
Abstract
We report our experience with congenital cholesteatoma over a span of 20 years with an emphasis on presenting characteristics and predictors of outcome.
We conducted a retrospective review from 1981 through 2000.
One hundred seventy-two congenital cases were identified in 167 patients. Five patients had bilateral disease. The majority (72%) were found in boys, with an average age of 5.0 years. Hearing loss was slight to moderate. When confined to 1 quadrant, cholesteatoma was anterosuperior in 82% of cases; 47% had cholesteatoma in 2 or more quadrants. Ossicular chain involvement was found in 43% of all cases, and mastoid extension was evident in 23%. The rate of recurrent disease was directly related to the extent and number of quadrants involved.
To our knowledge, this is the largest series of congenital cholesteatomas to be reported. This review confirms the male predominance and predilection for the anterosuperior quadrant. The extent of cholesteatoma and its relation to residual disease should be used as a guide for planning a second-look procedure.
Although cholesteatoma was first described in 1683 by DuVerney, Körner 1 was the first to hypothesize that there may be a congenital origin of cholesteatomas. Until recently, the importance of a congenital origin of cholesteatoma was called into question. Nager 2 and Fernandez et al 3 believed that it may exist but was too rare to be of clinical significance. Similarly, Juers 4 stated that in 30 years of practice, he had never encountered a case.
The distinction between congenital and acquired cholesteatoma was clarified in 1965, when Derlacki and Clemis 5 published a report that presented criteria for the diagnosis of congenital cholesteatoma of the middle ear. Subsequently in 1986, Levenson et al 6 modified the Derlacki and Clemis criteria for diagnosis and established the criteria that are now widely accepted. These include an intact tympanic membrane with no history of previous perforation or otorrhea and no prior otologic surgery. Previous acute otitis media no longer excludes the diagnosis of congenital cholesteatoma.
The embryonic cell rest 7 is the favored theory of pathogenesis and best describes the origin of congenital cholesteatomas involving the anterosuperior quadrant. Recently, however, some have noted a subset of cholesteatoma involving the posterosupe-rior quadrant while sparing the anterosuperior quadrant. 8 - 10
The number of reported cases of congenital cholesteatoma has increased dramatically since 1980, when House and Sheehy 11 reported an incidence of 3.7%. Possible reasons for this include a heightened awareness, leading to timely diagnosis. Improved otoscopes and microscopes have also aided diagnosis. Finally, prompt medical care for acute otitis media may avoid tympanic membrane perforation, which would otherwise make it impossible to rule out acquired cholesteatoma. In addition, a decrease in the number of acquired cholesteatomas may have resulted in a relative increase in the percentage of the congenital type. Friedberg, 12 however, suggests there may be an actual increase in the incidence of congenital cholesteatoma and that, in the past, inflammation may have aborted the process or otorrhea after spontaneous perforation may have expelled early cholesteatoma.
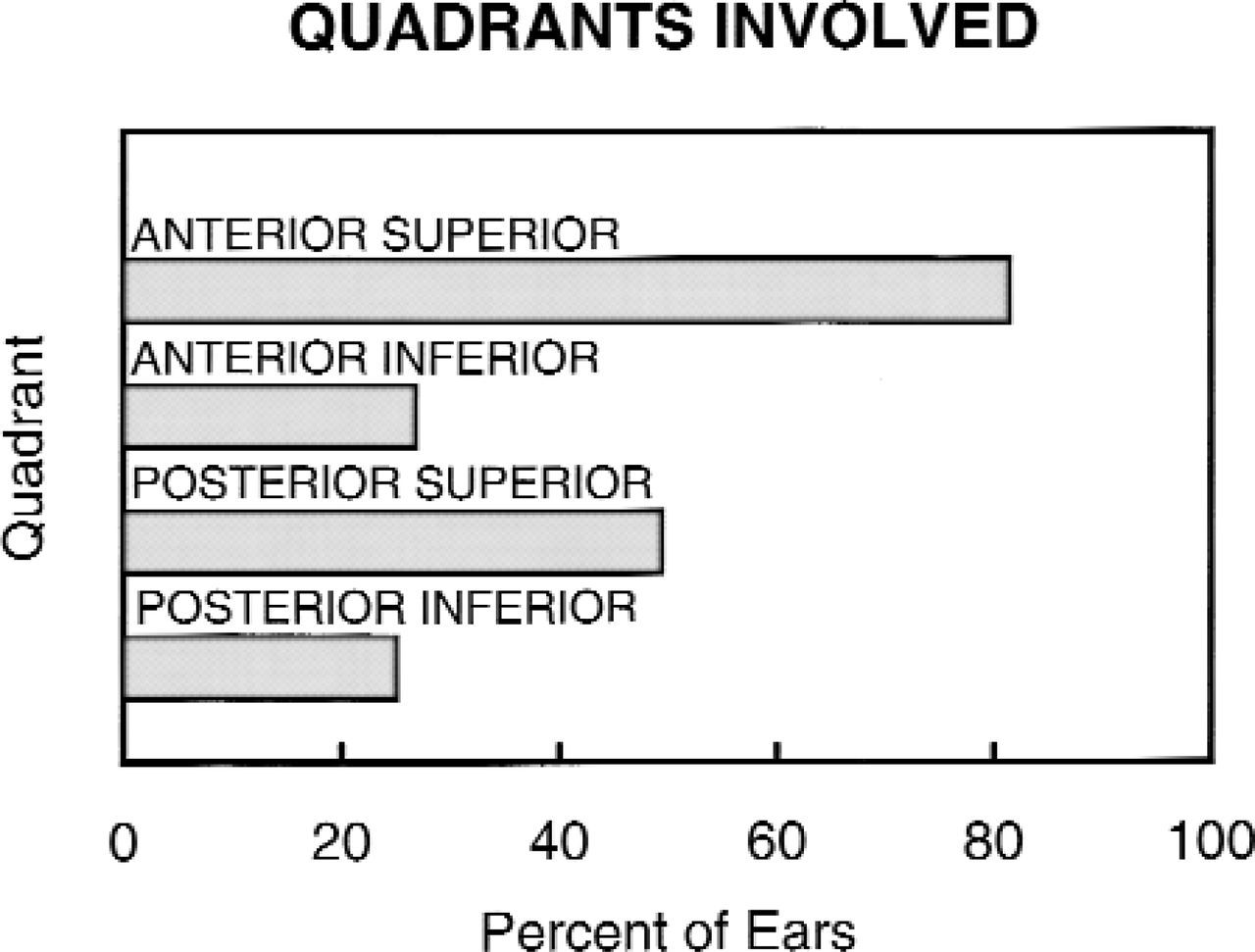
Middle ear involvement with cholesteatoma. Although the anterosuperior quadrant was by far the most commonly involved, disease often spread to and involved other areas. Disease often involved more than 1 quadrant, so the numbers do not equal 100% when added.
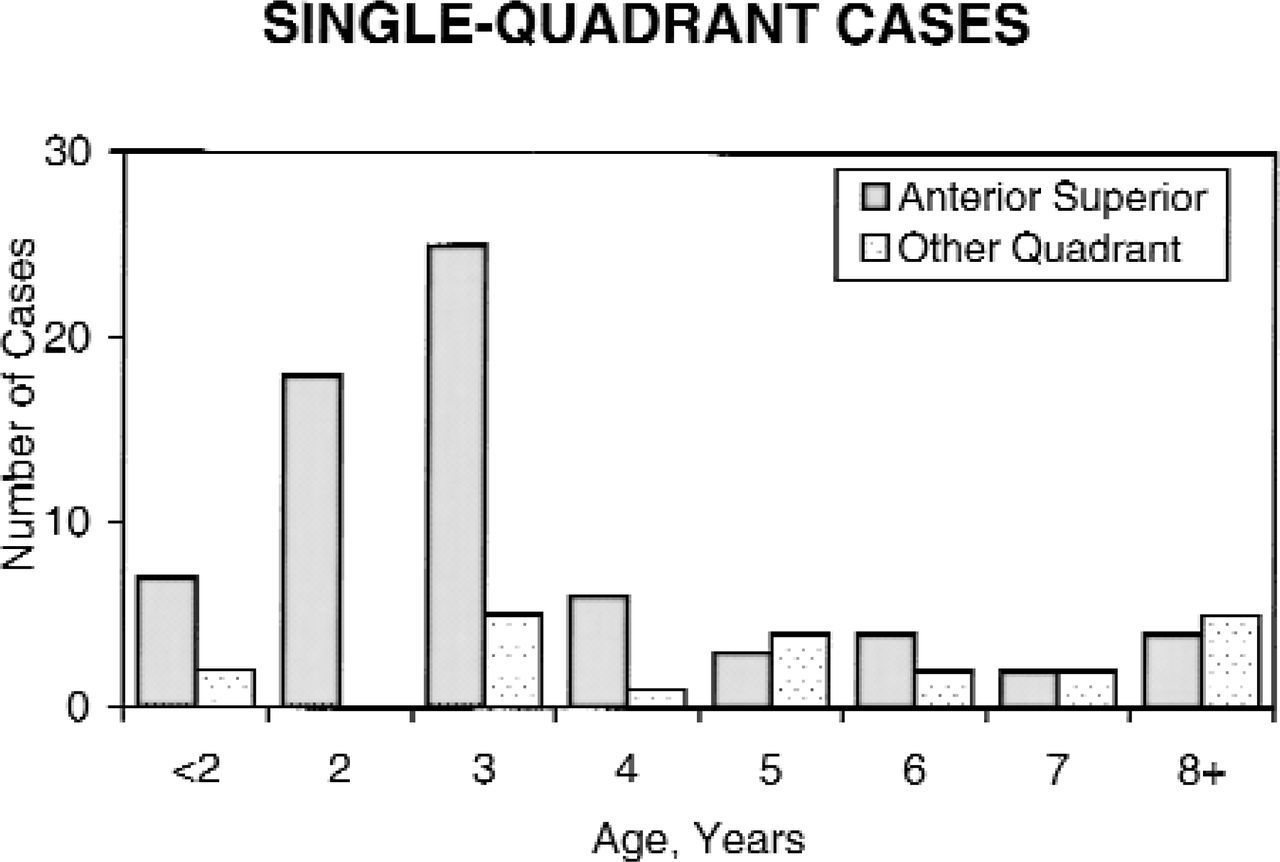
Site of cholesteatoma and patient age. Site of occurrence of congenital cholesteatoma in the anterosuperior quadrant primarily involves younger patients. In contrast, posterior disease occurs across a broad range of patient ages. This chart includes only those cases where cholesteatoma was confined to a single quadrant.
We report our experience with congenital cholesteatomas over the past 20 years, with an emphasis on predictors of outcome and presenting characteristics. This report extends the series reported previously for this institution in 199713 and offers new insights.
MATERIALS AND METHODS
After approval by the institutional review board of the hospital, we reviewed the surgical records and clinic charts of all patients seen at The Children's Hospital of Philadelphia who underwent surgery for cholesteatoma between July 1, 1981, and December 31, 2000. Cases were considered to be of congenital origin if there was an intact tympanic membrane with a normal pars tensa and pars flaccida without a history of previous perforation or otorrhea and there was no history of otologic surgery, including myringotomy tubes. In accordance with currently accepted standards, cases were not excluded if there was a prior history of acute otitis media without perforation or serous otitis media.
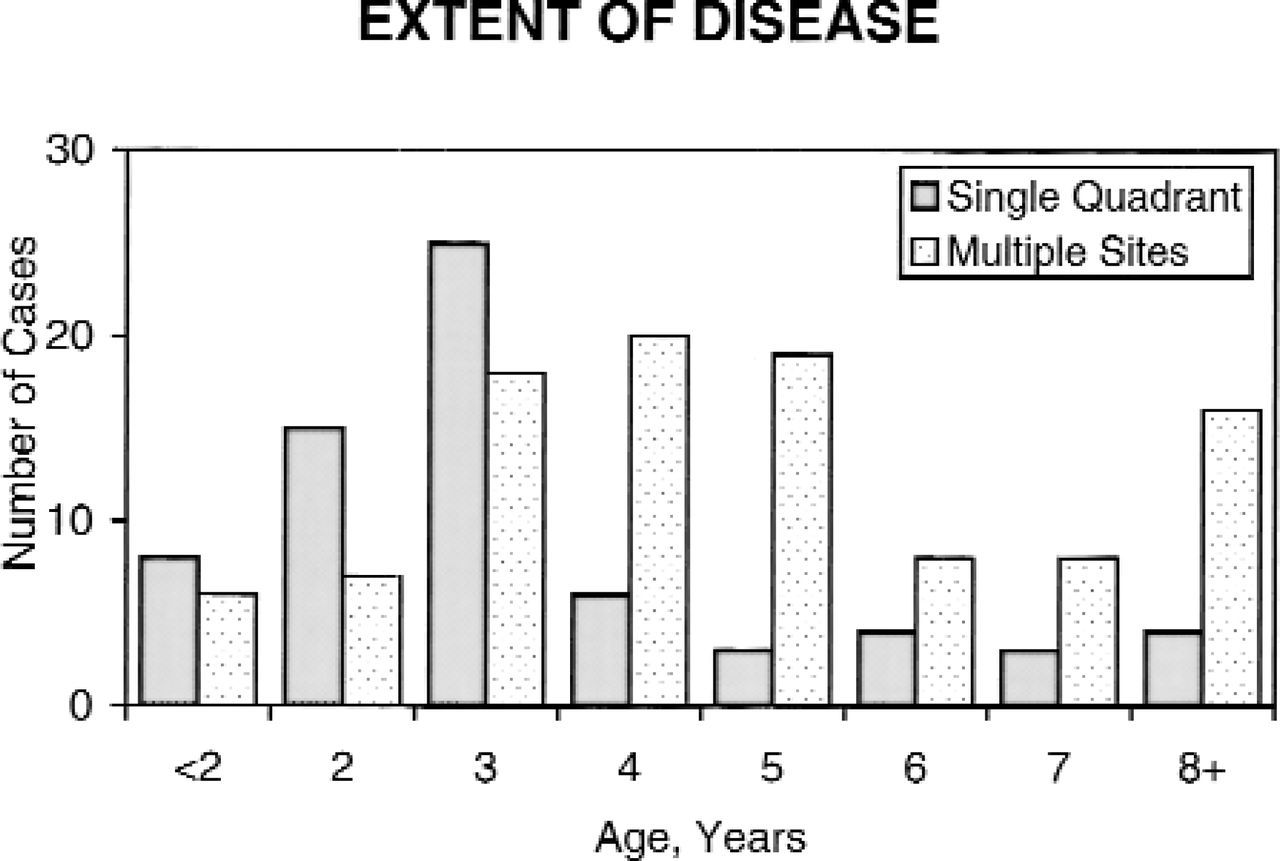
Extent of disease at time of surgery. The degree of extension of the cholesteatoma increases with the age of the patient.
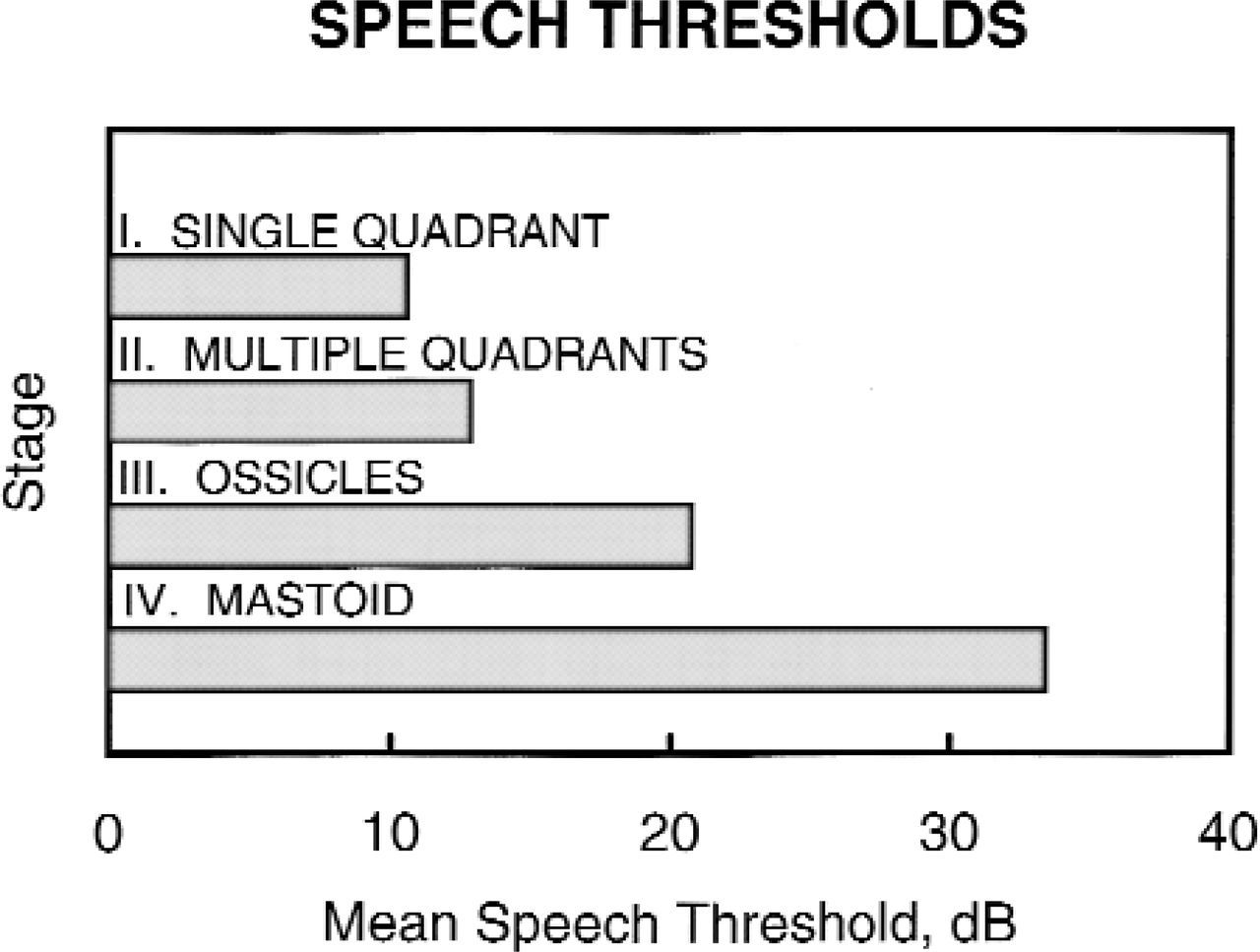
Preoperative hearing threshold. The first postoperative speech reception threshold changed by only 0.07 dB on average for all stages.
RESULTS
We identified 172 cases in 167 patients that met the inclusion criteria for congenital cholesteatoma. Bilateral cholesteatomas accounted for 10 cases, or 5 patients. The average age of the patient at time of the initial surgery was 5.0 years, and the average follow-up was 2.8 years. In line with previous reports, there was a high preponderance for boys (72%) and no predilection for either side (46% right and 54% left). Of 172 ears, all except 8 ears had undergone a canal wall up operation as the initial procedure.
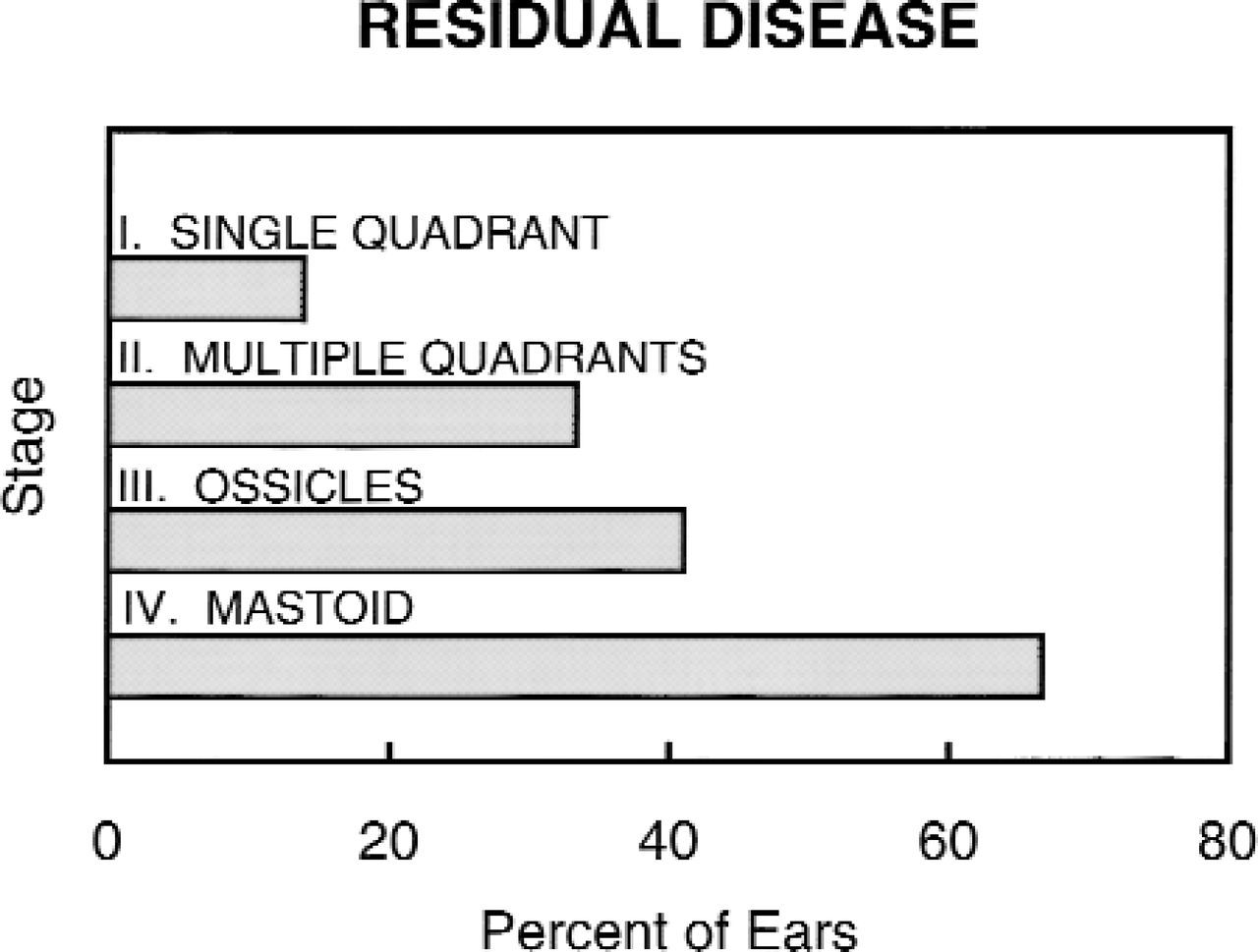
Residual disease. Congenital cholesteatoma confined to a single quadrant had a much lower rate of residual disease. The presence of ossicular involvement or mastoid extension was associated with much higher occurrences.
In early reviews, facial nerve paralysis was the most common clinical presentation 14 ; however, this is now rare and constituted only 2 cases (1%). An asymptomatic middle ear mass was the most common presentation (82%). Surprisingly, 13% were found at myringotomy for serous otitis media. Forty percent of patients had a prior history of at least 1 episode of acute otitis media, and 21% had a completely negative otologic history.
As mentioned in the articles just cited, the anterior superior quadrant of the middle ear was by far the most commonly involved site: 81% in this series. Of significance, 49% of the cholesteatomas involved the posterior superior quadrant at the time of surgery (Fig 1). As reported by others, there was a minority of cases that involved the posterosupe-rior quadrant, and not the anterosuperior. When the cholesteatoma was confined to 1 quadrant of the middle ear with or without mastoid extension, it was in the anterosuperior in 77% of the cases and the posterosuperior in 22% (Fig 2); 1 patient had posteroinferior-only disease. Cholesteatoma occurred in 2 or more quadrants in 48% of patients. Ossicular chain involvement was defined as any destruction of ossicles either by cholesteatoma or the necessity to remove ossicles during surgery to eradicate disease, and it was evident in 42% of all cases. Mastoid extension occurred in 23%. As illustrated in Fig 2, congenital cholesteatoma involving the anterior superior quadrant was found primarily at a young age, whereas posterior disease occurred at all patients ages.
Potsic and colleges proposed a staging system based on a subset of cases presented here (W. P. Potsic, D. S. Samadi, R. R. Marsh, and R. F.
Wetmore, A Staging System for Congenital Chole-steatoma, presented at the 16th annual meeting of the American Society of Pediatric Otolaryngology, May 10, 2001). In this system, cases involving only 1 quadrant, with no ossicular involvement or mastoid extension, are classified as stage I. This accounts for 40% of the cases. Stage II includes cases with involvement of multiple quadrants but not of the ossicles or mastoid, accounting for 14% of cases. Twenty-three percent had ossicular erosion but no mastoid extension, stage III, and an additional 23% were stage IV, with mastoid extension.
As might be expected, cases that were identified early were more likely to be confined to a single quadrant, whereas cases found later were more likely to have involved multiple sites. The mean age of the stage I patients was 3.9 years, and the mean age of the other groups, combined, was 5.6 years (Fig 3).
Patients with higher stages of disease had worse postoperative hearing and a higher probability of residual disease, which we defined as the presence of cholesteatoma at a planned second look or found at a follow-up visit. In children who were old enough to undergo ear-specific testing (n = 111), the preoperative speech reception threshold was directly related to the stage of disease: from 11 dB with single quadrant only, stage I, disease, progressing steadily to 33 dB with mastoid extension, stage IV (Fig 4). Preoperative hearing was preserved in the majority of patients. The preoperative speech reception threshold was virtually unchanged from the first postoperative audiogram, a difference of only 0.07 dB.
The staging system is a good predictor of residual disease. Initial involvement of a single quadrant had the lowest rate of residual disease, 14%, increasing to 33% when more than 1 quadrant was involved without ossicular or mastoid involvement. The presence of ossicular or mastoid involvement was associated with high rates of residual disease: 41% and 67%, respectively (Fig 5).
DISCUSSION
Congenital cholesteatoma is now an accepted entity and relatively common diagnosis. There are a number of possible reasons for the large increase in reports of congenital cholesteatoma during the past two decades. First is the recognition of the disease, both by otolaryngologists and by pediatricians. Also, earlier diagnosis and intervention identify these lesions before they progress to a stage where they perforate the tympanic membrane and can no longer be identified separately from acquired cholesteatoma. Finally, increased use of antibiotics and myringotomy tubes as well as access to better microscopes and otoscopes by primary care providers may have decreased the total number of acquired cholesteatomas and therefore increased the relative number of congenital cases.
We found 13% of congenital cholesteatomas at the time of myringotomy for otitis media with effusion. It may be that thick fluid prevented visualization of the middle ear space in the office and that the operating microscope provided better illumination.
In this series, as in other recent studies, 8 - 10 there is a subset of congenital cholesteatoma originating in the posterosuperior quadrant. This finding conflicts with the theory that congenital chole-steatomas arise from an embryonic cell rest in the anterior superior quadrant. It may be that their origin is multifactorial, perhaps explained by other well known theories of pathogenesis including metaplasia of the middle ear mucosa (Sade et al 15 ) and epithelial migration (Aimi 16 ). We have shown location to be related to the patients' age, with anterosuperior disease most common at a younger age, whereas posterosuperior disease is discovered over a broad range of ages. It may be that postero-superior disease was more difficult to detect, thus presenting later.
Finally, this series shows clearly how the location of the disease relates to postoperative hearing results and the likelihood of residual or recurrent disease. Our data indicate that involvement of the ossicles or mastoid extension warrants a planned second-look procedure and close observation for many years.
AVAILABILITY OF JOURNAL BACK ISSUES
As a service to our subscribers, copies of back issues of Otolaryngology–Head and Neck Surgery for the preceding 5 years are maintained and are available for purchase from Mosby until inventory is depleted. The following quantity discounts are available: 25% off on quantities of 12 to 23, and one third off on quantities of 24 or more. Please write to Mosby, Subscription Customer Service, 6277 Sea Harbor Dr, Orlando, FL 32887, or call 800-654-2452 or 407-345-4000 for information on availability of particular issues and prices. If unavailable from the publisher, photocopies of complete issues may be purchased from Bell & Howell Information and Learning, 300 N Zeeb Rd, Ann Arbor, MI 48106-1346; phone, 734-761-4700 or 800-521-0600.