
Abstract

Pathophysiology of migraine
The specific cause of migraine headache remains unknown. Current theories suggest the initiation of a migraine attack involves a primary CNS dysfunction with subsequent activation of the trigeminovascular system that gives rise to the headache pain. Studies in familial hemiplegic migraine (FHM), a rare subtype of migraine with aura, have led to the proposal that the primary deficit may be genetic abnormalities in neuronal ion channels (P/Q calcium channel) that alter the excitability of CNS neurones. These mutations have been shown in vitro to produce a broad spectrum of changes in channel function. As a result of these studies into FHM, it has been suggested that migraine could be a ‘channelopathy’. However, it is noteworthy that migraine with and without aura are likely to be very much more complex genetically than FHM. Nevertheless, it is still likely that a fundamental deficit within the CNS leads to neural dysfunction in the CNS of migraineurs that makes them unable to compensate adequately for the effects of migraine trigger factors when they are strong, frequent or convergent.
What evidence is there for abnormal brain excitability or responsiveness in migraineurs? Many studies have looked at cortical hyperexcitability as the possible cause of a reduced threshold and increased responsiveness to triggering of migraine attacks, while others focus on the spreading depression of the brain. The presence of cerebral blood flow changes, both oligemia and transient hyperaemia, have been interpreted as supportive of early CNS dysfunction in the initiation of a migraine attack and have been used to support a role for spreading depression, or indeed activation, in the pathogenesis of migraine with aura. During migraine without aura, no significant haemodynamic changes are observed. It is interesting to speculate that the presence or absence of aura may reflect a threshold phenomenon that is determined by the extent of the cortical dysfunction (e.g. spreading depression) produced by migraine trigger factors. Such an explanation could accommodate those patients who present symptoms of migraine with aura and migraine without aura, suggesting that these conditions are part of a single entity disease spectrum rather than being distinct disorders.
As a result of clinical observations showing the development of migraine-like episodes in patients undergoing surgery to implant electrodes in the periaqueductal grey and raphe nuclei for the treatment of chronic pain, it was hypothesized that CNS dysfunction early in a migraine attack could provoke changes in these brain stem nuclei. Diener and colleagues (1) used positron emission tomography (PET) to examine the changes in regional cerebral blood flow as an index of neuronal activity, during spontaneous migraine attacks. They found selective activation of the same monoaminergic brain stem regions (raphe nucleus: serotonergic neurones and the locus coeruleus: noradrenergic neurones) and the periaqueductal grey. Thus, during the migraine attack there was a clear dysfunction within the centres that are involved in cerebrovascular regulation and the endogenous control of pain. The anterior cingulate cortex, related to pain perception, was also activated during spontaneous migraine attacks and this was reduced concomitant with headache pain relief after administration of a 5-HT1B/1D receptor agonist antimigraine drug.
In an innovative study to test the whether this brain stem activation during migraine was simply a function of head pain or was inherent to the attack itself, an experimental head pain study was conducted in healthy volunteers (2). In these studies capsaicin was injected subcutaneously into the right forehead to evoke a highly painful burning sensation in the first division of the trigeminal nerve. No brain stem activation was found in the acute pain state compared with the painfree state. Thus, brain stem activation seems to be unique to migraine.
The central processes involved in the initiation of a migraine attack are poorly understood compared with the factors involved in the pathophysiology of migraine headache pain that is known to be mediated through activation of the trigeminal nervous system. A simplified flow diagram of an integrated neurovascular hypothesis for migraine is shown in Fig. 1.

Simplified flow diagram of an integrated neurovascular hypothesis for migraine.
Trigeminovascular system
Our understanding of the pathophysiological mechanisms involved in migraine pain has been driven by comprehensive studies from Professor Michael Moskowitz's laboratory, together with increased knowledge of the receptor distribution within the trigeminovascular system and the introduction of highly effective serotonergic acute antimigraine drug therapies. Understanding the pharmacology of antimigraine agents requires some background knowledge of the anatomy and physiology of the trigeminovascular system.
The brain has little sensory innervation and, like other viscera, it is the capsule structures (meninges) and large blood vessels that are the most significant pain-producing intracranial tissues. The ophthalmic division of the trigeminal nerve (V cranial nerve) primarily provides the sensory afferent innervation of the cerebral blood vessels but also has the potential to play an efferent role in pathophysiological conditions.
The trigeminal nerves arise from pseudo-unipolar neurones in the trigeminal ganglia and project to the intracranial extracerebral blood vessels in the meninges (peripheral terminals) and behind the blood-brain barrier to neurones in the trigeminal nuclei in the brain stem and upper cervical spinal cord (central terminals). These fibres are the pathway for nociceptive transmission from intracranial blood vessels to the brain where headache pain is registered and perceived at the level of consciousness. Many of the second order sensory relay neurones in the trigeminal (V) brainstem complex also receive convergent afferent inputs from other craniofacial tissues. This convergence may be an important factor in migraine as prolonged afferent input could cause changes in the sensitivity and receptive field properties of these neurones.
Figure 2 shows a diagrammatic view of several key steps thought to be involved in migraine pain: (a) intracranial blood vessel dilatation that activates perivascular sensory trigeminal nerves, (b) release of vasoactive neuropeptides from the activated trigeminal sensory nerves, and (c) the relay of pain impulses from the activated peripheral sensory nerves to second-order sensory neurones within trigeminal nuclei in the caudal brain stem and upper cervical spinal cord (Cl and C2) (trigeminocervical complex). Pain signals are then transmitted to the thalamus, via the quintothalamic tract which decussates in the brainstem, and on to higher cortical centres where migraine pain is registered and perceived. Symptoms commonly associated with a migraine attack such as nausea, phonophobia and photophobia, are produced when other associated central pathways are activated.

Peripheral and central targets in the trigeminovascular system for the antimigraine activity of triptans.
Mechanisms of antimigraine action of the 5-HT1B/1D receptor agonists
One of the major research objectives of the scientists at Terlings Park, was to demonstrate the mechanisms by which antimigraine compounds such as the ergots and, more particularly, the triptan 5-HT1B/1D receptor agonists are active antimigraine agents.
Receptor distribution
Longmore and coworkers used polyclonal antibodies to investigate the distribution of the 5-HT1B/1D receptors at which the triptans act (3). Their studies demonstrated that these receptor subtypes have a differential distribution within the human trigemino-cerebrovascular system. Only 5-HT1B receptor protein was detected in the medial layer of the middle meningeal artery, where it was colocalized with the actin present in the smooth muscle cells of this layer. The vessels were devoid of 5-HT1D receptors.
In contrast, only the 5-HT1D receptor was detected on the ophthalmic branch of the trigeminal sensory nerves that innervate blood vessels in the meninges and project behind the blood-brain barrier into the trigeminal nucleus caudalis, synapsing with second-order sensory neurones. 5-HT1D receptors were also present in the trigeminal tract and the solitary tract nucleus. This latter observation suggests that the efficacy of the triptans in reducing nausea may not only be due to the reduction of migraine headache pain, but also to a direct action at the level of the solitary tract nucleus which is intimately involved in the autonomic control of emesis and nausea.
This information on receptor distribution can be integrated into the scheme used to explain the genesis of headache pain (Fig. 1) to provide a mechanistic framework within which to investigate the actions of the triptan drugs. Thus, it was hypothesized (a) that the intracranial blood vessels that become dilated during the migraine attack are constricted by the stimulation of the 5-HT1B receptors, (b) by stimulation of 5-HT1D receptors, the triptans inhibit the release of the vasoactive peptides from the perivascular nerve terminals and thus interrupt the vicious vasodilator and inflammatory cycle that takes place in the meninges, and (c) at the level of the CNS, the activation of the same 5-HT1D receptor, interrupts pain signal transmission within the trigeminal nucleus caudalis. Thus, the 5-HT1B/1D receptors through which the triptans act are ideally placed within these nervous pathways to interrupt the headache pain (see Fig. 2).
The strategy of acting on prejunctional receptors is very convenient because it prevents the release of all the transmitters released by the sensory fibres. The post-junctional inhibition of the response only acts on a single component of the transmitter cascade. The 5-HT1F receptor that is being actively investigated at present, has a similar distribution to that of the 5-HT1D receptor since it is present on the same fibres both centrally and in the periphery.
Pharmacological properties of antimigraine compounds
The ergots were the first 5-HT agonists used for the treatment of migraine. They are not very useful when given orally due to their unpredictable absorption and their very low bioavailability. This is improved by intranasal or rectal administration. The peripheral vascular effects of the ergots were already known during the Middle Ages, when they caused the disorder known as Saint Anthony's fire due to the ingestion of bread infected by Claviceps purpurea, the ergot-producing fungus. This caused such a profound vasoconstriction of the extremities that patients felt as if they were burning and eventually became black. The victims looked as if they had been charred by fire and the epidemic got its name from this fact. Nausea and dizziness, the common side-effects of ergots, may be explained by the actions of these compounds on multiple monoamine receptors.
Ergots are potent agonists at the 5-HT1B/1D receptors and this explains their antimigraine effect. They also act on 5-HT1A receptors, an effect that is probably responsible for the production of nausea and dysphoria. The constriction of the peripheral vasculature is probably through alpha adrenoceptors and 5-HT2A receptors. Agonist activity at dopamine D2 receptor produces gastrointestinal disturbances, nausea and emesis. Thus, from their pharmacological activity at monoamine receptors, it is possible to conclude that although ergots are good antimigraine agents, they also have many other unwanted effects.
The triptans, like the ergots, have potent activity at the ‘antimigraine’ 5-HT1B/1D receptors, but have a much weaker action at 5-HT1A receptors. Indeed, these compounds can be considered as ‘super-selective’ ergots because they lack activity on the other monoamine receptors that bring many of the unwanted effects of the ergots. It is interesting that the different triptans have a variable affinity for the 5-HT1A receptor, and this may underlie differences in their ability to relieve nausea. Thus, although they are effective against the headache pain and thereby reduce associated nausea, they may also induce it directly, as a result of 5-HT1A receptor activation.
Pharmacokinetics and metabolism
The major route of rizatriptan metabolism is through monoamine oxidase A (MAO-A). A minor active N10-desmethyl metabolite circulates in plasma and comprises about 12% of the total circulating drug. Rizatriptan is excreted from the body as an indole-3-acetic acid that is inactive at monoamine receptors. This monoamine metabolism underlies the contra-indication for rizatriptan (and sumatriptan and zolmitriptan) use with monoamine oxidase inhibitors and the recommendation to reduce the dose of rizatriptan from 10 to 5 mg in patients receiving concomitant propranolol therapy. This metabolic interaction is not with propranolol itself but with one of its metabolites, and causes an increase in plasma rizatriptan Cmax of 60%.
The demonstration of a pharmacokinetic advantage for rizatriptan in comparison with sumatriptan was essential for the continued development of the compound during the drug discovery phase. Rizatriptan reached peak plasma concentrations at 1.0 h after administration compared with 2.5 h for sumatriptan, and its bioavailability was threefold better than that of sumatriptan. The half-lives of both compounds are very similar, between 2 h and 2.5 h (4). The reduced time to reach peak concentrations suggested that rizatriptan might produce faster antimigraine effects. In addition, its increased bioavailability could make it possible to reduce variability when an individual takes the drug on different occasions, as well as to diminish inter-individual variability: the higher the bioavailability, the more consistent the absorption.
There have been many discussions on the potential benefits of drug half-life and prolonged receptor activity when treating migraine. However, an important counter view is that when vasoconstrictor compounds are used for treating an episodic disorder, such as migraine attacks, it is better to use drugs with a relatively short half-life and administer a second dose only if needed. In this way prolonged vasoconstriction and any associated safety risks are minimized.
Migraine is known to alter drug absorption because gastrointestinal stasis is very common during the attacks. This can cause delayed and variable absorption of some antimigraine drugs. This is usually managed by administering gastroprokinetic agents, such as metoclopramide, to improve the absorption profile of these compounds. A surprising finding with rizatriptan, during the clinical pharmacology studies, was that the absorption of rizatriptan is not affected during migraine attacks: the absorption of a 5-mg dose of rizatriptan had similar kinetics during the attacks and between them (5). This indicates that the absorption profile and the performance of the compound were unaffected by the disease, as was later confirmed by the clinical experience. This consistent absorption leads to a reliable and predictable therapeutic effect. Rizatriptan is the only one of the currently marketed triptans whose absorption is unaffected by the disease process (see Table 1).
Clinical pharmacokinetic properties of triptans
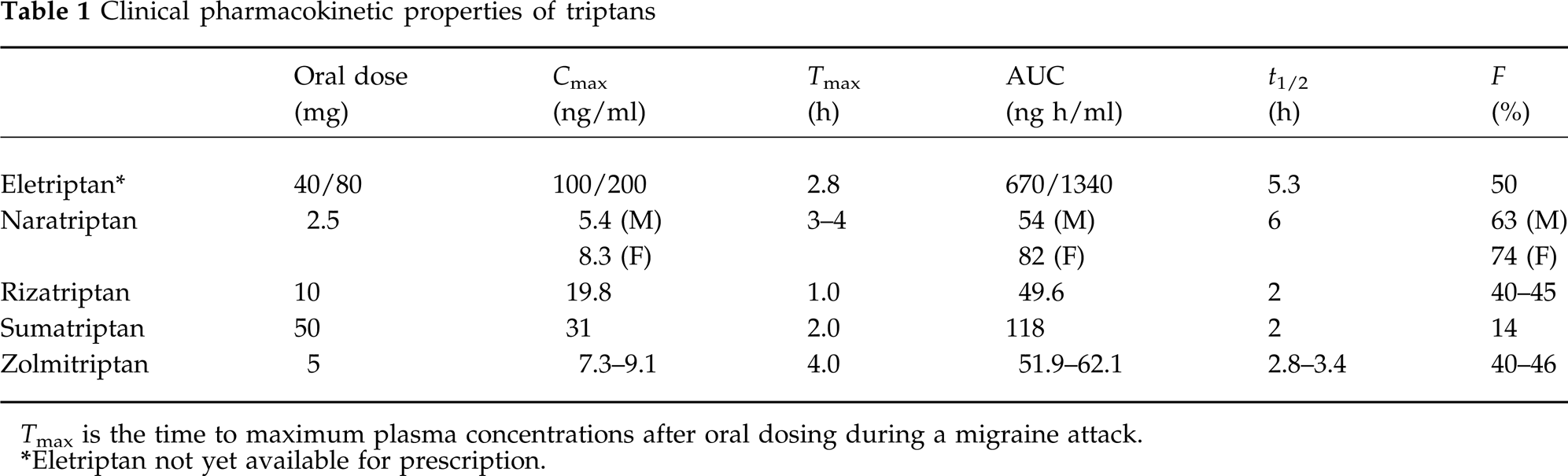
Tmax is the time to maximum plasma concentrations after oral dosing during a migraine attack.
Eletriptan not yet available for prescription.
Mechanisms of antimigraine action of rizatriptan
The trigeminovascular hypothesis was used in the laboratory to design a study programme to investigate the potential mechanisms of action of rizatriptan and other compounds in the triptan class. A secondary goal was to find new assays that could provide insights into migraine pathophysiology and antimigraine drug actions. The studies analysed the actions of the drugs on the cerebral vasculature, on the peripheral trigeminal nerves and on the central pain signal transmission within the trigeminal nucleus caudalis.
Vascular actions of rizatriptan
Comparison of the contractile response of the middle meningeal artery with serotonin and with rizatriptan show that both compounds produce a very similar maximal response (6, 7). Since rizatriptan has contractile activity only at 5-HT1B receptors, this indicates that predominant serotonergic contractile effect on this vessel is mediated by the 5-HT1B receptor (Fig. 3).

In vitro dose-response curves of human middle meningeal and coronary arteries to 5-HT and rizatriptan. Craniovascular selectivity comes from a higher density of 5-HT1B receptors in target middle meningeal blood vessels than in coronary blood vessels, and because all triptans are devoid of activity at 5-HT2A receptors at clinically relevant doses. Arrows represent therapeutic concentration range at which the compounds are virtually inactive in the coronary artery assay (modified from 6 and 7).
The demonstration that 5-HT1B receptors are present in the coronary arteries has caused concern over potential for adverse cardiac events with the ergots and the triptans. These events, however, are rare with all the triptans since the 5-HT1B receptor reserve appears to be lower in the coronary than the meningeal arteries, and collectively all triptans lack activity at 5-HT2A receptors which are the receptors that mediate most of the serotonergic contraction in coronary vessels. Taken together, these factors confer craniovascular selectivity for the class. Indeed Fig. 3 shows that in human isolated coronary artery there is little contractile response to rizatriptan at therapeutic concentrations and that the 5-HT1B-mediated section of the response is small relative to the 5-HT2A receptor-mediated component.
The actions of several antimigraine compounds (ergots and triptans) on human coronary arteries have been recently compared head-to-head in a single laboratory (8). The studies showed that, while these compounds can contract human coronary arteries in vitro, they are unlikely to cause myocardial ischemia at therapeutic plasma concentrations in healthy subjects. Any small differences between the triptan agents in their coronary contractility occurred at drug concentrations that are super-maximal clinically, and so irrelevant when they are used therapeutically against migraine. In the same study, the reversibility of the contraction of the coronary arteries was studied. The contraction produced by high concentrations of the triptans was very easily reversed by washing the artery with physiological salt solution. In contrast, the contractile response to ergot alkaloids was sustained. While it may be arguable that a long-lasting vasoconstrictor action is of potential therapeutic benefit in the middle meningeal artery, in the case of the coronary artery, it is preferable to avoid a long-lasting constrictor effect. In all considerations of triptan use, it should be remembered that these drugs are contraindicated in persons with cardiovascular disease, and careful assessment is needed before prescribing these compounds to those with risk factors.
Peripheral action on the trigeminal nerves
Activation of trigeminal nerve terminals in the periphery releases vasoactive neuropeptides such as substance P, neurokinin A and calcitonin gene related peptide (CGRP). CGRP is one of the most potent and long-lasting vasodilators known in the body. Receptors for this peptide are highly expressed in the smooth muscle layer of the human middle meningeal artery, making it a good candidate for involvement in the pathogenesis of migraine. Indeed, CGRP is known to be raised in external jugular venous blood during migraine, and normalized with successful treatment.
Experimentally CGRP may be given intravenously or released by electrical stimulation of perivascular trigeminal nerves to cause a reproducible vasodilation which can be monitored directly using intravital microscopy (9).
In these intravital studies, rizatriptan blocked the electrically evoked release of CGRP from perivascular sensory nerves in the meninges (Fig. 4), but did not inhibit the dilation to CGRP when it was given intravenously. This shows that rizatriptan is not a CGRP peptide receptor antagonist but a peptide release inhibitor. This action occurs through the stimulation of prejunctional 5-HT1B/1D receptors. As a class, all the triptan 5-HT1B/1D agonists are capable of blocking this neurogenic meningeal dilation (9, 10).

Dose-dependent inhibition by rizatriptan of the vasodilation produced by electrical stimulation, exogenous CGRP and substance P. The compound blocks electrical but not exogenous neuropeptide-induced vasodilation, suggesting a prejunctional inhibition of neuropeptide release. ∗P<0.05 from control (modified from 10).
Central sites of rizatriptan action
Electrical stimulation of the dura mater triggers the firing of neurones in the nucleus caudalis of the brain; a result of neurotransmitter release from the incoming trigeminal sensory fibres. This activation of these second-order sensory relay neurones in the brainstem can be monitored electrophysiologically by recording the firing activity of single cells (11). Rizatriptan, administered systemically, dose-dependently inhibited these neuronal responses (Fig. 5). This finding indicates that rizatriptan enters the brain and reduces pain signal transmission within the nucleus caudalis, probably by activating 5-HT1D receptors, thereby inhibiting neurotransmitter release.

Dose-dependent inhibition by rizatriptan of firing of a single pain relay neurone in the trigeminal nucleus caudalis caused by noxious stimulation of the dura mater (modified from 11).
Central activity and physicochemical properties of triptans
On the basis of its hydrophilic physicochemical characteristics, it has been argued that sumatriptan does not penetrate into the brain, but examination of its central adverse effects reveals a similar profile to triptans that are thought to enter more easily into the CNS. In contrast to sumatriptan, eletriptan is a lipophilic compound, suggesting that it should penetrate easily into the brain across the cell membranes of the blood-brain barrier.
A series of studies (12) was undertaken to compare the effects of the triptans (rizatriptan, sumatriptan, naratriptan, zolmitriptan and eletriptan) in the central activity assay, examining their effects on durally evoked firing of single neurones in the trigeminal nucleus caudalis of rats. The findings showed that rizatriptan, naratriptan and zolmitriptan, as expected, penetrated into the brain and inhibited durally evoked neuronal firing. The observations with sumatriptan and eletriptan were, however, of particular interest. First, sumatriptan showed similar inhibitory activity to the others in the class, suggesting that it too can access central sites of action. This fits with the presence of central effects with sumatriptan in clinical use that are similar to the so-called brain penetrant triptans. Second, unexpectedly, eletriptan was inactive over the same dose range as the other members of the class, despite its higher lipophilicity. This observation was even more striking since receptor binding studies show that eletriptan is one of the most potent agonists at rat and human 5-HT1B/1D receptors and, as such, was expected to be one of the more active in the class.
It is now becoming apparent that, in addition to lipophilicity, one of the major determinants of the entry of drugs into the brain is whether they are substrates for a multidrug efflux pump situated in the luminal membrane of the capillary endothelial cells that make up the blood-brain barrier. This strategic location of P-glycoprotein (Pgp) underlies its function which is to keep toxins (drugs) out of the brain. Pgp constitutes the major system for preventing access of chemotherapeutic agents to the brain and has a particular affinity for lipophilic compounds. It has also been suggested that Pgp has similar structure activity requirements to the CYP3A4 drug-metabolizing enzyme in the liver that is also predisposed to lipophilic compounds. Current thinking is therefore that the brain entry of any drug is the net product of passive influx (governed primarily by lipophilicity) and active efflux mediated by the mdrl Pgp efflux pump.
The brain penetration of the triptans was studied in mice with a homozygously disrupted mdrl gene that resulted in an absence of functional Pgp in the microvasculature of the brain (mdrl −/− mice) and compared with entry in wild-type mice with the pump present (mdrl +/+). The plasma concentration profile and protein binding of eletriptan was similar in mdrl −/− mice and mdrl +/+, but there was a 38-fold higher penetration of eletriptan into the brain of the mice deficient in the pump. A very minor increase of naratriptan and rizatriptan entry into the CNS was also detected in these mice in line with their relative lipophilicities.
It is interesting to speculate what impact this observation that eletriptan is a Pgp substrate in mice could have on the clinical profile of the compound. A comparison between the clinical pharmacokinetics of triptans at therapeutic doses (Table 1) indicates that the absolute dose and peripheral exposure (maximum plasma concentration Cmax ng/ml and AUC [area under the plasma curve]) to eletriptan is considerably higher than the other triptans. Thus, compared with the others in the class, there appears to be a mismatch with eletriptan between its potent receptor activity and the high plasma levels that are needed for antimigraine effects. This observation suggests (a) that peripheral mechanisms cannot alone explain the antimigraine efficacy of triptans since eletriptan plasma levels would have been expected to be similar or lower than the others in the class, and (b) that eletriptan needs a high dose to reach high plasma levels in order to provide a sufficient driving force to overcome the effects of the Pgp pump and gain access to the brain and central sites of antimigraine action. While it could be argued that eletriptan has a somewhat higher (approximately threefold) extent of plasma protein binding than the other triptans; this difference seems insufficient to account for the discrepancy in brain entry from others in the class. Most interestingly, these observations indicate that inhibition of pain signal transmission at central sites may be an important mechanism for optimizing the antimigraine efficacy of the triptans.
Sensitization of the trigeminovascular system in the maintenance and aggravation of migraine
Our recent studies have focused on the possibility that changes in central pain signal processing may also occur as a result of vasodilation during a migraine attack. In order to investigate this phenomenon, electrophysiological recordings were made from single convergent (receiving nociceptive input from the dura and non-nociceptive input from the vibrissae) wide-dynamic range sensory neurones in the caudal brainstem of anaesthetized rats in combination with intravital microscopy to monitor blood vessel calibre (13). The recordings showed that when the meningeal dural blood vessels were dilated by CGRP, the responses to the innocuous whisker stimulation was facilitated (Fig. 6a). This suggests that during a migraine attack in which there is a prolonged vasodilation, these neurones may become abnormally super-sensitive to all types of sensory stimuli. This is analogous to the phenomenon known as ‘wind up’ that is observed in the dorsal horn of the spinal cord. This sensitization process could be involved in the intensification of migraine during an attack and the transformation of the disorder and so could constitute another important target for the development of new antimigraine therapies. Interestingly pretreatment with a brain penetrant 5-HT1B/1D agonist (L-741,604), completely blocked trigeminal neuronal sensitization, an effect that could be involved in the therapeutic profile of these compounds (Fig. 6b). Sensitization induced by CGRP-dilation was also blocked by a glutamate NMDA antagonist suggesting a probable role for glutamate in this ‘wind up’ process.

(a) The responses of trigeminal neurones receiving dural inputs are facilitated when dural vessels are dilated by CGRP. (b) The 5-HT1B/1D receptor agonist L-741,604 inhibits the sensitization of trigeminal neurones produced by CGRP (modified from 13).
Conclusions
In summary, the triptan 5-HT1B/1D receptor agonists appear to have three key sites of antimigraine action:
The meningeal vasculature where they cause vasoconstriction thereby reducing the activation of trigeminal sensory nerves. All the triptans are selective for the intracranial over coronary vascular beds, because of the higher 5-HT1B receptor density in the meningeal blood vessels and the lack of 5-HT2A agonist activity in the molecules.
The perivascular trigeminal nerves, where they can block neuropeptide release, reduce inflammation and promote the normalization of swollen blood vessels, thus interrupting the cycle of vasodilation in the periphery.
The central trigeminal nerve terminals where the triptans interrupt neurotransmitter release and so inhibit processing of pain signals in the nucleus caudalis and block the sensitization of trigeminal neurones.
The triptans act simultaneously in concert at all of these sites to deliver their beneficial therapeutic effects, relieving the headache pain as well as its associated symptoms.
Footnotes
∗
Based on a presentation to the Symposium held during the IX Meeting of the International Headache Society, Barcelona, Spain, 24 June 1999.