1,2,4] Oxadiazolo[4,3-a] quinoxalin-1-one (ODQ), 8Br-cGMP, Rp-8Br-PET-cGMP, S-Nitroso-Nacetylpenicillamine (SNAP) were purchased from Tocris. Drugs were applied in the recording chamber for at least 10 minutes before OGD and maintained throughout the experiment. In some patch-clamp experiments 8Br-cGMP or Rp-8Br-PET-cGMP was added to the internal solution.
Statistical Analysis
Data analysis was performed off-line using Clampfit 10 (Molecular Devices) and Microcal Origin software. Values given in the text and figures are mean ± s.e., n representing the number of recorded neurons. Only one neuron per slice was recorded. Changes of EPSP or EPSC amplitude after i-LTP induction is expressed as percentage of the baseline, the latter representing the normalized EPSP or EPSC mean amplitude acquired during a stable period (10 to 15 minutes) before the OGD onset. Two-way ANOVA was used for statistical analysis. The significance level was established at *P < 0.05, **P < 0.01, and ***P < 0.001.
RESULTS
Nitric Oxide Synthase Inhibitors Prevent the Induction but not the Maintenance of Ischemic LTP in Striatal MSNs
Striatal MSNs are highly vulnerable to ischemia1–3 and a brief ischemic insult can produce i-LTP, the pathological form of long-term potentiation of synaptic transmission.4,22 In order to explore the mechanism by which i-LTP occurs in the striatum we recorded electrophysiologically identified MSNs with sharp electrodes. The main characteristics of these cells have been described in detail previously. 37 As shown in Figures 1A and 1B, they were silent at rest and showed tonic firing activity during a long-duration depolarizing pulse. Ischemic LTP was induced by in vitro ischemia obtained with a brief OGD episode. After obtaining EPSPs of stable amplitude (~10 minutes), the standard solution was switched to the ischemic solution (OGD) for 3 minutes. In this condition, 30 minutes after OGD onset, the EPSP amplitude was increased to 158.2 ± 9.6% of the baseline (n = 8) revealing the presence of i-LTP (Figures 1C–1F).
NO is known to be involved in the pathophysiology of brain ischemia 8 and we previously reported its pivotal role in mediating hippocampal i-LTP. 17 In order to test whether i-LTP was mediated by a NO-activated pathway in the striatum, we recorded EPSPs from MSNs from striatal slices in the presence of 50 μmol/L of the unselective NOS inhibitor L-Name or 10 μmol/L of the nNOS inhibitor 7-Nina applied 15 minutes before the administration of OGD and during all the duration of the experiment. We found that the EPSPs amplitude measured 30 minutes after OGD in the presence of L-Name (n = 5) or 7-Nina (n = 4) was 114.1 ± 11.5%, and 111.1 ± 3.5% of the EPSP measured in pre-OGD conditions, respectively (L-Name versus control, F(1,220) = 63.1, P < 0.001 and 7-Nina versus control, F(1,200) = 33.9, P < 0.001) (Figures 1C and 1D). Thus, OGD in the presence of L-Name or 7-Nina induced no i-LTP (pre- versus post-OGD, for L-Name and 7-NINA, t-test, P > 0.05). Conversely, i-LTP was not affected by L-Name (n = 4) or 7-Nina (n = 6) when they were bath applied 15 minutes after the induction of i-LTP, (L-Name versus control and 7-Nina versus control, P > 0.05) (Figures 1E and 1F).
Ischemic LTP Requires Nitric Oxide involving the Activation of a Postsynaptic Guanyl Cyclase, cGMP Dependent Intracellular Pathway, and PKG
In order to confirm that inhibition of NOS prevented i-LTP in MSNs by reducing striatal NO levels, we explored the role of NO on striatal i-LTP by recording MSNs in the presence of the NOS inhibitor L-Name co-applied with the NO donor, SNAP. We firstly measured the effect of 100 μmol/L SNAP on i-LTP applied in isolation and consequently in co-application with 50 μmol/L L-Name. As presented in Figure 2A, i-LTP was not altered by exogenous NO produced by SNAP alone (n = 4) (SNAP versus control, P > 0.05). However, the same concentration of SNAP (100 μmol/L) was able to restore i-LTP even in the presence of 50 μmol/L L-Name (n = 4) (L-Name + SNAP versus L-Name alone, F(1,140) = 146.4, P < 0.001, Figure 2A).
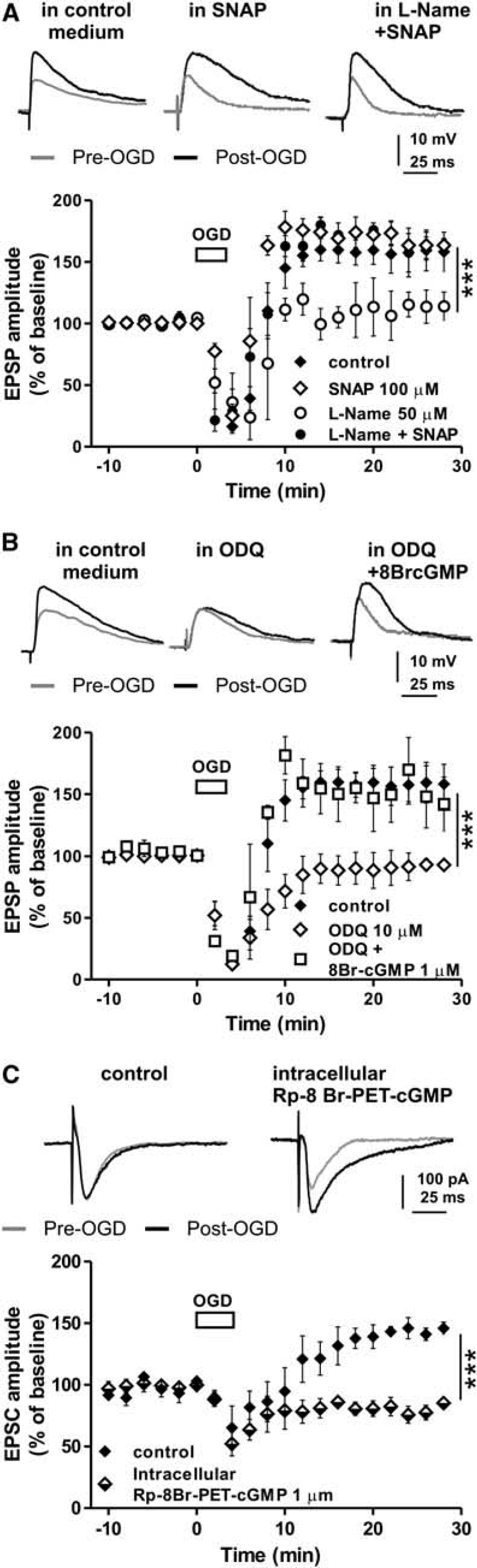
Exogenous NO or enhanced levels of intracellular cGMP restores striatal i-LTP abolished by inhibition of NOS or by inhibition of the soluble guanylate cyclase, respectively, whereas i-LTP is prevented by inhibition of PKG. (
Thus, NO seems to have a key role in the i-LTP induction, most likely by activating soluble guanylate cyclase (sGC) and increasing cGMP synthesis into neurons.17,38 Therefore, this intracellular messenger might be involved in the induction of striatal i-LTP. To test for this possibility, we recorded MSNs in the presence of the sGC inhibitor, ODQ. 38 Interestingly, we found that i-LTP induction was prevented by 10 μmol/L ODQ (n = 6) (ODQ versus control, F(1,240) = 178.4, P < 0.001; Figure 2B). To further support the involvement of the NO/cGMP pathway in the i-LTP induction, we tested whether the application into the bathing solution of the cGMP analog 8Br-cGMP, was able to restore i-LTP even in a condition in which sGC was blocked by ODQ. The application of 1 μmol/L 8Br-cGMP restored i-LTP in MSNs recorded in the presence of 10 μmol/L ODQ (n = 4, 8Br-cGMP + ODQ versus ODQ, F(1,160) = 231.2, P < 0.001; Figure 2B) further supporting the hypothesis that stimulation of NO/cGMP intracellular pathway in striatal MSNs is essential for i-LTP in this brain structure.
In addition, we tried to establish the possible target of cGMP during an ischemic episode. Since PKG represents a protein kinase that can be directly activated by cGMP, we tested whether this kinase could be activated by an OGD insult inducing i-LTP. In patch clamp experiments, 4 MSNs were therefore injected with the PKG inhibitor Rp-8Br-PET-cGMP via patch pipette. All patch clamp recordings for i-LTP analysis were performed in the presence of a Mg2+-free medium containing 50 μmol/L picrotoxin, applied 10 to 15 minutes before the onset of OGD solution and for all the duration of the experiment. This conditions produced no detectable seizure-like activity of the recorded neurons. After recording EPSCs of stable amplitude (~10 minutes), the standard solution was switched to the ischemic solution (OGD) for 3 minutes and the EPSC amplitude was monitored for subsequent 30 minutes. As shown in Figure 2C, in the recorded MSNs the inhibition of PKG by 1 μmol/L Rp-8Br-PET-cGMP completely prevented i-LTP induction (n = 6, Rp-8Br-PET-cGMP versus control, n = 4, F(1,160) = 89.7, P < 0.001) confirming the PKG as important downstream target of the NO/cGMP pathway mediating the i-LTP in MSNs (Figures 2C and 6).
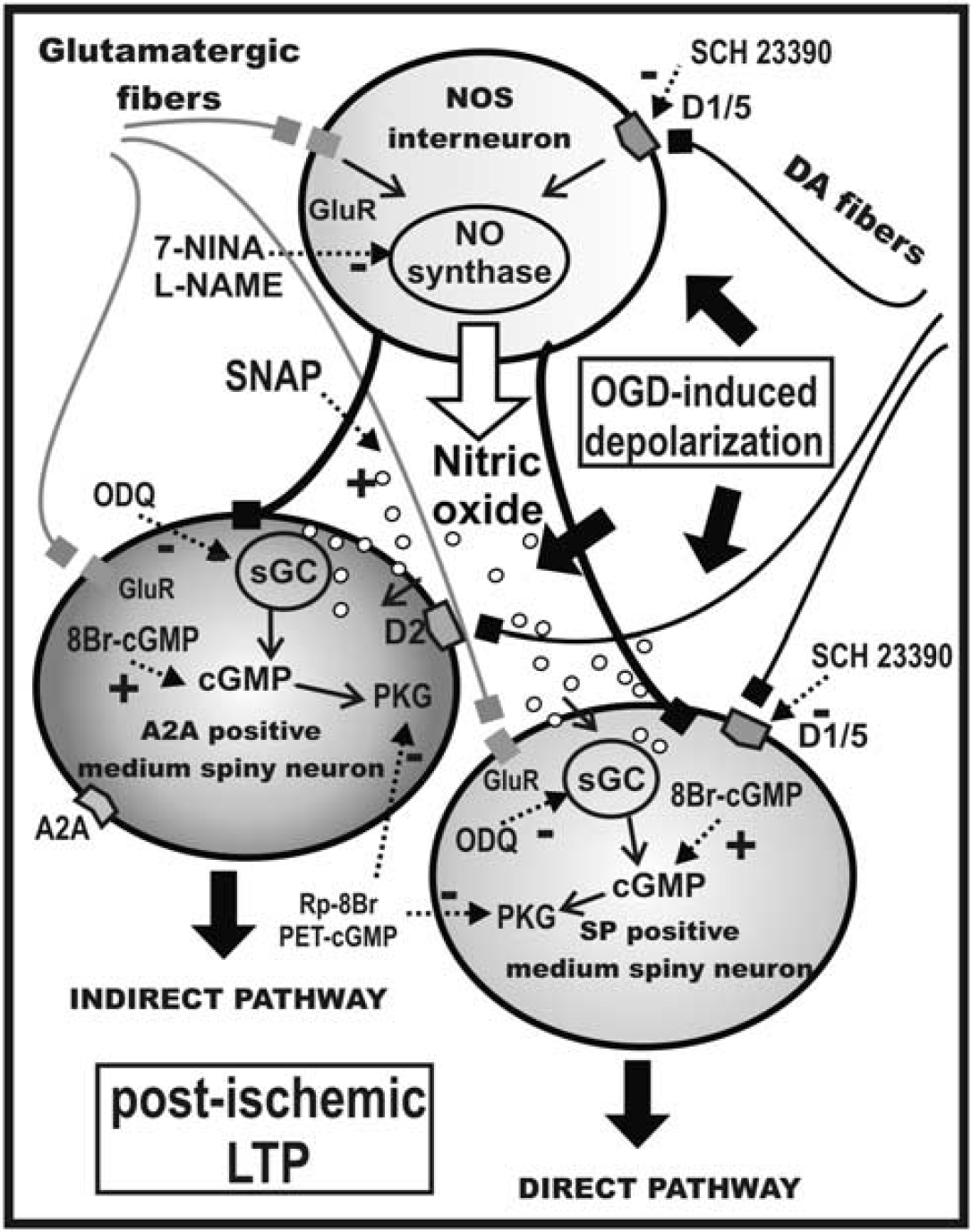
Model of the intrastriatal network during OGD. The scheme represents the involvement of the NO/cGC/cGMP signaling pathway in the ischemic LTP in the striatum. A D1-like-R-mediated activation of nitric oxide synthase (NOS) in NOS positive interneuron determines nitric oxide (NO) production. NO increases cGMP levels within MSNs of the direct and indirect pathways by activating intracellular sGC. D1-like-R antagonism (SCH23390) or inhibition of NOS (7-Nina or L-Name) prevented i-LTP induction, while a NO donor (SNAP) restores i-LTP of both the A2A- and the SP-expressing MSNs. Ischemic LTP is prevented by inhibiting sGC with ODQ, or inhibiting PKG with Rp-8Br-PET-cGMP, and restored by application of the c-GMP analog 8Br-cGMP in medium spiny neurons.
Ischemic-LTP is Prevented by D1-Like Receptor Antagonism in Both Substance P or A2A Receptor Expressing MSNs
Within the MSN striatal population D1-like-R and D2-like-R are thought to be segregated into distinct neuronal subpopulations and we have previously shown a critical role only for D1R stimulated pathway in striatal i-LTP, but not for D2R. 22 Therefore we explored whether a D1-like-R-dependent form of striatal i-LTP could be observed in MSNs of both the direct (expressing D1Rs) and indirect (expressing D2Rs) basal ganglia pathway.
After obtaining EPSCs of stable amplitude of patch clamped MSNs, the standard solution was switched to the ischemic solution (OGD) for 3 minutes. In order to distinguish direct and indirect pathway neurons, MSN were filled with biocytin during the recordings and immunofluorescence double labeling was performed a posteriori. Direct pathway MSNs were immunohistochemically identified as ‘substance P positive’ (SP +) neurons (Figure 3A), 32 whereas those from the indirect pathway were recognized as ‘A2A receptor positive’ (A2A +) neurons (Figure 3A).33,34 All MSNs injected with biocytin during control patch-clamp recordings showed i-LTP in response to 3 minutes OGD. Slices containing the recorded MSNs loaded with biocytin were then processed for SP and A2AR detection. Interestingly, the immunostaining of the slices revealed that biocytin labeled MSNs expressed either SP or A2A receptor. As presented in the time courses of Figure 3B, SP + and A2A + MSNs displayed i-LTP of similar features. The EPSC amplitude 30 minutes after OGD was 153.9 ± 10.4% in SP + MSNs (n = 5) and 146.0 ± 13.1% in A2A + MSNs (n = 5) (Figure 3B).
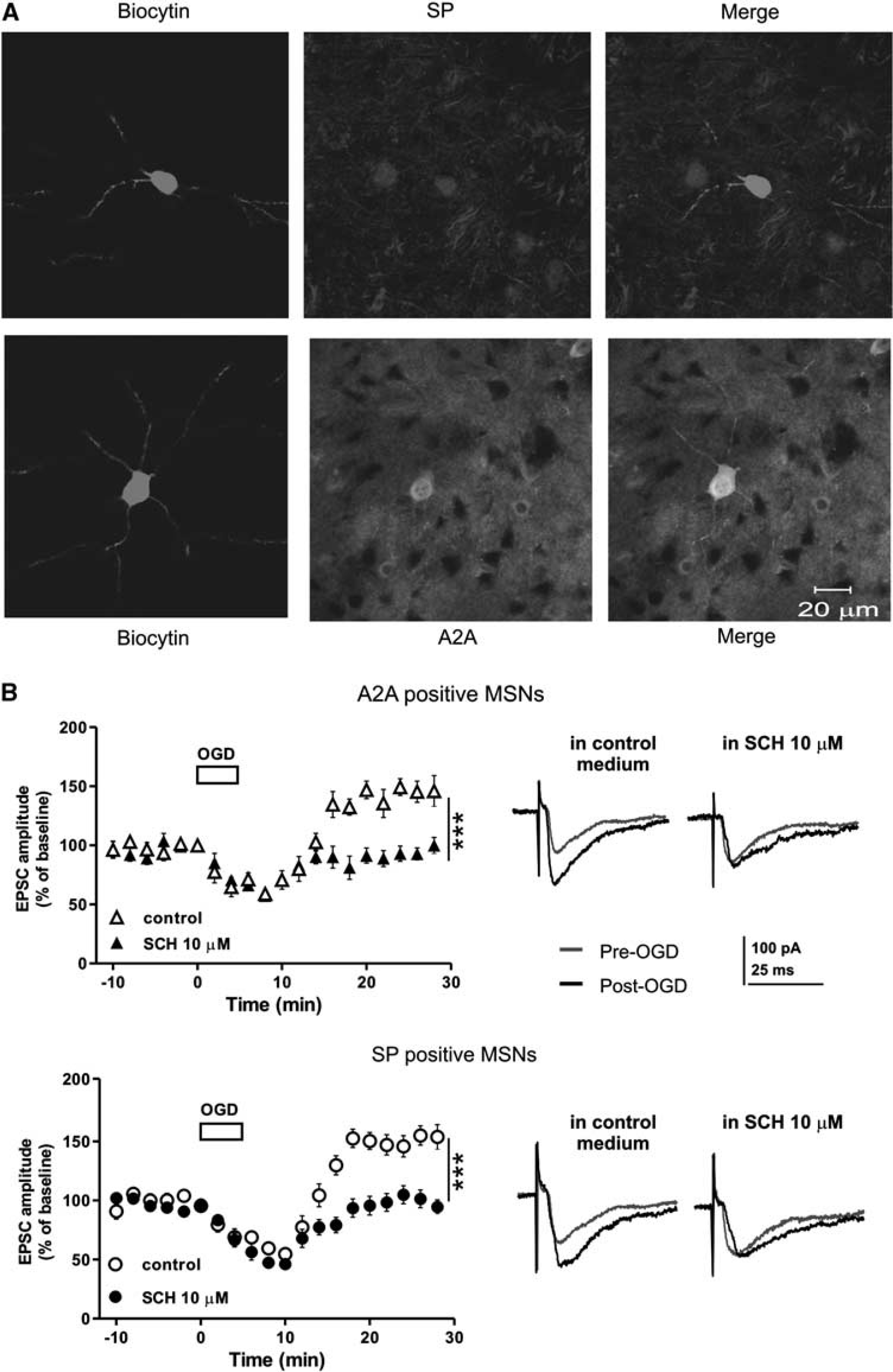
Antagonism of D1-like receptor blocks i-LTP induction in both A2A receptor- and substance P-expressing striatal medium spiny neurons. (
A group of MSNs loaded with biocytin was also recorded in the presence of 10 μmol/L of the D1-like-R antagonist SCH23390. This drug prevented i-LTP in all the recorded neurons and subsequent immunohistochemistry showed that these cells were both SP- or A2A-positive MSNs (Figure 3B). The EPSC amplitude measured 30 minutes after OGD in the presence of SCH23390 was 94.6 ± 5.9%, for SP + MSNs (n = 5) (SCH23390 versus control, F(1,160) = 122.4, P < 0.001) and 99.8 ± 6.8%, for A2A + MSNs (n = 5) (SCH23390 versus control, F(1,160) = 68.5, P < 0.001). Therefore, these experiments confirm that blockade of D1-like-R prevents i-LTP in MSNs of both direct and indirect pathways.
Exogenous Nitric Oxide or a cGMP Analog Restores i-LTP during Pharmacological Blockade of D1-Like Receptor or in DA-Denervated Slices
D1R stimulation of striatal tissue increases NO production and cGMP levels29,39 and we have shown that either inhibition of NO/cGMP pathway (Figures 1 and 2) or blockade of D1-like-R (Figure 3) prevented i-LTP of striatal MSNs. Thus, we explored whether the blockade of i-LTP by D1-like-R antagonism was mediated by a NO dependent pathway in striatal MSNs.
An OGD containing solution was applied for 3 minutes on striatal slices in the presence of either the D1-like-R antagonist SCH23390 or in the presence of SCH23390 plus the NO donor SNAP. Similarly to what observed in patch-clamp experiments (Figure 3B), 10 μmol/L SCH23390 prevented i-LTP induction in MSNs intracellularly recorded with sharp electrodes in fact, in these neurons, the EPSP amplitude measured 30 minutes after OGD was 104.9 ± 4.7% of the baseline (n = 4, Figure 4A). Conversely, the application of 100 μmol/L SNAP plus 10 μmol/L SCH23390 (n = 4) did not alter i-LTP induction. Under this experimental condition, in fact, the i-LTP was similar to that observed in control condition (157.6 ± 4.4%, P > 0.05, Figure 4A). This experiments suggest that NO has a pivotal role in mediating the D1-like-R effects on i-LTP in striatal MSNs.
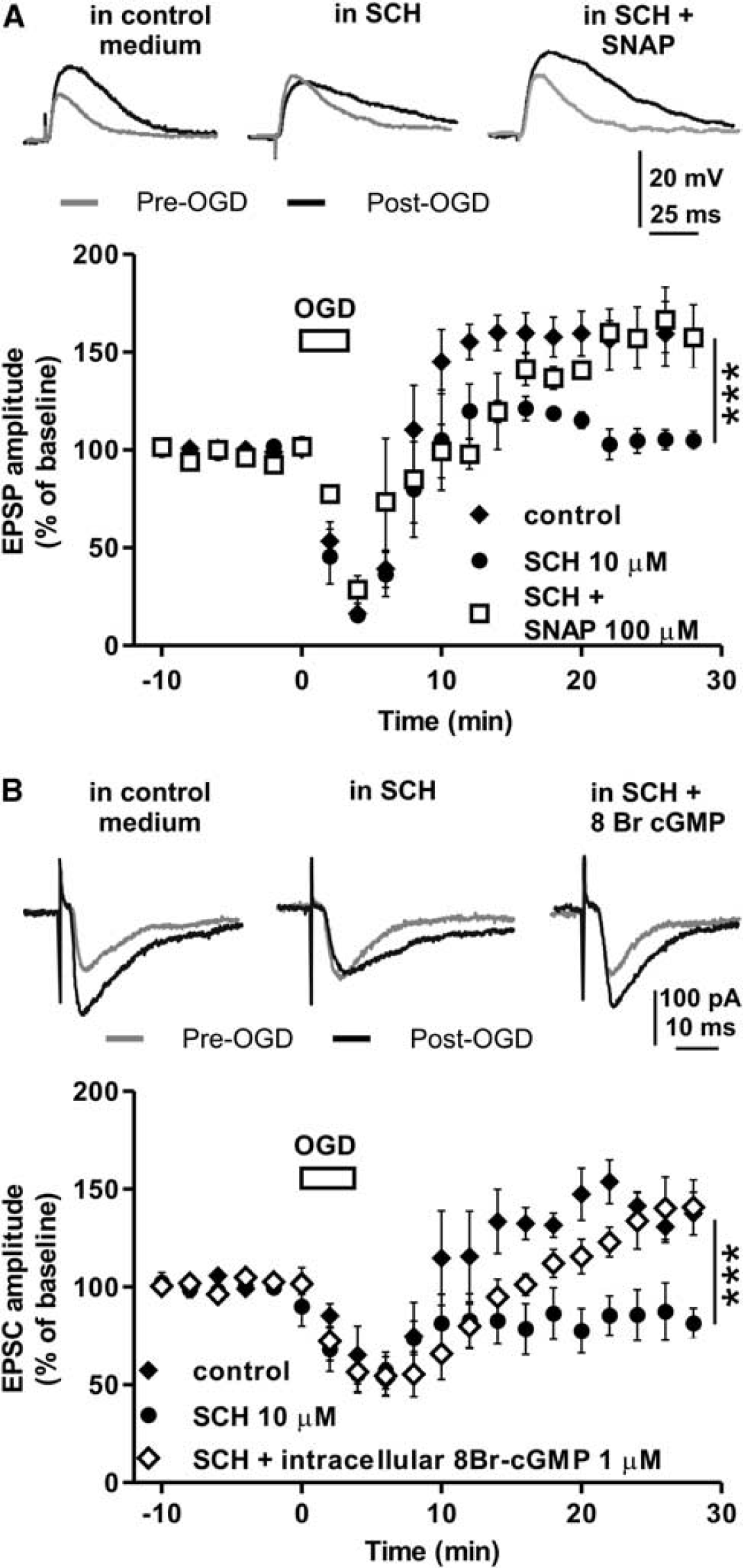
Exogenous NO or enhanced cGMP levels restore i-LTP abolished by D1-like receptor antagonism. (
To further confirm the role of NO in the D1-like-R mediated i-LTP we performed patch-clamp recordings of MSNs injecting 1 μmol/L of the cGMP analog 8Br-cGMP into MSNs through the recording pipette and we applied OGD in the presence of 10 μmol/L SCH23390 given in the bathing solution (n = 5). In these conditions OGD induced i-LTP in MSNs, in fact the EPSC amplitude measured 30 minutes after OGD was 140.7 ± 14.1% (Figure 4B). Therefore, cGMP production is involved in the D1-like-R mediated i-LTP, most likely via the increase of striatal NO levels.
In order to confirm the role of D1-like-R stimulation in NO mediated i-LTP in the striatum we also used DA-denervated rat slices obtained by injecting animals with 6-OHDA, a neurotoxin that destroys dopaminergic projections to the striatum by preferentially targeting DA neurons located in the substantia nigra pars compacta (SNc) (see methods). Intracellular recordings of MSNs performed in slices from sham-operated and 6-OHDA DA-denervated rats showed that, while 3 minutes OGD induced i-LTP in slices obtained from sham-operated rats (EPSP at 30 minutes after OGD 159.9 ± 8.4%, n = 3), in 6-OHDA slices i-LTP was absent (EPSP at 30 minutes after OGD 97.5 ± 1.7%, n = 3, Sham versus 6-OHDA F(1,80) = 180.4, P < 0.001, Figure 5). Taken together these data confirmed that i-LTP in striatal MSNs depends on endogenous DA and on the stimulation of D1-like-R. In order to establish whether striatal DA mediates i-LTP by a NO-dependent intracellular pathway, we recorded MSNs from 6-OHDA slices in the presence of the cGMP analog 8Br-cGMP (n = 3). In this condition OGD produced i-LTP in MSNs (154.1 ± 7.3%, F(1,80) = 124.5, P < 0.001, Figure 5) indicating that striatal DA is able to stimulate an increase of cGMP in MSNs. Thus, we suggest that striatal DA, released during an ischemic episode, stimulates the synthesis of NO in striatal NOS positive interneurons by activating D1-like-Rs. During energy deprivation NO might diffuse to all the striatal neuronal population, including MSNs of both the direct and indirect basal ganglia pathway, activating the sGC/cGMP biochemical cascade and contributing to i-LTP induction (Figure 6).
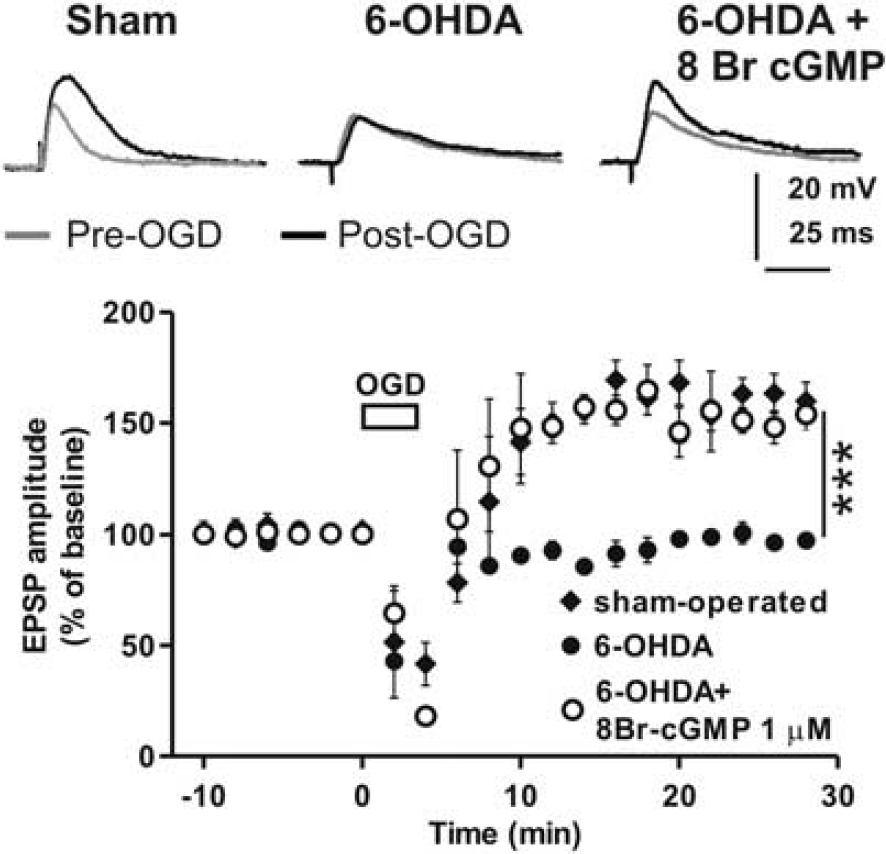
8Br-cGMP restores i-LTP in 6-OHDA DA-denervated slices. Time-course plots and example EPSP traces showing the effect of OGD in striatal MSNs recorded from sham-operated and 6-OHDA DA-denervated rats. Note the lack of i-LTP in the 6-OHDA DA-denervated group. Bath application of 1 μmol/L 8Br-cGMP restores i-LTP in slices from 6-OHDA animals. ***P < 0.001. Upper traces, representative of recordings from sham-operated (left), 6-OHDA DA-denervated (middle) and 6-OHDA DA-denervated slices treated with 8Br-cGMP (right), are acquired 5 minutes before (gray traces) and 30 minutes after the onset of OGD (black traces).
DISCUSSION
This study represents the first demonstration that NO/cGMP pathway plays a pivotal role in post-ischemic long-term potentiation of excitatory transmission in the striatum. We demonstrated that NO, possibly produced by striatal NOS-positive interneurons, via the stimulation of D1-like-Rs located on these cells, is critical for i-LTP induction. Our protocol for i-LTP induction involved the use of a nominally magnesium-free external solution in order to better unmask the NMDA receptor component for the measure of postsynaptic response. This condition was reported to initiate seizure-like activities in other brain structures such as the hippocampus. 40 However, in MSNs recorded in our slice preparation no seizure-like activities could be observed. Accordingly, MSNs have been characterized as ‘silent’ and ‘non-bursting’ neurons. 41 In fact, although they possess slow conductances for inward currents which they share with other mammalian central neurons, their oscillating and bursting activities are suppressed by the high resting membrane potential and the effective potassium currents present in MSNs. 41
Based on outcomes from double labeling studies, it is likely that i-LTP induction occurs in a majority of both direct and indirect pathway MSNs and that the magnitude of the effect is similar in these populations.
NO plays a physiological role in neuronal cell signaling, in particular for ischemic tolerance induced by transient ischemia, an effect involving the activation of sGC and the production of cGMP. 42 Moreover, NO overproduction may cause the formation of oxidant species, such as peroxynitrite, and neuronal energy impairment leading to neurodegeneration. 9
The overproduction of NO is involved in the pathophysiology of brain ischemia 8 as well as in the formation of activity-dependent synaptic plasticity.12,13 It has been suggested that NO may be protective or destructive depending on the stage of evolution of the ischemic process and on the cellular source of NO. 43 However, the mechanism by which NO is involved in i-LTP has never been explained so far. In the present study, by using an in vitro model of ischemia, we show that blockade of NOS by L-Name or 7-Nina prevents i-LTP induction. Furthermore, the finding that NOS inhibitors do not modify i-LTP after its induction reveals that NO production is necessary for the induction of this pathological form of synaptic plasticity, but not for its maintenance. Moreover, we observed that SNAP, a NO donor, is able to restore i-LTP in the presence of NOS blockers, further supporting the critical role of NO during an ischemic insult. In line with these experiments, blockade of NO synthesis markedly reduced the cortical and striatal infarct volume induced by cauterization of the middle cerebral artery in rats. 44 It is worth noting that in the striatum NO is able to trigger the induction of i-LTP, but not its maintenance. In line with this observation, it has been reported that in the hippocampus and entorhinal cortex NO is involved in the induction, but not the maintenance of seizure-like activity. 45
NO is a diffusible neuronal messenger that activates sGC and increases intracellular cGMP levels during an ischemic episode in the brain. 46 Accordingly, we found that inhibition of sGC by ODQ, and of PKG by Rp-8Br-PET-cGMP prevented i-LTP induction. However, this pathological form of synaptic plasticity was not prevented by the blockade of sGC when ODQ was applied in the presence of a cGMP analog such as 8Br-cGMP.
It is well known that a massive DA release occurs during ischemic events in the striatum and that endogenous DA amplifies neuronal damage caused by excitotoxicity and energy deprivation. 47 We have previously reported the involvement of D1R in i-LTP of MSN. 22 Interestingly, here we show that antagonism of D1-like-R by SCH23390 prevents i-LTP in both SP positive and A2A positive MSNs, as confirmed by the experiments utilizing the neuronal sub-type identification by immunofluorescence (Figure 3A). Thus, our study provides evidence that i-LTP is mediated by D1-like-R activation, NO production, increased cGMP levels and PKG activation in a majority of both direct and indirect pathway cells.
Considering that striatal MSNs represent a heterogeneous population in terms of DA receptor expression, and that the prevailing view is that D1Rs and D2Rs are segregated into separate subpopulations, we suggest a pivotal presynaptic role of D1-like-R in the release of NO by NOS positive interneurons. In fact, striatal NOS interneurons express DA D1/D5 mRNA and D1R protein26–28 and D1R agonist administration increases striatal NO efflux via a NOS- and D1R-dependent mechanisms.29,48 Interestingly, our study suggests that, during a brief ischemic episode, DA neurotransmission may modulate striatal NO signaling via D1-like-R mediated pathway expressed in NOS positive interneurons (Figure 6). In line with this hypothesis, exogenous NO was able to restore i-LTP even in the presence of a D1-like-R antagonist. NO produced by NOS positive interneurons might thus diffuse to striatal neurons, pass the MSN plasma membrane at dendritic level and increase intracellular cGMP levels by activating sGC.
NO/cGMP signaling has numerous effects on protein kinases and phosphodiesterases in striatal MSNs 49 which ultimately modulate membrane excitability and corticostriatal transmission. 30,50 Striatal MSNs have been shown to express high levels of intracellular sGC and proteins associated with cGMP signal transduction pathways 51 and we have shown that the cGMP-activated protein kinase (PKG) exerts a critical role in i-LTP induction. Thus, it is plausible that the release of NO, modulated by D1-like-R located on NOS positive interneurons, is involved in MSN i-LTP during an ischemic episode. Our experiments support this hypothesis, in fact, the cGMP analog 8Br-cGMP, applied into MSNs via patch pipette, was able to induce i-LTP even in the presence of a D1-like-R antagonist.
The role of DA stimulation and NO/cGMP pathway in striatal i-LTP were confirmed by the experiments performed in DA-depleted conditions obtained by utilizing slices from 6-OHDA DA-denervated rats. In these conditions we showed that i-LTP did not occur. Accordingly, striatal NOS activity was found to be depressed in 6-OHDA lesioned animals52,53 and patients with PD.54,55 Interestingly, our experiments show that i-LTP could be induced when the cGMP analog 8Br-cGMP, was applied in these DA-denervated slices. Therefore, we suggest that DA has a major role in striatal i-LTP by stimulating D1-like-Rs in NOS positive interneurons. We also suggest that the diffusion of NO would activate the NO/sGC/cGMP/PKG cascade in the MSNs of both the direct and indirect basal ganglia pathways (Figure 6). Further studies are required to elucidate the mechanism through which D1-like-R pathway modulate NO-cGMP signaling for the treatment of ischemic stroke and other neurological disorders.
DISCLOSURE/CONFLICT OF INTEREST
Dr Calabresi serves as an editorial board member of Lancet Neurology, the Journal of Neuroscience, Movement Disorders and Synapse; receives research support from Bayer Schering, Biogen, Boehringer Ingelheim, Eisai, Merck Chemicals, Novartis, Lundbeck, Sanofi-Aventis, Sigma-Tau, and UCB Pharma; and from Ricerca Corrente IRCCS, Ricerca Finalizzata IRCCS [European Community Grants SYNSCAFF and REPLACES], the Italian Minister of Health and AIFA (Agenzia Italiana del Farmaco). All other authors reported no biomedical financial interests or potential conflicts of interest.