
Abstract
In recent years, the hydrogen economy has been strongly favoured by governmental and industrial bodies worldwide. A tremendous number of papers are published every year on different aspects of protonic ceramic electrochemical cells (PCECs) due to their lower operation temperature, easier reversible operation, and brighter prospects for further development. While new progress is being made continuously, many critical challenges remain. The effort on PCEC investigation could be more aligned for greater collective impact, e.g. the academic community could devote more effort to overdue critical problems but less to incremental improvements. This review aims to provide some insightful perspectives on critical challenges facing the development of PCECs, to sort out priorities in future effort, and to suggest promising directions to pursue. In this way, it is hoped that the technical readiness level of PCECs might advance more quickly, toward field demonstrations and commercialization for a clean and sustainable energy era.
Introduction
As building an ecosystem with low-emission and sustainable energy has gained momentum, extensive studies have been devoted to hydrogen-related energy conversion/storage/utilization. Solid oxide electrochemical cells, as promising electrical-chemical conversion device, have been actively studied because of their high energy conversion efficiencies and potential applications in electrochemical reactors. However, the high operating temperature (700–900°C) of conventional oxygen-ion-conducting solid oxide electrochemical cells is an obstacle to commercialization, due to exacerbated degradation, uneconomical manufacturing and maintenance costs. For this reason, tremendous efforts have been devoted to lowering the operating temperature of solid oxide electrochemical cells. In the 1980s, solid oxide electrochemical cells using protonic ceramic materials (i.e. PCECs), were developed and soon considered promising candidates for ceramic electrochemical devices in the intermediate temperature range (400–700°C) due to their high ionic conductivity and low activation energy of proton transport [1,2]. After decades of development on protonic ceramic materials, the formation of proton defects and their migration mechanisms have been investigated [3–6], and tremendous discoveries have been made recently regarding these materials, especially the electrodes and electrolytes for PCECs. The application of highly conductive and electrochemically active materials has yielded exciting progress in PCECs. This critical review summarizes recent advancements in composition refinement on classic doped barium zirconate/cerate electrolytes and prospective proton conductors; significant achievements in air electrode catalytic activity and stability through advanced structural and compositional innovation; and microstructural engineering on Ni-based electrodes and strategies to increase their tolerance and performance. The remaining materials/structural design challenges regarding the three aspects of PCECs to achieve reliable and efficient operations are particularly discussed. Further, novel concepts for cell design and novel microstructures to improve functionality are discussed as well.
Applications of protonic ceramic electrochemical cells
Research using protonic ceramic materials started in the early 1980s. After decades of development, various applications have been explored, where most of the protonic ceramic devices share a similar theoretical background and material design. The basic electrochemical devices can be constructed in a sandwich-like structures, as illustrated in Figure 1. Sketch of the working mechanism of protonic ceramic (a) fuel cells (b) electrolysis cells (c) membrane reactors; WGS = water–gas shift.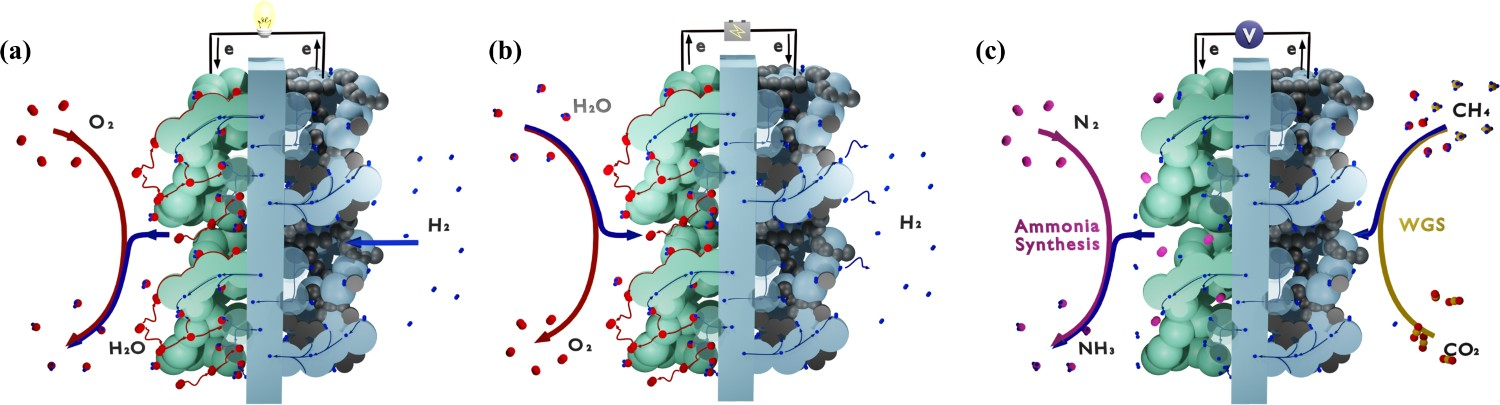
Fuel cells for power generation
Solid oxide fuel cells (SOFCs) represent a promising electricity generation technology with advantageous efficiency, fuel flexibility, and emissions. To make SOFC technology economically feasible and competitive with established energy conversion systems, its operating temperature must be further reduced from high to intermediate temperatures. Reduced operating temperatures would minimize long-term degradation, reduce chromium poisoning, enable the use of less expensive materials, and open the possibility of a rapid start-up. In this regard, protonic ceramic materials were proposed as a solution for intermediate temperature SOFC with an operating temperature under 700°C owing to the low activation energy (0.3–0.5 eV) of proton transport, which is approximately half of the typical value for oxygen ion transport.
Electrolysis cells for H2 production
Among different energy storage technologies, chemical storage, especially in hydrogen[7–9], is expected to be the basis for future energy technology and has been widely discussed for many years and gained significant interest during the past decade. It could serve as an attractive energy carrier, as a feedstock for synthetic gas, as well as a potential clean fuel for heating, electricity, and vehicle [10–12] applications. However, low-carbon hydrogen production capacity has remained relatively constant and is still not sufficient for the sustainable development scenario. The solid oxide electrolysis cell (SOEC) is an economically attractive and sustainable technology to efficiently convert electricity from abundant renewable energy sources to hydrogen fuel with zero carbon emissions.
Compared to proton-exchange membrane electrolysis cells that split water molecules at low temperatures (below 100°C), it is thermodynamically advantageous for electrolysis cells to operate at elevated temperatures. The total energy demand (ΔH) of the hydrogen evolution reaction (HER) can be expressed as ΔH = ΔG + TΔS, where ΔG is the electrical energy demand and TΔS is the heat energy demand. Figure 2 demonstrates ΔH, ΔG, and entropy change (ΔS) of steam electrolysis at a steam pressure of 1 atm. The ΔG drops significantly with the compensation of TΔS. Therefore, SOECs working at elevated temperatures can reduce costs for hydrogen production with decreased electricity consumption. Considering the balance of heat energy from unexploited heat sources from industry and the disadvantages of the high-temperature, intermediate temperature PCECs would be a promising, reliable, and practical choice for hydrogen production and utilization of renewable energies. Electric, thermal, and total energy demand for H2O electrolysis as a function of temperature. The electricity demand decreasing considerably by thermal energy compensation. Reproduced with permission from Reference [13].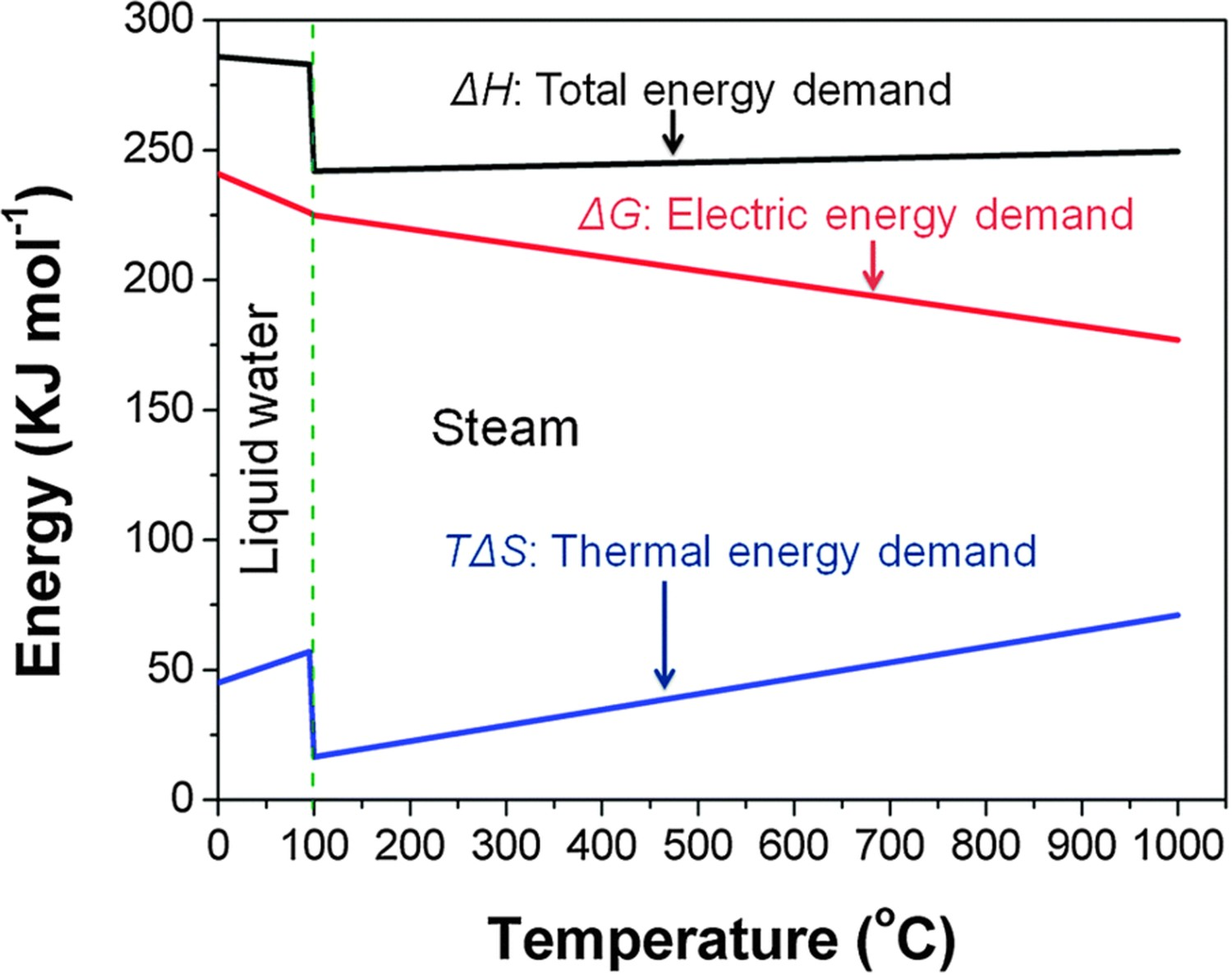
Despite the advantages of using proton ceramic materials, the commercialization of protonic devices for fuel cell and electrolysis cells is yet to be realized due to several barriers:
Many new materials and novel designs have been developed for achieving a manufacturing-ready device, while most of the available materials and strategies in the reported sources cannot solve all of these problems simultaneously, because the manufacturing-ready components must meet several strict requirements. For example, besides the essential requirement of high conductivity, the electrode and electrolyte should have a comparable thermal expansion to ensure high thermo-mechanical compatibility, negligible chemical reactions with other cell components to ensure sufficient chemical compatibility, and high chemical stability with reactants to ensure long-term operational stability.
Electrochemical reactors for chemical production
Purification
A hydrogen separation membrane based on dense proton conductors separates hydrogen and hydro-related species on an industrial scale. The separation of uncharged species in a typical membrane process is driven by a chemical potential gradient, Δμ, according to the equation: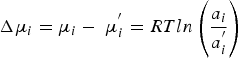
General membrane separation depends upon a pressure gradient to drive one component of the mixture. In an electrochemical separation, an electrochemical potential difference, Δµ–, can provide a driving force across the membrane: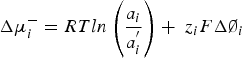
The electrochemical potential gradient across a proton-conducting ceramic membrane is determined by the activity (concentration), temperature, and electric field. When the electrochemical potential of hydrogen across the membrane is unequal, the diffusion of electrons and protonic defects takes place through the proton-conducting oxide membrane toward the low potential side of the dense ceramic membrane. Although highly conductive and electrochemically active materials have yielded exciting progress, the hydrogen oxidation and/or electron conductivity rate determine the hydrogen permeation rate of the proton-conducting membrane and achieving high hydrogen flux remains the greatest difficulty for developing proton-conducting separation membranes. The ceramic-based chemical separation membrane is still limited by its slow kinetics of exchange on the surface of the membrane and tendency to crack when compared to polymer and palladium-based devices. Its widest application to date is serving as a hydrogen pump/compressor for membrane reactors and ammonia synthesis.
Membrane reactors
Proton-conducting membranes equipped with different catalysts can be used for methane steam reforming and other hydrocarbons hydration/dehydration. The water–gas shift (WGS) for methane reforming favours low temperature, and its dominant route of hydrogen production is the following: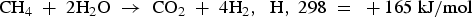
The diminished energy demands and cheaper reformer construction materials, due to lower operating temperatures, can significantly reduce the overall cost. On the other hand, the low temperature thermodynamically suppressed the kinetics of methane conversion. A possible solution to this problem would be to continuously remove hydrogen and shift the methane steam reforming equilibrium. Proton-conducting solid electrolytes have been successfully used as membranes for the methane steam reforming (MSR) process at 450–650°C and a significant increase in methane conversion and hydrogen yield was observed [17]. In addition, pressurized CH3OH production under 10 MPa at 450°C was also evaluated as an integrated electrolysis-synthesis system with a tubular PCEC [18]. Further, catalytic conversion to benzene and hydrogen with molybdenumzeolite catalysts from benzene was also achieved by combining the fundamentals of heterogeneous catalysis and solid-state electrochemistry [19].
Ammonia synthesis
Ammonia is one of the most important and widely produced chemicals worldwide. Its synthesis by the Haber–Bosch process usually employs Fe-based catalysts at temperatures between 400 and 500°C with pressures between 130 and 170 bar. The newer KAAP (Kellogg Advanced Ammonia Process) process uses a Ru-based catalyst at relatively lower pressures (<100 bar). The need for high operation pressure is a trade-off for achieving high reaction rate and conversion efficiency simultaneously, which adds high cost and complexity. Numerous researchers have contributed to the development of synthesis technology at lower temperatures and lower pressures. PCEC with specific catalyst is promising in enabling ammonia synthesis at atmospheric pressure because of the counterbalance from electrochemical potential to the high pressure by the consumption of electrical energy [20,21]. As shown in Figure 1(c), gaseous hydrogen was electrochemically dissociated and pumped through the electrolyte, where they reacted with gaseous nitrogen or/and other intermediates to produce ammonia.
High-temperature protonic ceramics
The discovery of proton-conducting oxides is one of the critical steps allowing cells to operate at intermediate temperatures. After decades of development, despite new alternatives being found, the doped barium zirconate/cerate perovskite oxides are still the best performing proton-conducting electrolyte materials. Previous research was devoted mainly to the proton conductivity of protonic ceramics. The issues of faradic efficiency and stability did not receive much attention until recently. As the focus is gradually shifting from fuel cell operation for power generation to electrolysers for fuel production, current leakage and degradation could be much more significant.
The state-of-the-art
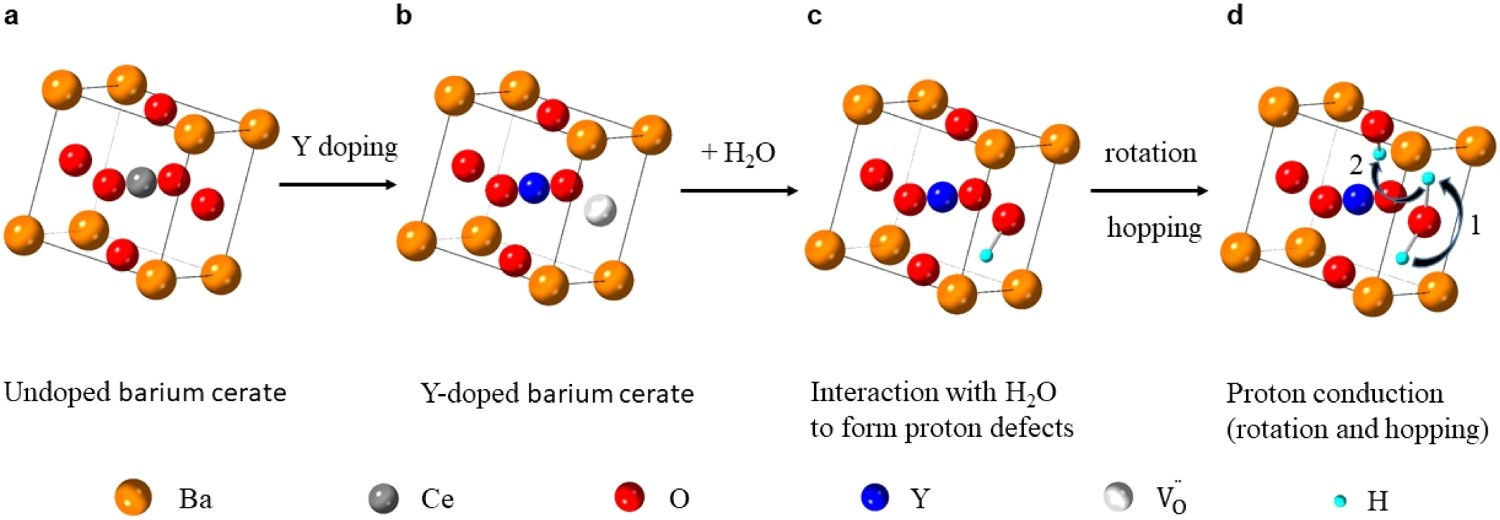
Doped perovskite barium zirconate materials and doped barium cerate-zirconate solid solution (BaCe1−xZrxO3-δ) are the most widely used proton-conducting electrolytes, especially the doped BaCe1−xZrxO3-δ family, owing to their high proton conductivity, high ionic transference number, and low sintering temperature [13,22,23]. Proton conduction requires the formation of protonic defects in the ABO3 perovskite structure through dissociative adsorption of water (Equation 4) or proton uptake (Equation 5). To obtain appreciable proton conductivity under humidified conditions, it is necessary to create excess oxygen vacancies for the incorporation of proton, and acceptor doping is the most common strategy that has been used to enhance the defect concentration. Taking Y as an example, one oxygen vacancy is formed by replacing two cerium with two yttrium (Equation 6). Subsequently, the oxygen vacancy can interact with a proton source to produce protonic charge carriers, followed by rotation and hopping of the charge carriers from one oxygen site to another (Figure 3). However, conductivity depends on both the concentration and mobility of the charge carriers. As all trivalent dopants are likely to introduce a similar amount of oxygen defects, the conductivity variation mostly comes from proton mobility differences [24]. As mentioned before, protonic species need to rotate and jump within the lattice, but different trivalent ions trap protonic charge carriers at different degrees, resulting in differences in proton mobility [25]. Moreover, lattice expansion and minimal deviation from the ideal cubic structure benefit proton mobility due to enlarged unit cell volume [26]. Although lattice expansion can be achieved by introducing trivalent dopants with a larger ionic radius, it is important to note that larger ions also have a greater tendency to occupy the divalent sites and, therefore, create fewer oxygen vacancies, which could be detrimental to proton incorporation [27]. (a) Cubic perovskite structure of undoped BaCeO3. (b) Defect perovskite structure in Y-doped barium cerate. (c) Representation of hydrated Y-doped barium cerate. (d) Schematic illustration of the proton conduction mechanism. Step (1) represents the proton rotation, and step (2) shows the proton hopping towards a neighbouring oxygen atom.
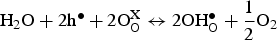
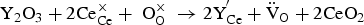
To date, the substitution of B-site cation by a lower-valent cation has been widely studied. Much of the data concerning B-site doping available before 2014 have been summarized by D. Medvedev [28]. Among all compositions, some lanthanide (Y, Ho, Er, Tm, Yb) doped [29–31] materials possess the highest ionic conductivity. The materials that were the most effective, both as the electrolyte and as a component of composite electrodes, were found to be BaZrxCe0.8-xY0.2-yYbyO3-δ (BZCYYb) [32]. However, as the atmosphere requirements of SOFCs and SOECs are getting harsher, the stability of the most widely applied electrolyte BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb1711) against high concentrations of contaminants such as steam and carbon dioxide is raising concerns [33]. Some results [34–38] suggest that the chemical stability of BZCYYb1711 is still questionable for practical operation in high humidity conditions (e.g. p(H2O) > 40 vol.-%) and the current leakage (low faradaic efficiency) becomes a significant concern when electrolysis potential is applied. As such, the development of novel electrolyte materials with enhanced properties is crucial for the commercialization of proton-conducting fuel cells and electrolysis cells.
Alternative candidates
Although complex oxides such as Ba3CaNb2O9 [39] and LaNbO4
10 have been developed, these materials have been rarely studied in fuel cells or electrolysis cells due to their relatively low conductivity. A few other prospective electrolytes were reported in recent years. A new class of Hf-based electrolyte material, BaHfxCe0.8-xY0.2-yYbyO3-δ (BHCYYb), with enhanced stability while maintaining high conductivity was developed by replacing Zr from BZCYYb with Hf [40]. The improved stability was attributed to the higher reaction-free energy between BaHfO3 and contaminant species such as CO2 and H2O than the BaZrO3 counterparts, as shown in Figure 4. (a) Schematic of the degradation reactions with CO2 and H2O. (b) Gibbs free energy of the reaction between BaMO3 and CO2 to form BaCO3 and MO2 where M = Zr, Hf. (c) Van’t Hoff plot of the reaction between BaMO3 and CO2. Reproduced with permission from Reference [40].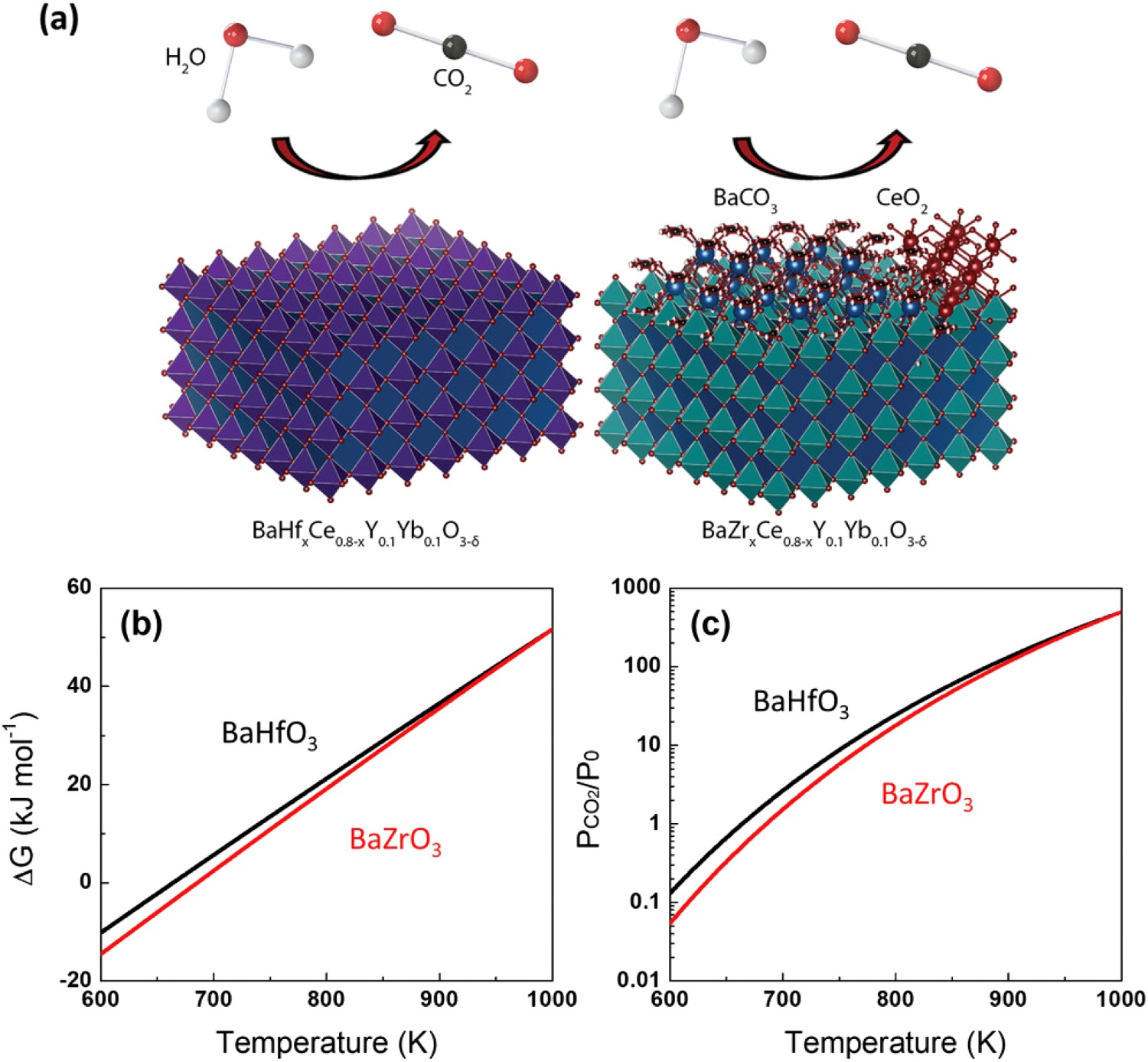
Another promising candidate is a hexagonal derivative perovskite Ba7Nb4MoO20. In this structure, corner-sharing octahedrons start to share faces instead because of the large size difference between the cations, thus forming a hexagonal perovskite that intrinsically has ionic vacancies [41]. As a result, acceptable proton and oxide ion conductivity were observed in undoped Ba7Nb4MoO20 (4 mS cm−1 at 510°C) with limited electronic contribution. Although its conductivity was about one order of magnitude lower than that of BZCYYb1711, Ba7Nb4MoO20 was stable against pure CO2 up to 800°C, making it a promising candidate for operation on hydrocarbon fuels and CO2 electrolysis. However, cell tests of Ba7Nb4MoO20 were not reported due to its chemical incompatibility with NiO, a standard fuel electrode component. In addition, a Sc doped barium zirconate with a composition of BaZr0.4Sc0.6O3-δ was developed, achieving conductivity as high as 0.01 S cm−1 at 396°C [42]. Unlike conventional Y-doped barium zirconates, where the optimal doping concentration was well documented to be around 20%, above which the conductivity quickly drops, heavy doping of Sc up to 60% continued to increase the conductivity, likely due to the higher solubility of Sc in the barium zirconate system than Y. Researchers discovered that heavy doping of Sc simultaneously promoted proton concentration, proton mobility, and grain growth. Most importantly, BaZr0.4Sc0.6O3-δ showed remarkable phase stability in pure CO2 for more than 240 h. However, the application of this material on cells was not reported either, likely due to Sc's scarcity and high cost. Accordingly, it is highly desirable to develop new electrolyte materials with enhanced stability against various contaminants, excellent phase compatibility with electrode materials, cheaper raw precursor sources, while maintaining adequate ionic conductivity.
Challenges: chemical stability, sinterability, compatibility with electrode materials, and electronic leakage
Chemical stability
Since the inception of this material family, the chemical stability of state-of-the-art protonic electrolyte BCZYYb in steam and CO2-rich atmosphere has been a practical concern [36–38]. A high concentration of steam in the feed gas is crucial for highly efficient operation, and trace CO2 is unavoidable under practical conditions. Decomposition of doped barium cerates will lead to the formation of BaCO3 or Ba(OH)2 and various insulating oxides, resulting in degradation in cell performance. In contrast, doped barium zirconates are thermodynamically stable under CO2 or H2O-containing atmospheres, but their low electrical conductivity and poor sinterability often limit their use. Optimizing the ZrxCe0.8-x
ratio of BaZr
x
Ce0.8-x
Y0.2-y
Yb
y
O3-δ is an effective strategy to balance conductivity and chemical stability. Taking BZCYYb as an example, the low Zr content (10%) can improve the chemical stability to an acceptable level for its application in mild environments. Intermediate Zr doping (∼40%) can offer reasonable stability against aggressive conditions, such as CO2-containing atmospheres (Figure 5) [40,43]. Although this strategy increases the stability, the conductivity and processability of doped barium cerates are sacrificed as a tradeoff for introducing Zr [44–47]. (a) XRD pattern of BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb4411) before and after exposure to 100% CO2 at 500 °C. (b) TGA profile of BZCYYb4411 on exposure to 60% CO2 (balance N2) at 500°C. Reproduced with permission from Reference [43].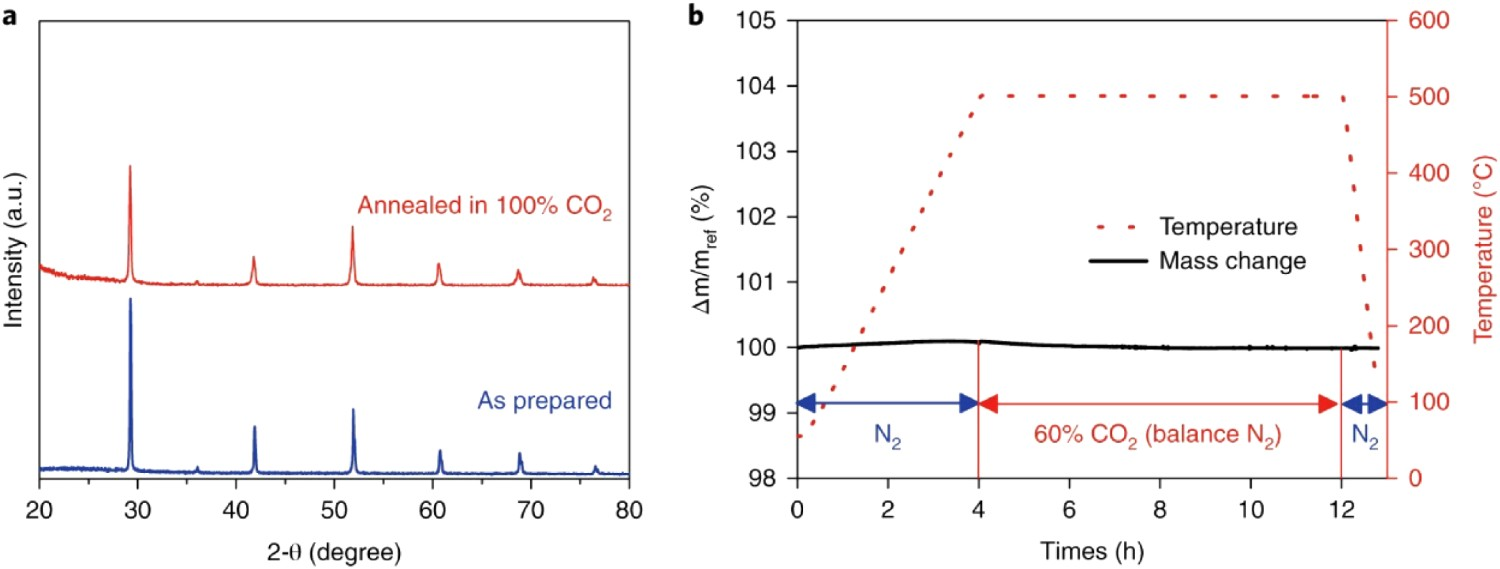
Certain acceptor (In3+, Ga3+), iso-valent (Ti4+, Sn4+) and donor (Nb5+, Ta5+, Bi5+) co-dopants can offer, as in the case of Zr-substitution, better chemical stability than other trivalent dopants [27,48]. However, it is difficult to increase conductivity and stability by dopant modification simultaneously. For example, the conductivity of In-doped samples is significantly lower than that of traditional Y-doped samples. When a more electronegative element is used, more acidity is added to the crystal lattice, stabilizing the overall structure. At the same time, conductivity is sacrificed due to weaker hydration and slower proton transport kinetics [49]. To further push the performance limit of proton-conductor-based cells, novel structures or materials systems other than doped barium cerate/zirconate need to be developed. Besides cation dopants, it is reported that halogen (F, Cl and Br) doping [50] enhances the chemical stability of BaCe0.9Gd0.1O3-δ in CO2 and steam effectively by decreasing the materials basicity. For example, Br doping has little influence on the proton conductivity of BaCe0.9Gd0.1O3-δ , but substantially improves its chemical stability. However, the properties of this material in an electrochemical cell have not yet been reported.
Another strategy to improve the stability of highly conductive electrolyte materials is to protect them with a thin film coating. This bilayer structure would consist of a more stable but less conductive phase on a highly conductive but vulnerable bulk phase. The protective layer should be as thin as possible to ensure that the bilayer electrolyte resistance is sufficiently small. On the other hand, it should be adequately thick to be gas tight if pinholes are present. Several methods such as magnetron sputtering, atomic layer deposition (ALD), and pulse laser deposition (PLD) can be used to fabricate the protective layer, among which magnetron sputtering and PLD are usually utilized for micron-level films, and ALD are desirable for 1–100 nm films. Traversa et al. [51] fabricated a 1 μm BaZr0.8Y0.2O3-δ (BZY20) on a BaCe0.8Y0.2O3-δ (BCY20) substrate by PLD. The BZY20-BCY20 film maintains the same chemical stability of BZY20 while providing 2.2 times ionic conductivity compared to a BZY20 film of similar thickness. In another study, Nurka et al. [52] developed a highly dense and pure (BaZr0.8Y0.1O3-δ) BZY10 layer of 0.7 μm thick on BaCe0.8Y0.1O3-δ (BCY10) using a reactive magnetron sputtering method without sintering at higher temperatures. The prepared membranes with BZY10 protective layer were chemically stable in CO2 at 700°C. To obtain optimal properties, it is essential to carefully reduce the protection layer thickness to minimize the ohmic loss. The technical complexity of ultra-thin film fabrication via sputtering and PLD is high. With ALD, however, pinhole-free films in thickness of 1–100 nm can be fabricated over large areas. ALD of proton-conducting yttrium-doped BaZrO3 has been reported [53,54].
Sinterability
The electrolyte layer needs to be sufficiently dense to achieve theoretical conductivity and prevent any gas leakage. Typically, Zr-rich compositions require sintering at >1500°C for densification and grain growth. Specifically, the required sintering temperature of BZY20 is as high as 1600°C [55]. Such a high sintering temperature will cause nickel diffusion and barium evaporation, changing the final stoichiometry of the electrolyte material. Although the barium loss can be partly overcome by covering the electrolyte parts with powders of the same electrolyte composition during the sintering process, high sintering temperature can still be problematic as it might result in unwanted reactions between the fuel electrode and electrolyte components during co-firing of electrode-supported structures.
NiO is an effective sintering aid for acceptor doped barium cerate/zirconate systems [56–58]. In the case of BZCYYb1711, a liquid BaY2NiO5 phase is formed at high temperatures to facilitate ion diffusion, promoting the densification process. Afterward, this phase will partially decompose as the temperature drops. As depicted in Figure 6, the use of NiO as a sintering aid enables >95% relative density of BZCYYb1711 at 1350°C, which is 200°C lower than without the addition of NiO. Other additives such as CuO, LiF and ZnO can also function as a sintering aid for doped barium cerate/zirconate [59–61]. However, the addition of reactive additives as sintering aid introduces extra impurities to the material system and has a detrimental effect on the hydration ability, ionic conductivity, transport number and chemical stability of electrolytes [62–65]. Therefore, the amount of these additives should be carefully controlled. The relative density and average grain size of NiO-containing BZCYYb1711 (a and b) and unmodified BZCYYb1711 (c and d) pellets sintered at different temperatures for 3 h. Reproduced with permission from Reference [56].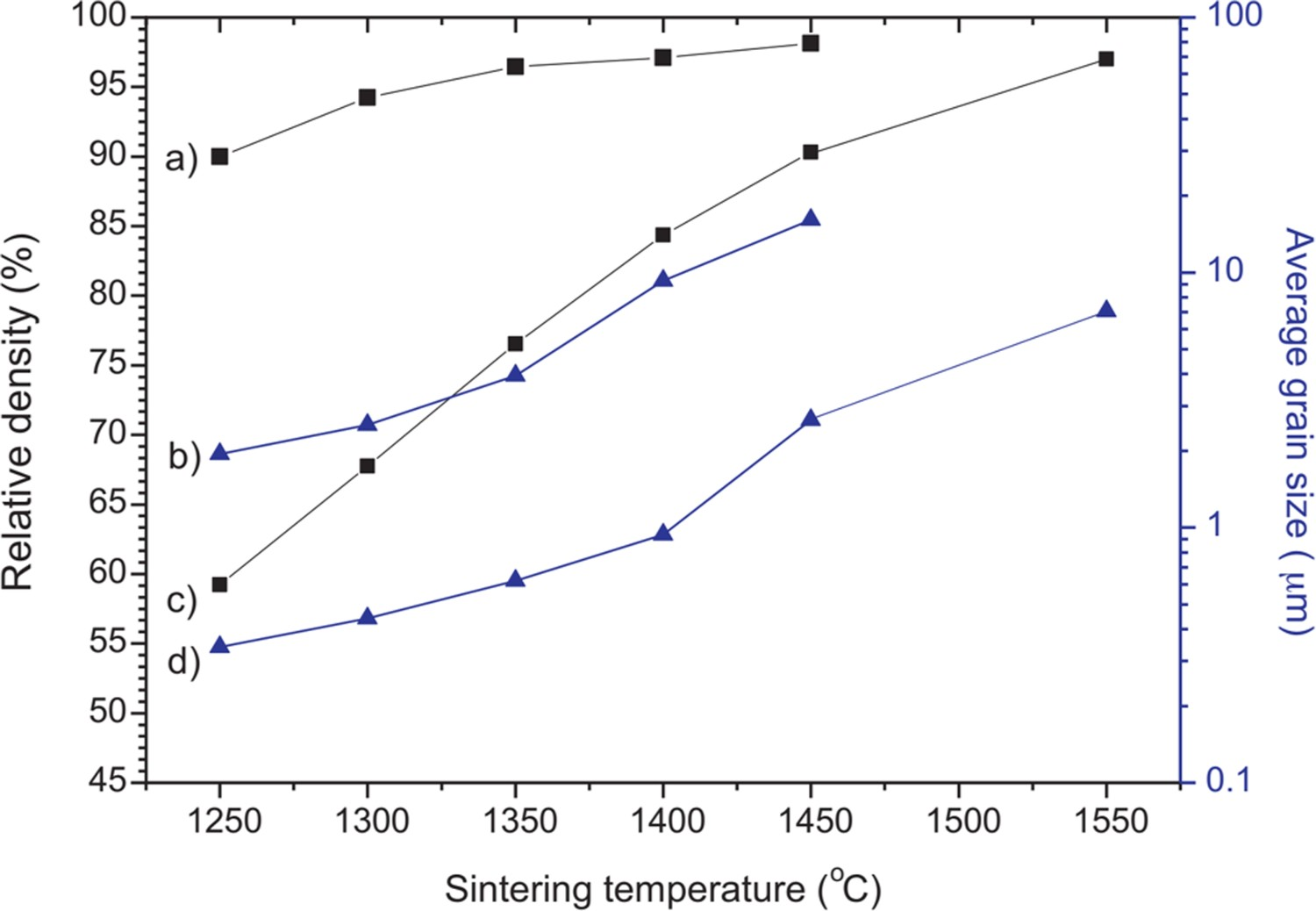
One alternative way of lowering the sintering temperature of Zr-rich recipes is cation replacement with certain elements. It is reported that light Sr substitution and Sm, Nd, La, Gd substitution can effectively improve the sinterability at an acceptable expense of ionic conductivity. Among these Gd appears as a promising dopant that balances high ionic conductivity and good sinterability [30,66]. It is also helpful for sintering if the initial size of starting powders is sufficiently small. The high surface area of nano powders can facilitate the sintering process. Fabrication of nano powders can be achieved by high-energy ball milling, but contamination from grinding media becomes possible if the friction force gets too high at fast milling speeds. In comparison to solid-state reaction, sol–gel and combustion syntheses usually produce powders of smaller particle size [67]. However, not all materials can be synthesized with the liquid-phase method as metal nitrate precursors are not readily available for many commonly used elements.
Another strategy is to use a novel sintering method. Conventional furnaces can only heat up and cool down at a fairly slow rate, and grain growth of particles becomes significant at intermediate temperatures, diminishing the driving force for solid-state sintering. Recently, several techniques were developed that are able to achieve heating rates up to ∼2000°C in seconds, which could be very helpful for sintering ceramics [68]. However, it is unknown if electrode-supported cells can sustain such an aggressive heating profile as the thermal expansion mismatch of different cell components could be significant.
Compatibility with electrode materials
Electrolyte materials must be chemically compatible with both fuel electrode and air electrode components during fabrication and operation. Although incompatibility between the electrolyte and air electrode materials has been frequently reported in the literature, it is now less of a concern as many high-performance air electrode materials are available for use [69,70]. Also, the air electrode fabrication temperature is often around 1000°C, at which the reaction kinetics are relatively slow. In contrast, the co-sintering of an fuel electrode-supported electrolyte bilayer takes place at temperatures >1400°C, which is possible only if the electrode and the electrolyte materials are chemically compatible under the co-firing conditions.
The chemical compatibility between the most widely used fuel electrode component NiO and electrolyte materials has not received enough attention in the past. Taking Y-doped barium zirconate as an example, the reactivity between electrolyte and NiO depends sensitively on the Y content. As shown in Figure 7, the solubility of Y in the Ba–Zr–Y–Ni system at temperatures above 1500°C is about 12%. BaY2NiO5 will form during the firing process to facilitate sintering, but this phase will not fully dissolve or decompose after cooling if the Y content exceeds the solubility limit [71]. The residual of this secondary phase at grain boundaries will increase electronic conduction and hamper the hydration capability of the electrolyte [72], thus reducing the efficiency of the protonic ceramic electrochemical cells, especially the Faradic efficiency in the electrolysis mode. Also, the formation of this phase means the extraction of Ba and Y from the bulk electrolyte phase, resulting in a deviation from the desired electrolyte composition. Although Han [73] demonstrated a temperature reduction cleaning (HTRC) process to mitigate the effect of Ni containing secondary particles segregated at the grain boundary, Ni contained secondary phase is still undesirable since it may decrease mechanical strength and introduce other unknown risks. Therefore, it would be beneficial to tune the electrolyte composition for optimal phase compatibility with NiO, as an alternative material to replace Ni in the fuel electrode is yet to be developed. BZY20 compositions in each equilibrium and the map of secondary phases expected to form when BZY20 is in contact with NiO. Reproduced with permission from Reference [71].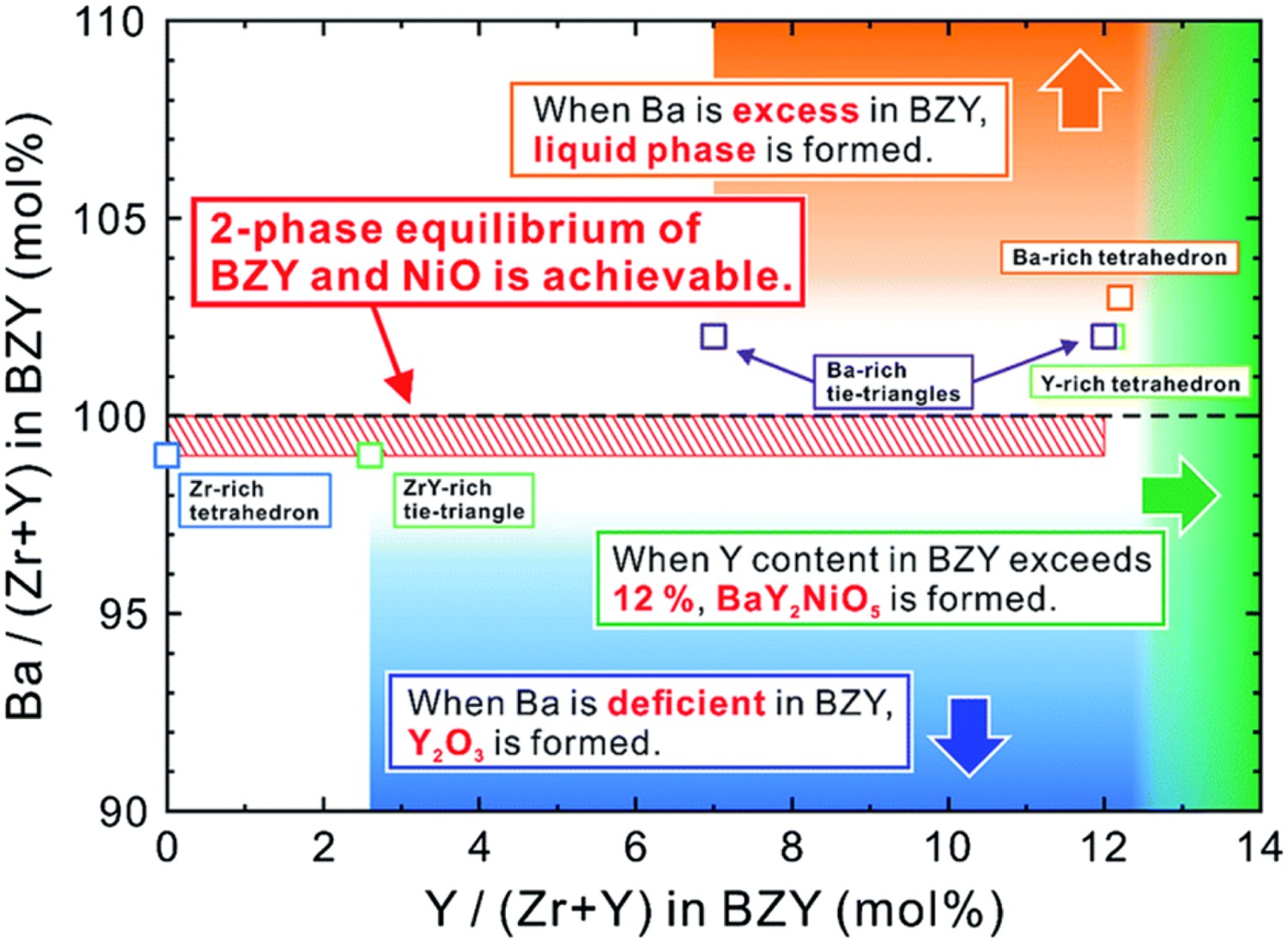
A possible way to mitigate the formation of BaM2NiO5 (M = Sc, In, Lu, Yb, Y or Gd) is to avoid using Gd and Y as it is reported that BaM2NiO5 was not produced in the case of M = Sc, In, Lu, and Yb [74]. However, Y is characterized by its highest ionic conductivity among all dopants; the absence of Y will hinder the development of high-performance proton-conducting electrolytes.
Electronic leakage and low faradaic efficiency
The most important challenges for the application of proton conductor-based SOECs are electronic leakage and low Faradic efficiency. Barium cerate/zirconate-based materials are not pure ionic conductors but mixed ionic and electronic conductors, especially at temperatures higher than 600°C [75]. The electronic conductivity in BaCeO3 based proton-conducting materials originates from the trivalent dopants R and trivalent Ce; the process can be represented as follows: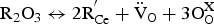
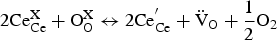
The electron holes arise as a result of the interaction of vacancies with the oxygen in the gas phase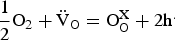
In a wet atmosphere, the above reaction competes with Equation (4). Combining Equations (4) and (9) gives Equation (5). In this case the electroneutrality, in competition between the above reactions, can be expressed as [76,77]: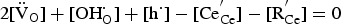
Therefore, although it is predominantly a proton-conducting electrolyte, oxygen vacancies and hole conductivity are also mobile defects. The conduction behaviour within the electrolyte depends strongly on composition, gas atmosphere, operating temperature, and applied bias. First, the composition has a significant effect on the conduction behaviour. Doped barium cerate has a higher ionic transference number than doped barium zirconate under humified atmospheres, likely due to its better hydration capability [76,78]. The electronic conductivity was found to be relatively high under reducing conditions in barium cerate, while almost negligible in the zirconates [79]. The use of different trivalent dopants can have a noticeable effect on electronic conductivity as some of them may present more oxidation states than the others [80–82]. Furthermore, the gas composition can have a dramatic impact on the production of electronic defects. It was observed that the electronic leakage of BaCeO3 based materials becomes much more severe under dry oxidizing atmospheres due to the increase of electronic holes [83–85], and the presence of water is important in improving the ionic transport numbers, as more water will promote hydration (Equation 4) and suppress the oxidation reaction (Equation 9). Operating temperature is another critical factor to take into consideration. Dehydration of proton conductors at higher temperatures (>600°C) will significantly suppress the activity of Equation 4 and enhancing the activity of Equation (9) [26].
Under experimental conditions, high faradaic efficiency is observed when a large current density is applied to the cells in fuel cell mode, where a more reducing atmosphere is created. In contrast, a decreased faradaic efficiency was observed in electrolysis mode with increased electrical bias [86,87]. The electrostatic bias applied to the electrolyte plays a vital role in determining the faradic efficiency. The electronic conduction through the electrolyte is driven by the cell voltage. In the fuel cell and the electrolysis cell modes, the cell voltage is given by Equations 11 and 12, respectively.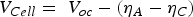
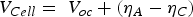
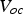
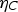
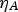
Mixed conducting air electrode materials
The primary role of the PCECs air electrode is to promote H2O-splitting in SOEC mode (H2O association in SOFC mode) and to facilitate the migration of protons between electrode surface and electrolyte. In the early-stage of development, air electrodes for proton-conducting cells were mixed ion (O2–) and electron (e–) conductors (MIEC) that are borrowed from O2–-conducting systems, which confines the reactions to standard triple phase boundaries (TPB). To significantly enhance the performance of PCECs, air electrode materials of triple conducing (mixed H+/O2–/e–) properties have been developed for enabling the electrochemical reactions to occur over the entire electrode. Therefore, the overview of air electrode materials in this section is composed of materials designed for proton-conducting systems, different strategies for improving catalytic activity, and state-of-the-art challenges.
Advancement from double to triple-conducting air electrode
In early-stage research, the air electrode used in proton-conducting system is traditional MIEC electrode materials. The proton-related reactions on MIEC were confined to the TPB because of the limited access to protons. Later, composite electrode [88,96–100] by mechanically mixing or self-assembly (in situ synthesis) of a proton-conducting phase (usually electrolyte material) with a MIEC was applied. However, the electrochemically active site is still limited to the interface between the air electrode and electrolyte (Figure 9(a)). He et al. [98] and Poetzsch et al. [101] studied the elementary reaction steps of the air electrodes for proton-conducting systems, suggesting that introducing proton conductivity instead of increasing oxygen-ion conductivity in the air electrode is crucial for improving the performance of PCECs. Since then, the concept of triple conducing oxides (TCOs) has emerged. Different water-splitting pathways on two categories of PCEC air electrode in electrolysis mode: (a) mixed O2– and e– conducting air electrode shows limited reactive points, (b) H+, O2–. and electron triple conductive air electrolyte shows entire active surface [102].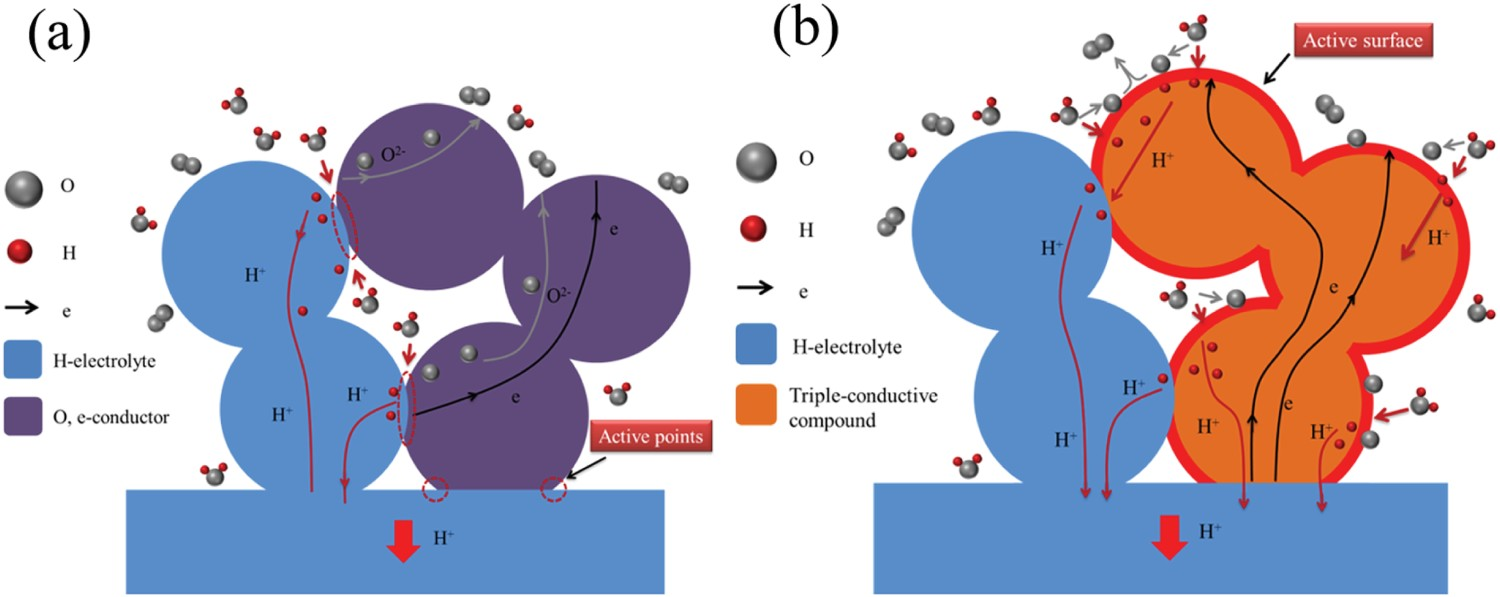
As shown in Figure 9(b), TCOs enable the electrochemical reactions to occur anywhere on the entire surface of the air electrode, and the resulting protons can incorporate into the bulk phase of the air electrode and diffuse through the air electrode interior and finally transport to the electrolyte.
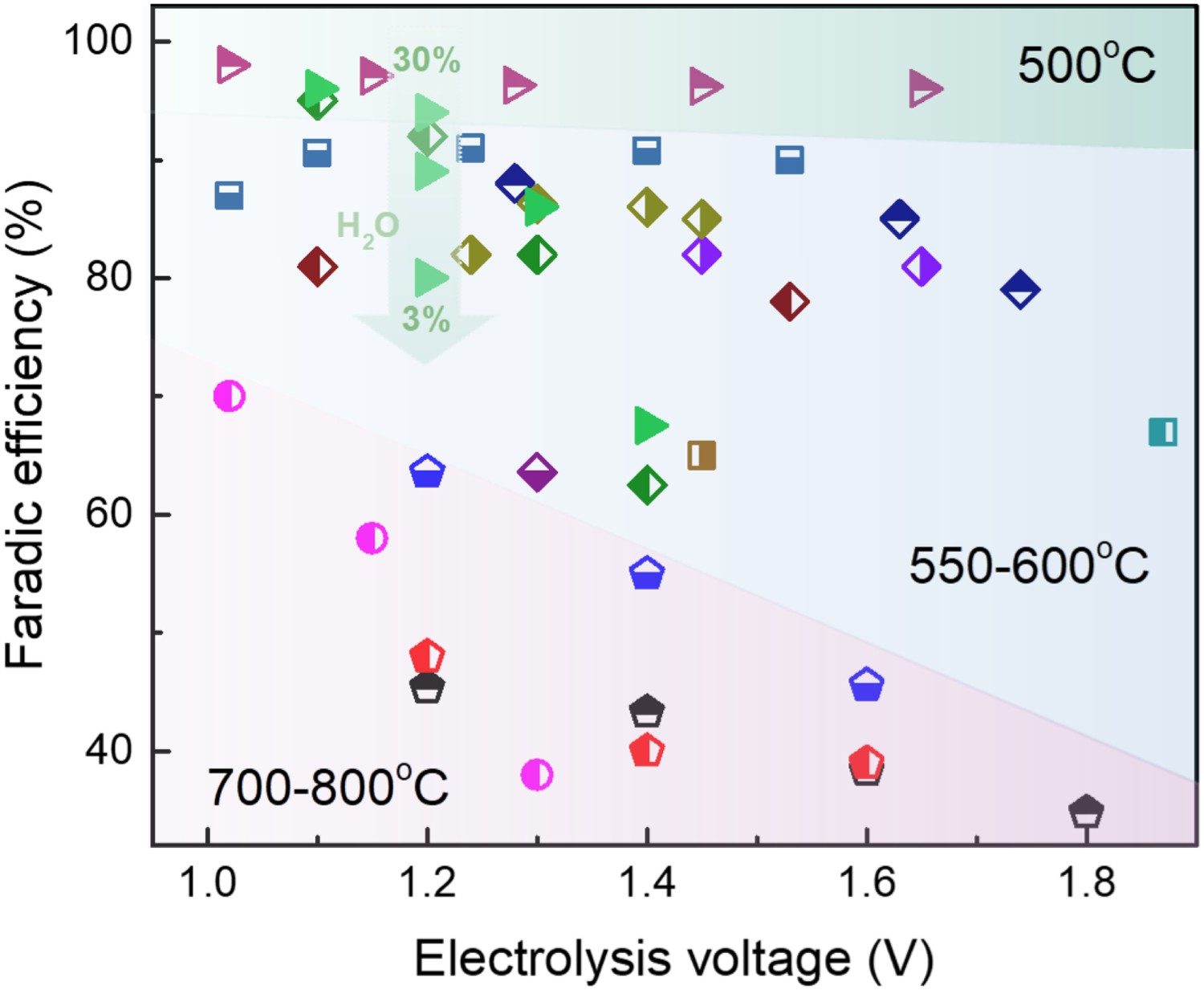
The superiority of TCOs over traditional MIECs in a proton-conducting system has been investigated and confirmed during the past decades. A. Grimaud [103] studied the electrochemical response of La0.6Sr0.4Fe0.8Co0.2O3−δ (LSCF), Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF), PrBaCo2O5+δ (PBCO), and Pr2NiO4+δ (PNO), pointing out that the protonic conductivity, induced by water insertion, of the last three materials improves electrode performances as increasing p(H2O) increases. Zhou et al. [104] compared the conductivity of Sr2Sc0.1Nb0.1Co1.5Fe0.3O6-δ (SSNCF) with that of BSCF, suggesting the non-conflicting dual-ion (H+ and O2–) diffusion channels within SSNCF bulk increased the proton conductivity and significantly improved the performance. Li et al [105] investigated Pr2-xBaxNiO4+δ (PBNO) materials, demonstrating the increased proton conduction from Ba substitution was crucial to performance improvement. Staring from BSCF, PBCO, and PNO, extensive efforts have been devoted to increasing the electrochemical performance of these TCOs. Figure 10 presents the representative polarization resistances (R
p) of MIEC and TCO air electrodes applied in proton-conducting system and the representative single-cell performances are listed in Table 1 with their specific configurations. These promising results suggest that TCO electrodes are already highly active towards oxygen reduction reaction (ORR) and water oxidation reaction (WOR) at 600°C. The electrolyte resistances (R
s) are comparable to R
p or even dominate the total cell resistance, depending on the practical conductivity and thickness of the electrolyte (typical value of 0.15 Ω cm2 for 10 µm BZCYYb 1711). Representative polarization resistances of MIEC and TCO air electrode applied in the proton-conducting system. The figure is compiled from data published in ref. NBSCF(NdBa0.5Sr0.5Co1.5Fe0.5O5+δ) [106], SFM(Sr2Fe1.5Mo0.5O6) [107], SLF(Sr2.8La0.2Fe2O7−δ) [108], PNO [102], LSN(La1.2Sr0.8NiO4) [109], PSN(Pr1.2Sr0.8NiO4) [110] BSCF [111], NBN(Nd1.95Ba0.05NiO4+δ) [112], BCFZY [89], SSNCF [113], PBSCF (PrBa0.5Sr0.5Co2−xFexO5+δ) [114], PBNO [105],PNC(PrNi0.5Co0.5O3-δ) [115],PBCC(PrBa0.8Ca0.2Co2O5+δ) [90]. The electrochemical performances of PCEC single cells in fuel cell mode and electrolysis mode from recent publications at 600°C.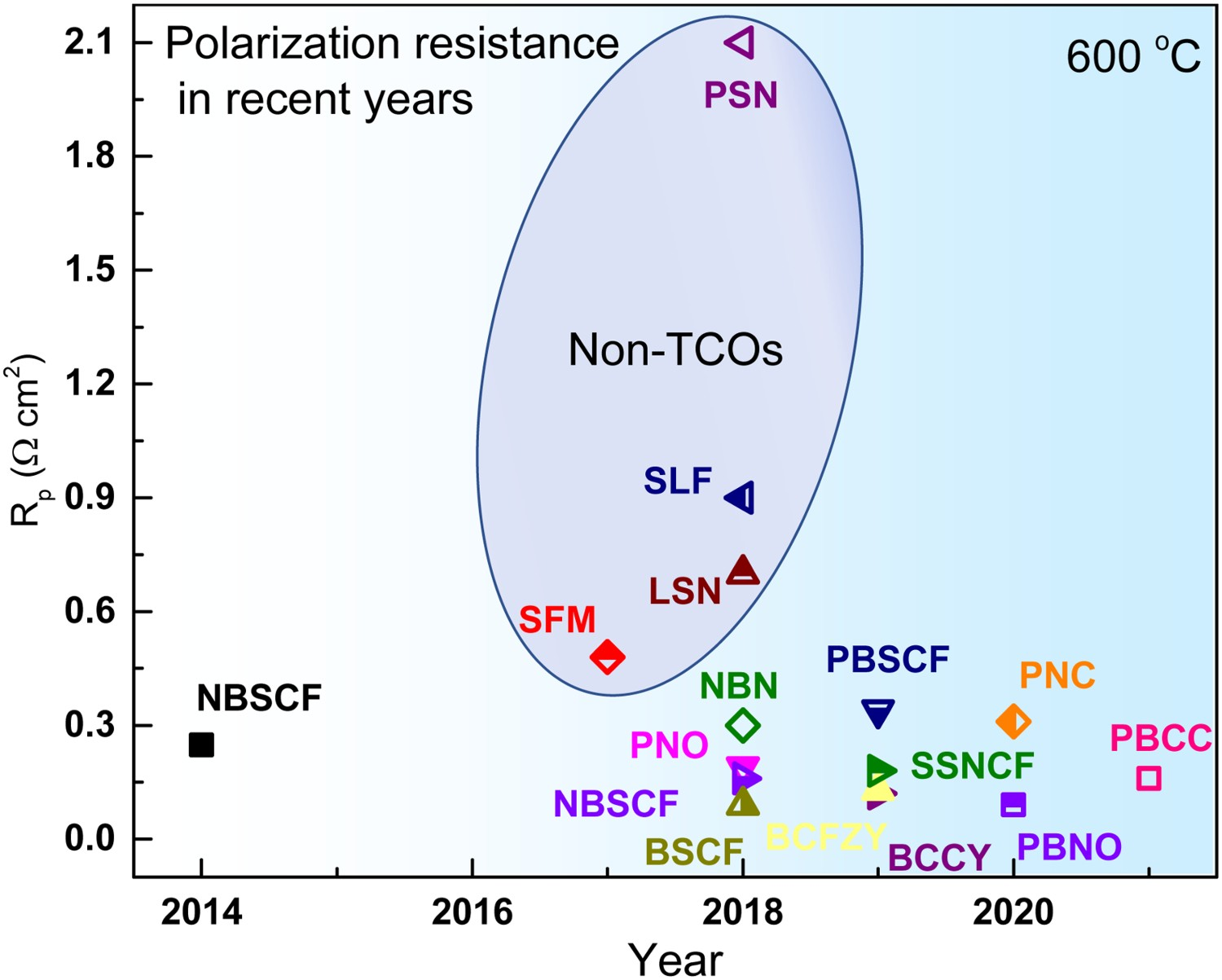
As mentioned, the importance of the proton conductivity of TCOs has been repeatedly addressed, while the role of oxygen ion conduction of the triple conductivity has received less attention. The question of whether oxygen ion conduction is necessary seems to be a pseudo proposition since most of the electrode materials are unavoidably TCOs rather than H+ and e– dominated conductors at 600°C. With the reduction of electrolyte thickness and the discovery of more active air electrodes, there is a noticeable trend of pushing the operation of PCECs to lower temperatures (≤ 500°C). The oxygen ion bulk diffusion, which exhibits considerable activation energy, could unavoidably become the rate limiting step at lower temperature. As such, understanding the evolution of TCOs reaction mechanism is crucial. According to reports of Zhou et al. [104] and Li et al. [105], the electrode performance can be improved with increased proton conductivity even at the expense of bulk oxygen ion conductivity expense. It seems that, although TCOs are capable of triple conducing, the conducting species do not evenly contribute to the performance. Recent mechanistic studies of reaction details from Tian [121] proved that, at 500–650°C, the expansion of the reaction network of PBNO is established mainly by bulk proton conduction (Figure 11(b)); the oxygen ion bulk diffusivity does not practically contribute to the reaction kinetics. This result is promising and further investigation on perovskite materials at temperatures < 500°C would be highly valuable; it needs to be verified whether the sacrifice of oxygen conductivity in some cases is accompanied by a decrease of oxygen in surface exchange kinetics of the oxygen-related species. Schematic diagrams of micro reaction on TCO with conducting of H+ and O2– via (a) bulk pathway and (b) H+ via bulk while O2 via surface pathways. Reproduced with permission from Reference [121].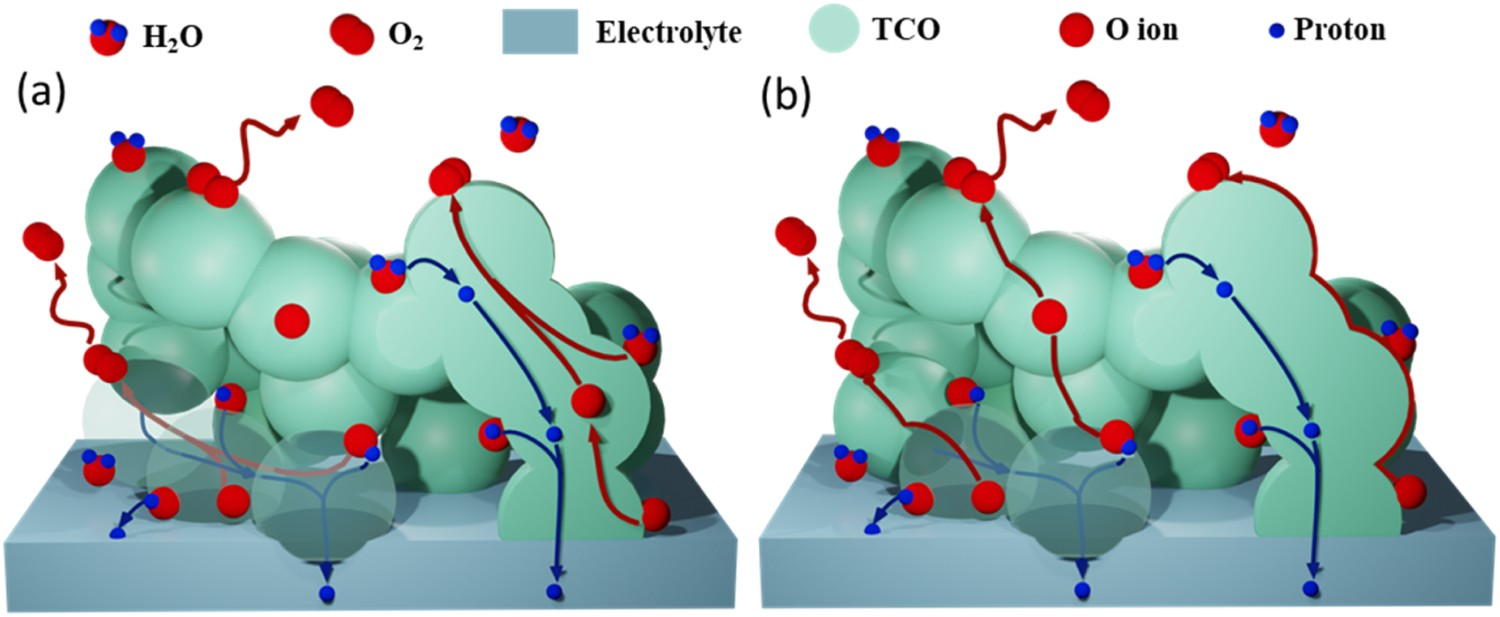
Surface modification to improve activity
Continuous efforts have been made to further push the operating temperature towards the intermediate to even low-temperature range, at which the performance of SOFCs and SOECs is limited by the activity of the air electrodes rather than the ohmic resistance from the electrolytes. According to published data, the R p increases by roughly 2.5 times while the R s increases by 1.3 times with a temperature decrease of 50°C [103,105]. Therefore, electrode catalytic activity limit may be unavoidable as the temperature is being pushed to a lower range as enabled by the high activation energy of electrode reaction. Therefore, air electrodes with higher catalytic activity at low temperatures are necessary.
The systematic development of TCOs is still challenging because of the difficulties of characterizing protonic behaviour in TCOs. Direct electrical measurement (for example, electromotive force) of proton conductivity requires the sensing current to be purely protonic. For TCOs, however, heavily doping of transition metal and creation of oxygen vacancies for water insertion inevitably introduce oxygen and electron conductivity into the system. In addition, the complexity of the water-splitting reaction chain involving three charge carriers (O2–, H+, and e–) simultaneously, renders techniques for interpreting the electrochemical responses insufficient. Similar to proton-conducting electrolyte materials, TCOs
To better serve the SOCs at lower temperature, surface modification, which improves the electrochemical activity while maintaining the bulk properties, for promoting the catalytic activity may effectively boost the performance in the near future.
Surface decoration by impregnation
Nano-catalysts, including transition metals, perovskites, fluorites, etc., have been used to lower the operating temperature of traditional MIEC electrodes. Modifying the surface composition via infiltration can develop nano-metal catalysts decorated electrodes without changing the proton-conducting properties. Depending on the morphologies, there are three distinct infiltration coating types, film-like coating, discrete coating, and concentrated coating, determined by the metal ions adhesion within the precursor solution and the substrate capillary force [126]. To improve the surface electro-catalytic activity while maintaining the protonic properties of the electrode, discrete coating or concentrated coating are preferred. Fluorite-type doped ceria infiltrates have been widely used as catalyst materials due to their ionic conductivity, excellent surface exchange, and oxygen storage capability. Samarium-doped ceria in the form of nanoparticles (loading amount 0.198 wt-%) has been applied to enhance the transport of oxygen-related species of triple conducting BaCo0.4Fe0.4Zr0.1Y0.1O3+δ (BCFZY). As a result, the R p significantly decreased by 36% at 600°C and both the SOFC and SOEC performances were improved [127]. Further, in situ formation of triple conducting Ba(Zr0.4Ce0.4Y0.2)1-xCoxO3-δ, decorated by spinel Co3O4 and fluorite (Ce, Zr, Y)O2 via infiltrating was also demonstrated [128]. Besides promoting performance, the underlying principle also provides helpful guidance for the design of catalysts.
Surface termination atomic layer tailoring by atomic layer deposition
Surface chemical and geometric composition are essential for the electrochemical performance of perovskite and related oxides. ALD is an ideal method for tuning atomic-level surface composition on various electrodes since this method only changes the outmost layer without affecting other properties. Its application in heterogeneous single-atom catalysis has achieved great success [129]. In the case of traditional MIEC, monolayer / sub-monolayer surface tailoring has been proved to significantly promote the surface O2 adsorption and dissociation by creating surface catalytic-active centres and defects that offer a wider variety of structural arrangements of cation and anion sites. For example, Rahmanipour et al. [130] reported repeatable decreased polarization resistance on La0.8Sr0.2FeO3 electrode with monolayer La2O3 and increased resistance at higher layer coverages. Julian M.Paige [38] reported that Co monolayer could improve the ORR of a Co-free electrode, where the La0.8Sr0.2FeO3 with monolayer Co coverage achieves the same performance as pristine La0.8Sr0.2CoO3.
It is worth mentioning how water dissociates on the oxide surface. Based on the results of studies on SrTiO3, BaTiO3, and TiO2 [131–137], the H2O molecule’s oxygen atom first needs to be chemisorbed on the highly undercoordinated surface cation sites, which translates being more acidic and thus more energetically favourable as binding sites for molecules like water. Then, a reasonably short bonding with an adjacent O site from the substrate will facilitate proton transfer from the water molecules to the substrate surface (Figure 12(a)). Furthermore, defects have long been believed to be the most active sites for water dissociation on oxides. As shown in Figure 12(b), defects (irregular surface sites) offer a wider variety of structural arrangements of cation and anion sites, which is particularly important for water dissociation since this process requires the concerted effort of a cation site binding water and an anion site abstracting a proton. Many defects also involve lower oxidation state cations, which opens the possibility of charge-transfer interactions with adsorbates. (a) (c) Chemisorption of water for the most favourable adsorbate configuration for one water molecule above surfaces of planar SrO and TiO2 terminated 2 × 2 supercells. (b) (d) Geometries with irregular surface sites formed by removing a sociometric SrO or TiO2 unit. Reproduced with permission from Reference [131].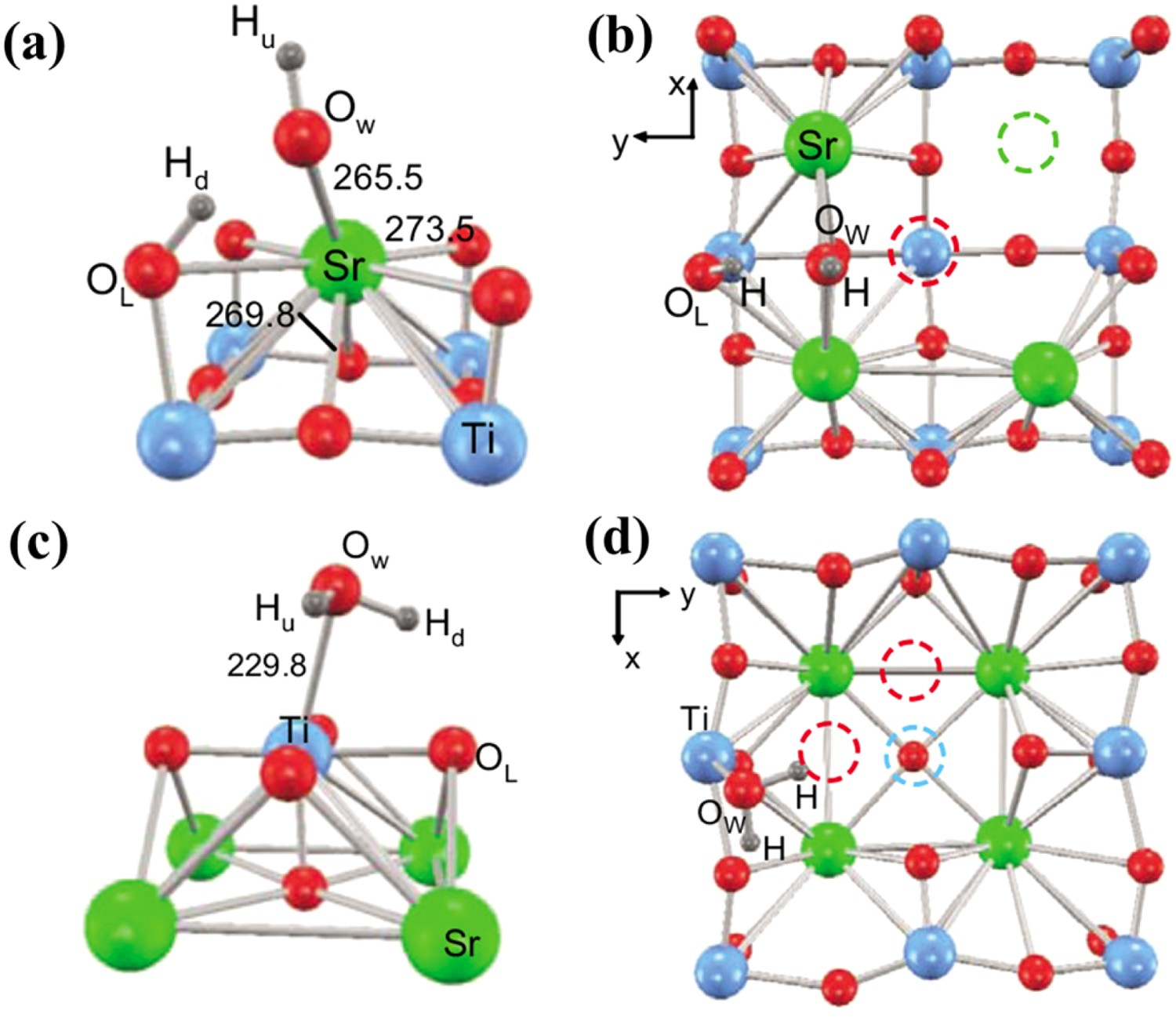
As the critical factors in water dissociation activity, the nature and concentration of surface and geometric arrangement of cation and anion sites can be delicately modified by atomic decoration technology. Surface atomic tailoring is expected to have bright a future in developing efficient and durable PCEC air electrodes. However, such work for a proton-conducting system is scarce. Only Tian [121] has used ALD as a tool for studying the reaction mechanism of TCOs. Considering the high electrochemical activity demands and the feasibility of a thermodynamiclly stable atomic layer at low temperature, it is reasonable to expect that surface atomic modification would be an efficient approach to meet such requirements.
Challenges
Long-term reliability
Besides catalytic activity and conductivity, air electrode materials need to be chemically stable under moist air during operation. The triple conducting electrode materials can be sorted into three structures, Cubic-type perovskites (ABO3), double perovskites (AA′B2O6) and Ruddlesden-Popper phase (K2NiF4). Cubic-type perovskites and double perovskites that are triple conductors usually contain alkaline earth elements, such Sr and Ba. However, the basic nature of alkaline earth elements makes them thermodynamically favourable to decompose in H2O–CO2 conditions. For the first generation TCOs, BSCF, and PBCO have been found to decompose after experimental treatment in humified environment at 600°C [138].
Comparison of the cell configuration, experimental condition and degradation rate of solid oxide electrolysis cells-based proton-conducting electrolytes. BCZY81 = BaCe0.8Zr0.1Y0.1O3-δ.
One effective strategy to improve the stability of perovskite oxides in a humid atmosphere is to partially replace the alkaline earth elements with elements with higher acidity, such as lanthanide elements. For example, Ba1−xGd0.8La0.2+xCo2O6−δ is chemically stable in 1.5 bar of steam for 100 h at 873 K, when x
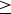
Characterization of the protonic properties
The proton concentration can be measured in situ by thermogravimetry in humidified gas as a function of temperature and p(H2O). This has been performed for measuring the proton uptake of the perovskite family (Ba,Sr,La)(Fe,Co,Zn,Y)O3-δ [49]. It can also be determined from the weight change of hydrated samples after switching from H2O to D2O atmosphere at fixed T and H2O and D2O partial pressure [141,142], while the proton uptake occurs either by H2O or H incorporation and the small H/D weight difference limits the accuracy for samples with low proton concentration. Ex-situ measurements [143] are applicable for low proton concentration samples and work well for H2O or H incorporation, but are time-consuming and tedious to cover an extended T and p(H2O) range.
The measurement of proton conductivity of triple-conducting oxides is more challenging. Generally, measuring the secondary ion mass spectrometry D/H isotope concentration profiles of quenched samples is a well-established method for determining ion diffusivities [144]. However, for triple conductors, the distribution of the measured signal seems nonhomogeneous, which may be due to the hydroxide layers on the surface or in the interior walls of pores.
The indirect measure of proton conductivity was unachievable until very recently. Song et al. [119] employed an effective method where an O2– blocking Pd layer is deposited (via PLD) as a proton-selective layer on both sides of the dense triple conducting membrane to avoid the direct contact of the membrane with gaseous hydrogen and block the transportation of oxygen ions, and the conductivity can be extracted by measuring the hydrogen permeation flux (Figure 13a). This method is challenging as the growth of a dense Pd film via PLD requires accumulated experience.
A simple alternative is to use electrical conductivity relaxation(ECR) measurement to investigate the chemical diffusivities of protons in an H2O/O2/N2 mixture [121]. By controlling the experimental conditions, proton uptake occurs predominantly via H2O or H incorporation. The chemical diffusion coefficients can be extracted from the transient behaviour of conductivity from the partial pressure change (Figure 13b). Similar to the proton concentration measurements by thermogravimetry, the ECR measurement may also have limited accuracy for samples with low proton concentration.
Fuel electrode materials
The fuel electrode of PCECs acts as a catalyst for fuel oxidization/hydrogen generation, electron collection, proton-conducting, and even mechanical supporting. An ideal fuel electrode should possess the following properties: high catalytic activity for various fuels, high electronic and proton conductivity, stable microstructures under operating temperatures and atmospheres, good chemical compatibility with the electrolyte, and favourable coking resistance [85,145–147].
Durable, coking and sulfur tolerant, fuel-flexible electrode
Robust perovskite electrode towards coke deposition and poisoning
Perovskite oxides represent a large category of complex oxides with the characteristics of flexible composition and rich properties. Many perovskites show mixed H+, O2–, and e– conductivity, possess good catalytic activity for various fuels, including H2 and hydrocarbons, and good resistance to coke deposition and sulphur poisoning. Some perovskites oxides, including La0.8Sr0.2Cr0.5Mn0.5O3-δ [148], La0.5Sr0.5Fe0.9Mo0.1O3-δ (LSFM) [149], La0.7Sr0.3V0.9O3-δ [150], Sr2Fe1.5Mo0.5O3-δ [151], etc., have been investigated as fuel electrode for PCECs. For example, Joo et al. [152] proposed a SOFC with BaZr0.3Ce0.5Y0.2O3-δ (110 μm) as an electrolyte, and PrBaFe2O5+δ as both air electrode and fuel electrode. The cell achieved a peak power density (PPD) of 0.30 W cm−2 at 700°C in H2 fuel. Luo et al. [149] proposed an BaZr0.1Ce0.7Y0.2O3-δ electrolyte-supported SOFC with LSFM as fuel electrode, which can be fed with H2 and C2H6 as fuels. In addition, SrFe0.75Mo0.25O3-δ was also characterized as a sulphur-tolerant electrode [151]. Some strategies, including optimizing morphology and introducing noble catalysts or nano-catalysts via infiltration, were proposed to improve electrode performance. For example, Park et al. proposed a nano (La0.7Sr0.3)V0.90O3−δ , CeO2, and Pd catalysts co-decorated fuel electrode via infiltration [150]. However, limited by low electronic conductivity and chemical incompatibility with electrode materials, the perovskite fuel electrode introduces significant challenges to fabricating high-performance fuel electrode-supported cells. The power outputs of present cells are too low for practical application.
Ni-based cermet electrode
Nickel (Ni) is the most promising fuel electrode catalyst due to its low cost, high electronic conductivity, good thermal conductivity [153–155], and excellent thermal/chemical compatibility with ceramic proton conductors, including BaZrO3-δ/BaCeO3-δ-based perovskite [156–159], SrCeO3-δ-based perovskite [160], LaNbO4 [161,162], etc. For introducing mixed conductivity into the electrode and increasing thermo-mechanical compatibility with the electrolyte, Ni is always composited with a proton-conducting ceramic phase.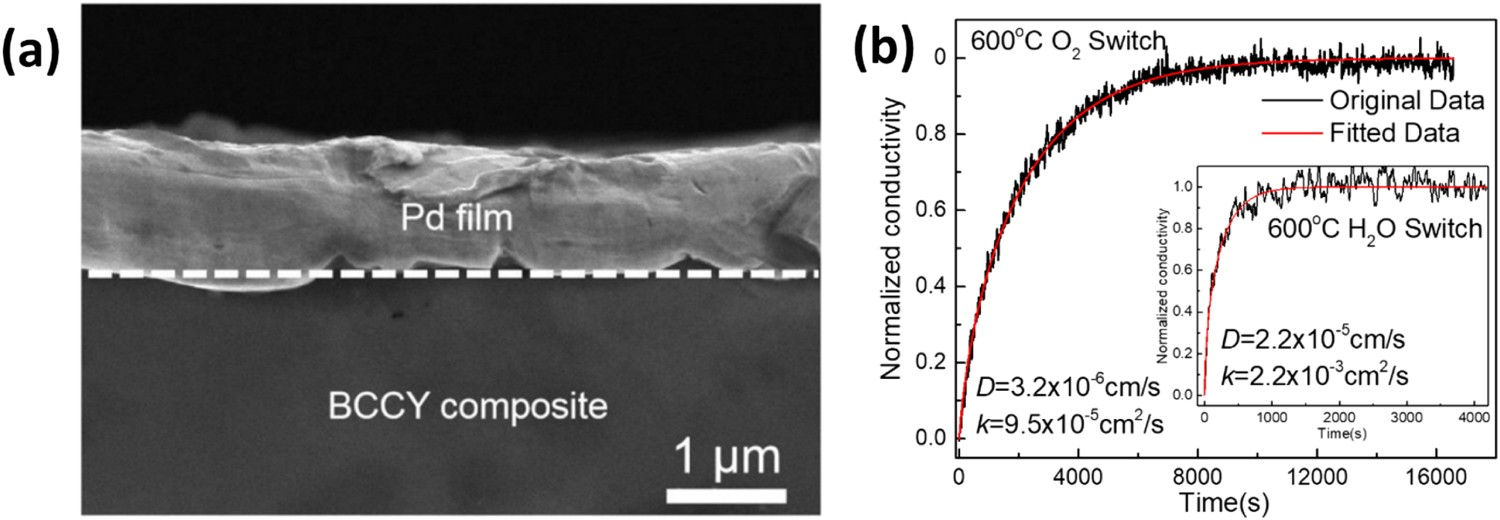
The Ni-BZCYYb1711 composite electrode was found to possess high resistance to coke formation and sulphur poisoning [32]. O’Hayre et al. investigated the practicality of different Ni-cermet electrodes, including Ni-BZY20, Ni-BZCYYb1711, and Ni-BaZr0.3Ce0.6Y0.1O3-δ (BZCY361), for operating in H2 and hydrocarbon fuels. It was found that Ni-BZCYYb1711-supported PCEC with BZCYYb1711 electrolyte and BCFZY air electrode (cell 1) (Figure 14(c)), achieved PPDs of 0.65 and 0.41 W cm−2 at 600 and 500°C, respectively, in H2 fuel (Figure 14(a)), while maintaining robust stability of 1100 h at 500°C (Figure 14(b)). The Ni-BZY20//BZY20//BCFZY (cell 2) (Figure 14(f)), achieved high PPDs of 0.235 and 0.105 W cm−2 at 600 and 500°C, respectively, in CH4-steam fuel (Figure 14(d)), while remaining robust stability for 1400 h at 500°C (Figure 14(e)). The above mentioned reported research results suggest that Ni-BZO/BCO-based fuel electrodes are promising for operation in sulphur/carbon-containing fuels in PCECs [33]. (a) I-V and I-P curves of cell 1 operated with H2 at 350–600°C. (b) Operational stability of cell 1 operated with H2 at 500°C. (c) SEM image of cell 1 after long-term stability. (d) I-V and I-P curves of cell 2 operated with 20% CH4+50 H2O+30% Ar at 500–600°C. (e) Operational stability of cell 2 operated with 20% CH4+50 H2O+30% Ar at 500°C. (c) SEM image of cell 2 after long-term stability. Reproduced with permission from Reference [33].
Strategies to improve performance
To enhance the performance of Ni-based electrodes, various strategies have been exploited, including infiltration, substitution and exsolution of nanoparticles, and insertion of a catalyst layer or functional layer. Infiltration or exsolution can produce finely dispersed nanoparticles to maximize the active surface area. The insertion of a catalyst layer above the fuel electrode layer can protect the fuel electrode from coking and thus enhance reforming activity. The insertion of a functional layer between the fuel electrode and electrolyte significantly increases the TPB length and reduces interfacial resistance.
Surface modification via infiltration
Surface modification via infiltration can be used to develop nano-metal catalyst decorated fuel electrodes to further enhance the catalytic activity for various fuels. Nano-catalysts, including Pd [163], Fe [164], Ni [165], Co, Cu, Rh [166], etc., have been infiltrated into the fuel electrode, which greatly enhanced hydrocarbon reforming reaction activity. For example, Park et al. proposed a Pd metal nanoparticles – infiltrated Ni-BaZr0.5Ce0.3Y0.1Yb0.1O3-δ fuel electrode, and corresponding SOFCs achieved higher PPDs of 0.59 and 0.28 W cm−2 in H2 and CH4 fuels, respectively, at 600°C, than that of non-Pd decorated cells (0.41 and 0.23 W cm−2) [163]. Rational design of infiltrated nano-catalysts can efficiently suppress coke deposition in the fuel electrode. For example, Hong et al. [166] infiltrated Ni–M (M = Ni, Co, Cu, Rh) nanoparticles into Ni-BZCYYb4411 electrode to enhance the activity for CH4 reforming. It was found that Ni-Rh nanoparticle decorated fuel electrode displayed stable CH4 conversion in long-term testing due to the higher activation energy of the C–C bond on Ni–Rh alloy compared to Ni, which prevented coke deposition, and lower C–O formation activation energy, thus removing carbon deposited on catalysts surface. The underlying principle also provides useful guidance for the design of alloy catalysts. In another study, Hua et al. developed a bifunctional Ni–Co-anchored PrBaMn2O5+δ catalyst in a Ni-BZCYYb1711 based fuel electrode by two-step infiltration (Figure 15). Because of the geometric blocking effect and strong metal–support interaction, NiCo nanoparticles are selectively monodispersed on PrBaMn2O5+δ nano-aggregates. Such well-designed architectures exhibited >97% CO2 conversion and good sulphur tolerance, and the corresponding SOFC delivered a stable output of 1 A cm−2 with 50 ppm H2S-containing CH4–CO2 fuel for 36 h [167]. Schematic preparation procedures of constructing NiCo/PBM bifunctional nanoarchitecture on BZCYYb1711 in the porous nickel cermet fuel electrode. Reproduced with permission from Reference [167].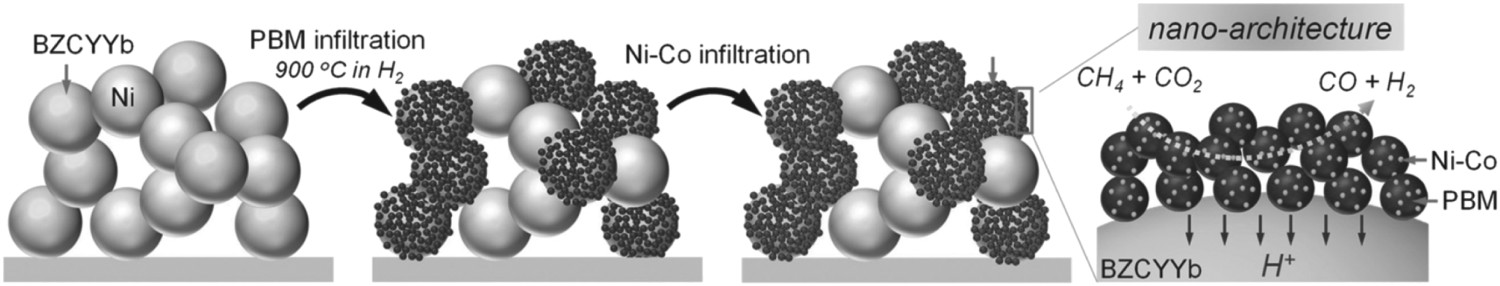
Substitution and exsolution
Compared to infiltrated nano-catalysts, exsolved metal nanoparticles with stronger interaction with the perovskite substrate have also been proposed to inhibit the delamination of catalysts and perovskite substrate resulting from coke deposition [168]. The strong interaction will also change the surface electronic structure of metal catalysts, thus inhibiting sulphur adsorption and poisoning [169,170]. For example, Shao et al. developed a novel Ni-doped proton conductor, Ba(Zr0.4Ce0.4Y0.2)0.8Ni0.2O3-δ (BZCYN), as a highly active and sulphur-tolerant fuel electrode for SOFCs. After treating BZCYN in H2 at 800°C, BZCYN decomposes into BaZr0.4Ce0.4Y0.2O3-δ (BZCY442), amorphous BaO, and Ni nanoparticles. Exsolved Ni nanoparticles promoted H2 oxidization activity, while the adsorbed water on BZCY442 and BaO surface immediately reacted with the adsorbed sulphur to inhibit sulphur poisoning [169].
In situ exsolved nano-catalysts can also be introduced to modify Ni-cermet fuel electrodes. Liu et al. used a Ni-doped proton conductor, Ba0.96Ce0.66Zr0.1Y0.2Ni0.04O3-δ (BZCYN96), as the ceramic phase in the fuel electrode. At high-temperature reducing conditions, Ni nanoparticles were exsolved from the lattice and enriched on the perovskite surface, thus enhancing catalytic activity for H2 oxidization. A SOFC with the Ni-BZCYN96 fuel electrode exhibited a higher PPD of 0.91 W cm−2 at 700°C in H2 fuel, which is much higher than the non-Ni nanoparticle modified fuel electrode (0.73 W cm−2). The surface-enriched Ni nanoparticles were still anchored on the BZCYN96 surface without noticeable sintering after 120 h operation, suggesting the good resistance to sintering capability of the exsolved Ni nanoparticles [171].
Rational substitution of proton conductors can optimize the catalytic activity and ionic conduction of fuel electrodes simultaneously. He et al. proposed a noble metal Pd doped proton conductor, Ba(Zr0.1Ce0.7Y0.1Yb0.1)0.95Pd0.05O3-δ (BZCYYbPd), as both the ceramic phase in fuel electrode and the electrolyte of the PCEC. In fuel cell operation, Pd was exsolved from the perovskite lattice and enriched on the Ni-BZCYYbPd fuel electrode surface in the form of Pd nanoparticles. The Pd nanoparticles modified fuel electrode displayed higher NH3 decomposition and H2 oxidization activity than the pristine Ni-BZCYYb fuel electrode (Figure 16(a,b)). Furthermore, the partial substitution of Pd in BZCYYb also promoted proton conduction due to the B-site deficiency resulting from Pd exsolution and surface modification of Pd nanoparticles, which accelerated proton generation. Finally, a Ni-BZCYYbPd fuel electrode-supported SOFC with BZCYYbPd and BCFZY as electrolyte and air electrodes, respectively, achieved high PPDs of 0.94 and 0.72 W cm−2 in H2 and NH3 fuels, respectively, at 650°C (Figure 16(c)), while also maintaining robust stability for 205 h in H2 and 130 h in NH3 at 550°C (Figure 16(d)) [172]. (a,b) Schematic of fuel electrode modified with Pd Nanoparticles operated with NH3 fuel. (c) I-V and I-P curves of Ni-BZCYYbPd fuel electrode -supported SOFC operated with NH3 fuel at 550–650°C. (d) Operational stability of the cell operated with H2 and NH3 fuels at 550°C. Reproduced with permission from Reference [172].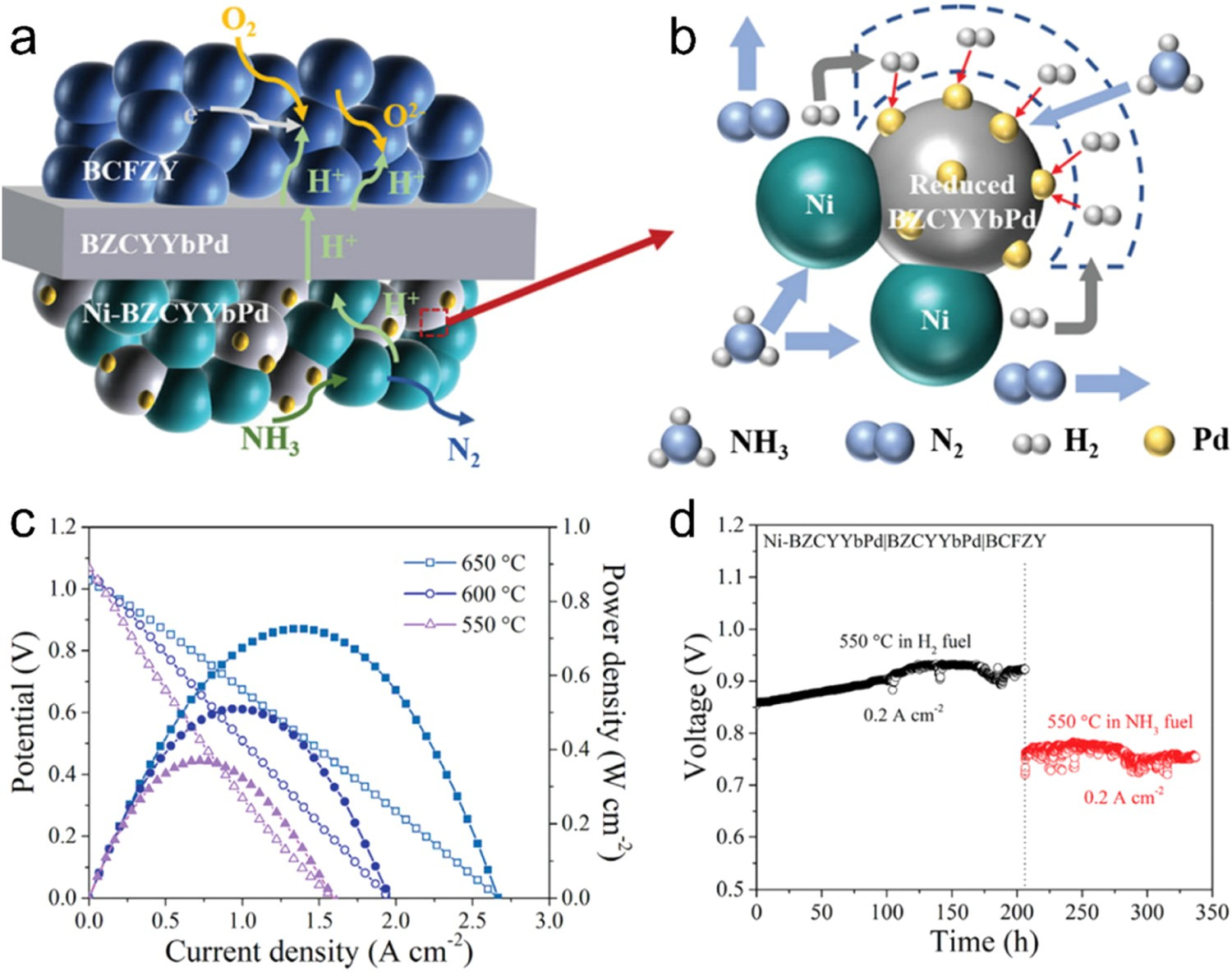
Recently, O’Hayre et al. systematically studied the long-term durability of SOFCs using exsolved Ni nanoparticle fuel electrodes and a BZY20 electrolyte [44]. The as-fabricated cells achieved attractive PPDs in various fuels, such as hydrogen, ammonia, methane, methanol, butane, propane, etc. (Figure 17(a)). Thousands of hours of performance stability and stable electrochemical impedance after switching fuels suggested that the Ni-BZY20 fuel electrode possessed high resistance to coke deposition and sulphur poisoning, highlighting the promise of SOFC technology and its potential for commercial application (Figure 17(b,c)). The SOFC displayed similar Nyquist plots in sulphur-containing and sulphur-free fuels, further confirming excellent sulphur-resistance of the Ni-BZY20 fuel electrode (Figure 17(d)). In situ exsolved Ni nanoparticles played a significant role in fuel catalysis and suppressing sulphur poisoning. As seen in Figure 17(e), exsolved Ni nanoparticles promote hydrocarbon reforming/water–gas shift processes with anti-fouling properties due to the intimate contact between Ni nanoparticles and the BZY20 support substrate. Furthermore, Ni nanoparticles also provided additional surface area to adsorb sulphur and suppress sulphur poisoning of the physically mixed Ni catalysts. The adsorbed sulphur could be immediately removed due to the high water-storage capacity of BZY20 and the intimate contact between Ni nanoparticles and the BZY20 substrate. (a) I-V and I-P curves of Ni-BZY20 fuel electrode-supported SOFCs operated with H2 (F1), NH3 (F2), Methanol (F3), iso-butane (F4), n-butane (F5), natural gas (F6), 19.5 ppm H2S-natural gas (F7), propane (F8), methane (steam/carbon ratio S:C = 2, F9), ethanol (F10), methane (S:C = 2.5, F11), and iso-octane at 600°C. Long-term stability of SOFCs operated with (b) iso-butane, propane, iso-octane, methanol, (c) ethanol, and natural gas fuels. (d) Nyquist plots measured under sulphur-containing, sulphur-free natural gas, and methane fuels. (e) Schematic illustration of SOFC and mechanism of hydrocarbon reforming, water–gas shift reaction, sulphur and carbon cleaning. Reproduced with permission from Reference [44].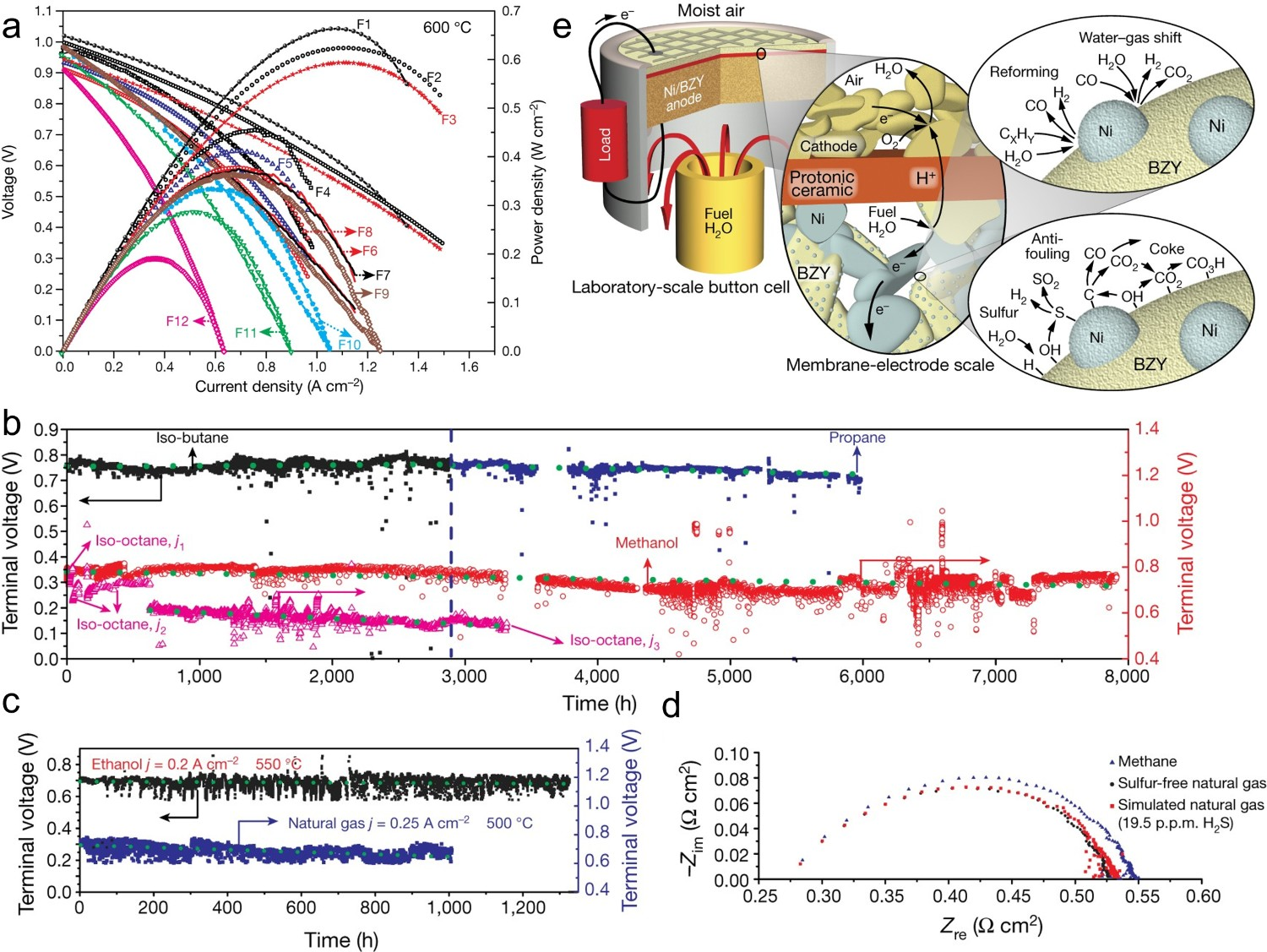
Introducing functional layer
The introduction of an internal functional layer between the fuel electrode and the electrolyte aims to maximize the TPB length for fuel catalysis and reduce interfacial resistance by creating more contact area between the electrode and electrolyte [173,174]. Hwang et al. introduced an internal functional layer (Ni-BaZr0.2Ce0.7Y0.1O3-δ (BZCY271)) between the highly porous fuel electrode and dense electrolyte via tape-casting, indicating increased performance from fuel electrode/electrolyte contact and extended TPB length for fuel catalysis [174]. Shim et al. further introduced internal micron and nano-functional layers between the conventional fuel electrode and electrolyte via tape-casting and pulsed laser deposition, respectively, as seen in Figure 18(a). In addition to the advantages mentioned above, the nano-functional layer also reduced the defects and roughness of the electrode surface, which is preferable for thin-film electrolyte growth. The nano-AFL maintained intimate contact with the micro-AFL and electrolyte via rationally optimizing the Ni content in the nano-AFL and the annealing temperatures, thus ensuring high performance of the SOFC. Finally, the resulting SOFC with La0.6Sr0.4CoO3-δ achieved high PPDs of 740, 563, 457, and 342 mW cm−2 at 600, 550, 500, and 450°C, respectively, in H2 fuel (Figure 18(b)). The high power output of the cells prepared via the special process demonstrated the great potential of those SOFC [175]. (a) Schematic image of BZY20 electrolyte-based fuel electrode-supported PCEC with micron/nano-AFLs, and thin-film electrolyte. (b) I-V and I-P curves of the SOFC operated with H2 at 450–600°C. Reproduced with permission from Reference [175].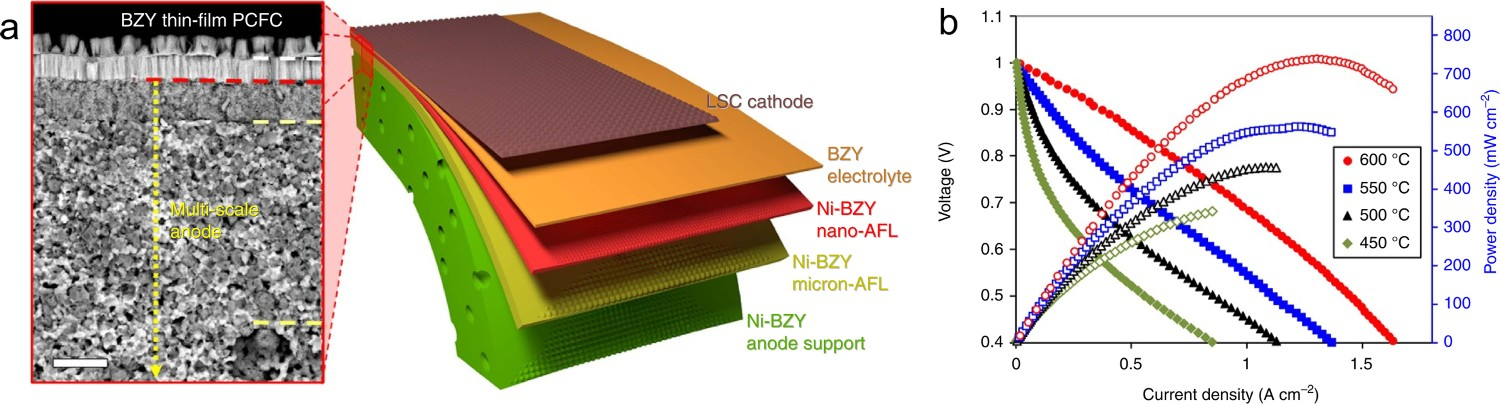
Introducing catalyst layer
An external catalyst layer is also often applied to promote fuel reforming and suppress coke deposition [176,177]. Lu et al. proposed a novel perovskite, (Pr0.3Sr0.7)0.9Ti0.9Ni0.1O3-δ (PSTN), as a catalyst layer. In reducing atmosphere, Ni nanoparticles were exsolved from lattice in situ and enriched on the PSTN surface. Highly dispersed Ni nanoparticles created more active sites for fuel catalysis and reforming, greatly improving the propane conversion rate, thus depressing the carbon deposition. A Ni-BaZr0.3Ce0.5Y0.2O3-δ (BZCY352) fuel electrode-supported cell with such a catalyst layer achieved PPDs of 0.45, 0.25, and 0.10 W cm−2 at 750, 700, and 650°C, respectively, operated with propane-10 vol% H2O fuel. Furthermore, the cell also displayed good operational stability for 50 h, which was much better than the cell without the catalyst layer [178]. Similarly, Guo et al. proposed La2NiO4 (LNO) as an ACL for SOFCs operated with CH4 fuel. Ni nanoparticles were enriched on the LNO surface in operation, promoting the reforming reaction of CH4/CO2. Finally, a SOFC with such an ACL achieved similar PPDs compared with non-layer ones in H2 fuel, but higher PPDs and longer operational stability in CH4/CO2 mixture fuel, confirming the superior reforming activity of the exsolved Ni nanoparticles [178]. Similarly, Yoon et al. also proposed a LaNi0.6Co0.4O3-δ perovskite ACL to promote the CH4/H2O reforming reaction, enhanced performance and durability were demonstrated [179].
As mentioned above, in situ exsolved metal nanoparticles could promote hydrocarbon reforming, however, the exsolved metal nanoparticles may suffer severe sintering during long-term operation in reducing conditions at high temperatures. Chen et al. introduced an external Ce0.9Ni0.05Ru0.05O2-δ (CNR) ACL to promote the hydrocarbon reforming reaction. Under operation conditions, Ni and Ru atoms were evenly distributed in the crystal lattice, where Ni atoms mainly participated in the activation of C–H to form intermediates, while Ru atoms, coupling with surface oxygen vacancies, mainly participated in the activation of H2O. The synergistic effect between Ni and Ru atoms greatly promoted the reforming activity of CH4/steam [180]. Similarly, Wang et al. also proposed a Ni and Ru co-doped perovskite catalyst, Ba(Zr0.1Ce0.7Y0.1Yb0.1)0.9Ni0.05Ru0.05O3-δ (BZCYYbNR), to promote the reforming reaction of n-butane/steam and enhance performance and durability, where Ni and Ru active sites were evenly distributed in the oxides in the form of single atoms, providing both high activities for hydrocarbon catalysis and good anti-sintering capability [181].
Challenges: performance at lower temperatures
The main function of the fuel electrode is to efficiently catalyse fuel cracking and reforming reactions with high resistance to sulphur poisoning and carbon deposition. Lower temperature operation (from 500 to 650°C) can accelerate the coke deposition on the fuel electrode in direct methane fuelled SOFCs [182]. Lower operating temperature also favours shifting the water–gas shift towards CO2 in steam reforming of methane in PCECs, leading to a low CO/CO2 ratio to reduce boundary reaction and mitigate coking [183]. However, the biggest challenge resulting from low operating temperature is the insufficient activity for fuel catalysis. Even if some reported cells with H2 fuel can operate at 350°C, the use of readily available hydrocarbon fuels will be more attractive in the future.
One of the key limitations for intermediate temperatures SOFCs with hydrocarbon fuels is the fuel electrode polarization resistance. Incorporating catalyst layers or nano-catalysts into fuel electrodes is promising, but these additional layers or particles may also increase the resistance to diffusion and current collection. Thus, there is a tradeoff between catalytic activity improvement and current and diffusion loss. Although the coarsening and grain growth of the nano components can be mitigated by reducing operation temperature, the durability of nanomaterials remains a critical challenge in practical application. To better understand the specific reaction mechanisms, in situ monitoring and characterization techniques are encouraged to probe the active sites in the TPB, surface/interface chemistry, and microstructure change as a function of time and temperature. Moreover, sulphur poisoning issues might be more significant at lower temperatures since its absorption is more dramatic [184,185]. However, little attention has been paid to date. There are various promising strategies to develop highly sulphur tolerant fuel electrodes, such as increasing oxygen flux from the electrolyte to the fuel electrode, incorporating more sulphur-tolerant additives (Sn, Mo, or BaO), and using ceramic fuel electrodes.
Novel cell designs and microstructures
Although extensive progress has been made recently on the development of protonic ceramic materials with promising electrochemical properties for intermediate temperature PCEC operation, the air electrode process remains the most rate-limiting factor for the future electrochemical system at lower temperature. Technical challenges arising from the air electrode side calls for innovative strategies in regard to the smart design and optimization of electrode microstructure/architecture in order to achieve the high performance, durability, and scalability requirements towards advanced industrial energy application. This section will briefly discuss the recent advances on the transition of air electrode structure from a convoluted particle-packing network with a random distribution of pores and electrochemically active sites into rationally designed and derived novel electrode architectures and features that allow versatile microstructural/surface-electrochemical tuning with intricate dimensional control over active electrode phase/pore morphology, alignment, and distribution.
Nanocomposite/hetero-conjunction
The triple-conductive interface with active sites that can provide electronic, oxygen ionic, and protonic conduction paths for H2O redox reaction is critical for the air electrode performance at low temperature. Thus, various reactive/non-reactive synthesis routes have been seeking to introduce TCOs with desirable microstructural and electrochemical properties to enlarge the active area and improve air electrode kinetics.
Non-reactive formation of nanocomposite
The conventional strategy of extrinsic incorporation of nanostructured secondary-phase particles into the backbone electrode via infiltration/impregnation is effective but time-consuming and applicability limited [126,169,186]. New preparation methods based on fibrous synthesis and precursor freeze-drying/spray pyrolysis have thus been developed as a more efficient, reproducible, and scalable replacement of the infiltration technique to obtain nanocomposite electrode structure with a high surface-to-volume ratios. Lee et al. have created a 1-D nano-architecture by embedding pre-synthesized BaCe0.5Zr0.35Y0.15O3−δ nanoparticles onto LSCF fibres electro-spun from nitrate precursor [187]. Compared to powder-mixture composite, the LSCF nanofibrous electrode showed not only 30–40% lower polarization resistances, but also distinct reduction of activation energy for electrode polarization, which can be attributed to the high BET surface area(>10 m2 g−1), less tortuosity and mass/charge-transport distance, and mitigated residual stress between LSCF fibre and BaCe0.5Zr0.35Y0.15O3−δ electrolyte after calcination. Alternatively, Santos-Gómez and co-workers have explored freeze-drying and spray-pyrolysis as industrially-viable techniques with high single-step loading for deposition of single-phase BCFZY onto the electrolyte scaffold to create nanostructured composite [188]. Compared to freeze-drying, pray-pyrolysis was more efficient at retaining electrode microstructure with 30–80 nm nanoparticles and a fine dispersion on the electrolyte grain after annealing at 950°C. Thus, a high electrochemical performance of 1 W cm−2 and ASR value of 0.067 Ω cm2 were reported for the cell with BCFZY air electrode at 600°C. Although promising results have been shown for the above nanocomposite electrodes, the absence of any long-term stability tests raises questions about their reliability because for extended electrochemical operation, loss of surface-active area and morphological evolution due to sintering can be a concern for nanostructured electrode under redox/thermal cycles [189].
In-situ formation via phase-transition
Usually, interfacial reactions during electrode manufacturing or subsequent operation are considered unwanted and should be avoided to attain the desired phase structure of each component. On the other hand, they can be utilized to obtain secondary phases with beneficial physiochemical characteristics if the interfacial reactions are selectivly controlled [190–192]. Borrowing this strategy, Geng and co-workers developed a triple conducting Sr3Fe1.8Nb0.2O7−δ Ruddlesden-Popper phase from surface-treatment and the in-situ reaction of SrFe0.9Nb0.1O3−δ perovskite with Sr nitrate to form a core–shell nanostructure triple conducting junction [193]. Aside from junction formation via extrinsic surface/cation reactions, Song and Zhao have derived nanocomposite electrode constituents more efficiently and directly from facile one-spot sol–gel synthesis and self-phase-evolution of a BaCo0.7(Ce0.8Y0.2)0.3O3 parent material during subsequent calcination [119,194]. HRTEM (Figure 19) analysis clearly indicates local-coupling of twin perovskites with different conduction natures of varied Co concentrations at the vicinity of nanograins converted from the stoichiometric precursor phase. Single cells with oxygen ion-electron–proton-conducting BaCexYyCozO3-δ (BCCY) and oxygen ion-electron conducting BaCoxCeyYzO3-δ nanocomposite as air electrode have delivered R
p as low as 0.11 Ω cm2 at 600°C using BZCYYb1711 as the electrolyte [193]. Also, the BCCY nanocomposite electrode showed no degradation over 800 hrs at 550°C, indicating high durability of the self-assembled nanostructure while retaining electrochemical activity. Despite metastable phase transformation showing such promises in obtaining active nanocomposites, it is found that, in certain cases, exsolution of nano-derivative phases from a precursor phase has not distinctively improved ORR kinetics compared with electrodes made by direct mixing of constituent phases of similar compositions [195]. This may suggest that self-assembled nanocomposite electrode activity can be highly sensitive to derived phase composition and interphasial contact morphology, and thus may pose a challenge for processing parameter control. (a) Schematic diagram of conventional oxygen ion-electron–proton triple conducting electrode (Left) and self-assembled oxygen ion-electron–proton triple conducting nanocomposite electrode (Right); (b) STEM and HR-TEM images of a BCCY composite particle. Reproduced with permission [119].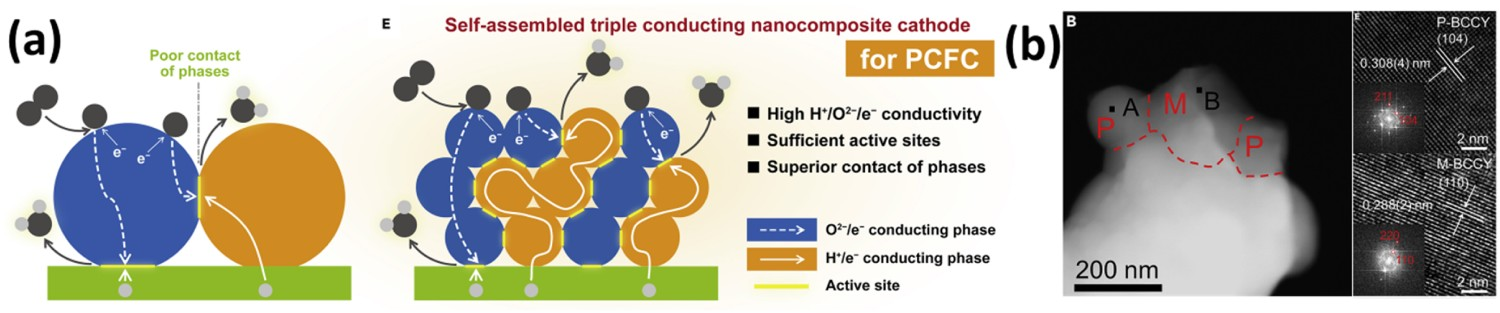
In summary, the performance and stability optimization achieved by nanocomposite formed in-situ via parent phase transformation can be attributed to aspects such as intimate nanocontact between derivative phases, alleviated CTE mismatch between electrode and electrolyte, and better physiochemical compatibility between parent, derivative, and electrolyte phases. Furthermore, it has been pointed out that the nano-coupling of two derivative phases may lead to certain synergistic effects that affect both oxygen surface change kinetics and bulk mobility for nanocomposite electrodes [193]. However, such hypotheses remain to be examined in a detailed study, and the potential phase/microstructure evolution of derived nanocomposites still needs further investigation after extended operation.
Triple-conducting 3D fibrous architecture
The largest obstacle at lower temperature for PCEC operation is the lack of electrode catalytic activity. One of the most effective ways to compensate for the loss of catalytic kinetics at reduced temperature is to increase the effective electrode surface area by architecture optimization. Nanofiber morphology was found to be one of the most effective strategies in tremendously increasing the surface area. When TCO, such as PBSCF, takes a nano-fibrous form, their mass activity shows a linear relationship with surface area. An unexpected intrinsic activity enhancement, due possibly to a favourable eg electron filling, presents as the diameter is reduced to ∼20 nm [196] (Figure 20(a)). Borrowing the advantages of nanofiber morphology, 3D fibrous architectures have been rationally designed and viably fabricated through a fabric-template-based technique to construct highly active PBSCF and PNC air electrodes for PCEC operation below 600 [115,118]. The nano-engineered hollow PBSCF and PNO fibres bearing 3 µm wide through-pores and 20–50 nm-sized surface-cladding nanoparticles lead to ultra-high bulk porosity of nearly 60 vol%, allowing full expansion of the effective surface area to the entire 3D nano-architecture (Figure 20). Such a hollow-fibrous strategy has proven to enhance the surface oxygen exchange kinetics more effectively than nano-powders or solid-core fibres for air electrodes [197,198]. As a result, PBSCF and PNC hollow-fibrous electrodes both showed remarkable (more than doubled) kinetic improvement compared to regular electrodes consisting of the same material, and the overall electrochemical performances are among the top ones reported. (a) Mass activities and BET surface area-normalized intrinsic activities of PBSCF sample with surface areas of 1.52, 9.09, 14.72, 18.81 m2g−1, respectively. (b) Cross-sectional view of the cell with PNC single layer of nanofiber-structured mesh adhered on the electrolyte. (c) Scanning electron microscopy images of the electrode mesh showing hollow-fibre-like string self-architecture. (d) Transmission electron microscopy images of a single nanofiber composed of ∼50 nm particles Reproduced with permission from Reference [115,196].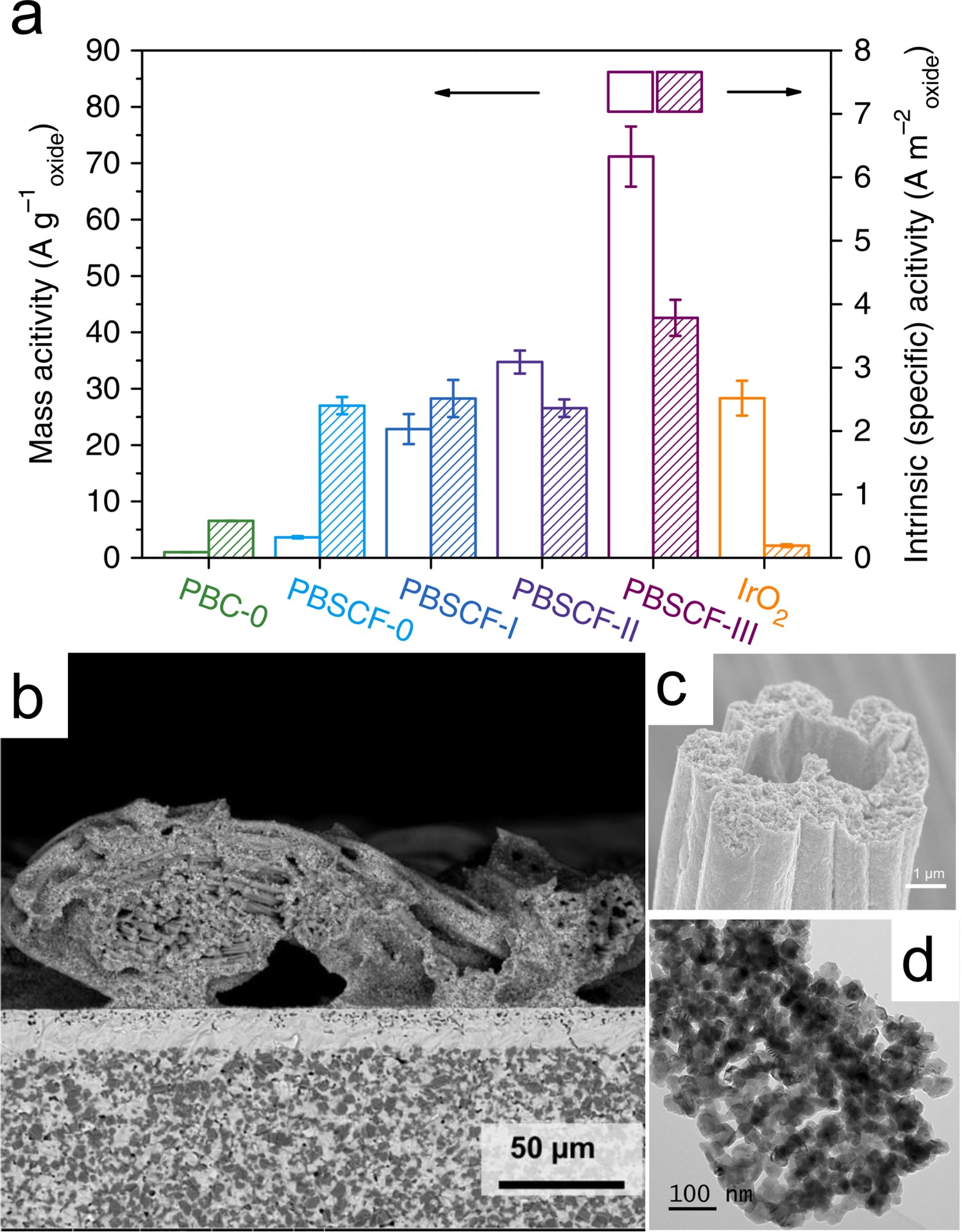
The functional successes achieved by 3D fibrous microstructure set new guidelines and criteria for advanced air electrode architecture design for next-generation PCECs. However, it should be noted that after a certain period of stability testing, wide-range reconstruction of 3D fibrous architecture occurred for the PBSCF/PNC electrode, with nanoparticles droping-off the hollow fibres and filling up pores in the mesh-like network, forming a continuous thin layer along with electrode/electrolyte interface. Although such morphological evolution was not found to adversely impact electrode kinetics in the given test duration according to AC impedance analysis, but rather reduce the cell ohmic resistance due to improved electrode/electrolyte contact, the reliability of such electrodes under practical redox/thermal cycles should be addressed. A comprehensive modelling effort that addresses individual contributions from mass transport, reactant/electrode surface exchange, intermediates incorporation, and bulk charge transfer to the overall electrode kinetics may be necessary to predict the effect of morphological evolution from the continuous reconstruction.
New cell concept
New concepts on cell configurational and operational design have been advised and tested recently to promote the applicability of PCEC system as advanced energy conversion/storage technologies. Kim et al. first proposed a novel ‘Hybrid-solid oxide electrolysis cell’ (Hybrid-SOEC) concept (Figure 21(a)) from theoretical consideration that with dual oxygen-ion proton-conducting electrolyte membrane, H2O could be electrolysed on both sides of PCEC and thus improve electrolysis efficiency [117]. The corresponding PCEC demonstrated much higher performance under hybrid-electrolysis with 10 vol% H2O presence on both sides. The achieved current density in hybrid-electrolysis under 1.5 V at 700°C is 15–45% higher than conventional electrolysis due to the co-production of H2 and O2 on both electrodes. However, a special system design may be needed for such a concept as separating and collecting the H2/O2 after co-electrolysis would become a challenge. On the other hand, some recent research efforts have been dedicated to using PCEC for direct in-cell or indirect out-of-cell CO2 conversion. These arise from the incentives of recycling CO2 as greenhouse gas while generating valuable products such as syngas, methane or methanol with higher volumetric density than pure H2 for energy storage. Danilov et al. reported that an increase of pCO2 in wet H2 mixture from 0 to 0.5 atm led to higher electrolysis current density and hydrogen production rate at 700°C (Figure 21(b)) [113]. According to AC impedance analysis, this can be ascribed to enhanced proton conductivity at the electrolyte surface by extra water vapour formed from the water–gas shift reaction due to the presence of CO2. Such a finding is generally consistent with Kim's conceptual work on co-electrolysis PCECs, though with a potentially different explanation for the performance enhancement. Parallel work by Shi et al.'s with BZY-based PCEC also verified CO2-mixing on the fuel electrode facilitated steam electrolysis at 550°C with a high current of 1.23 A cm−2 obtained at 1.5 V [199]. Although such CO2 in-cell conversion can improve PCEC performance, the Faradaic efficiency obtained from gas chromatograph for the above system is only 42%, indicating considerable optimization is needed to render the PCEC efficient for simultaneous H2 generation and CO2 conversion. Additionally, based on a new concept of power-to-methanol conversion, Schwabe et al. have devised and theoretically evaluated an integrated electrolysis-synthesis system with a tubular PCEC for H2 generation at 700°C and a catalytic chemical reactor for pressurized CH3OH (Figure 21(c)) production under 10 MPa at 450°C [18], for which a test platform remains to be built with funding support for proof-of-concept experiments. (a) Schematic diagram of Hybrid-SOEC system working principle [117]. (b) Dependence of V-I characteristic of PCEC on pCO2 at the Ni–BCZDy fuel electrode side at 700°C [113]. (c) A conceptual design of an integrated PCEC-electrolysis/catalytic-synthesis system for methanol production [18]. Reproduced with permission.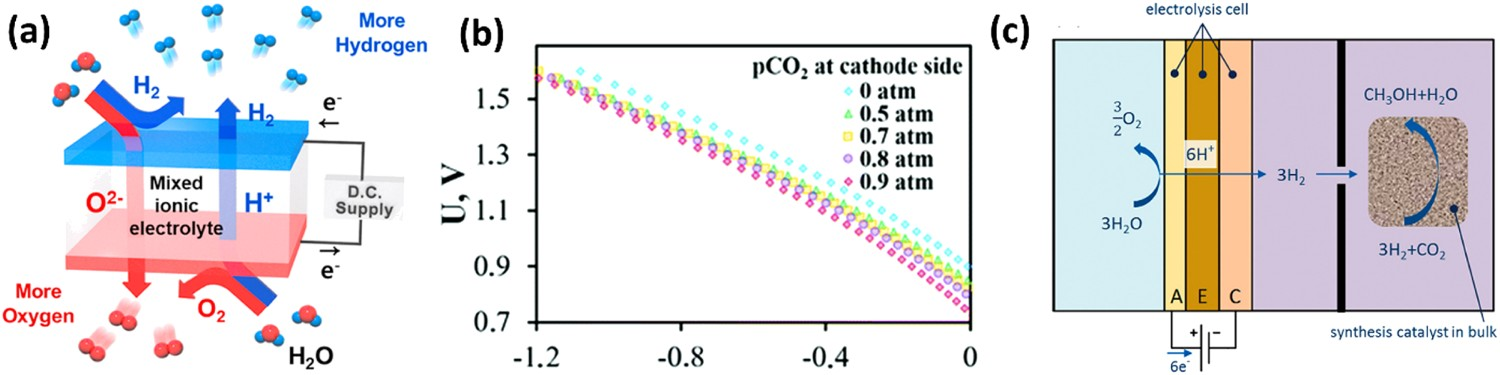
Conclusions and prospects
Protonic ceramic cells have attracted tremendous attention in the past decades due to their unique advantages. Most of the efforts in protonic ceramics have so far focused on electrolyte and air electrode materials. Recent progress regarding materials innovation and novel technologies are highlighted and discussed to provide essential perspectives on crucial aspects. After decades of optimization, despite new alternatives being found for electrolyte, doped barium zirconate/cerate perovskite oxides are still the best performing proton-conducting electrolyte materials, while promising progress has been made on the optimization of protonic conductivity. As attention gradually shifts from fuel cell operation for power generation to electrolysers, faradaic efficiency and stability are brought under spotlight. To advance the development of proton conductor-based cells, rational design of high-performance electrolyte materials with novel compositions and structures are urgently needed.
In term of air electrode materials with triple conducing (mixed H+/O2–/e–) properties have been developed. Their superiority over the traditional MIECs in a proton-conducting system has been investigated and confirmed over the past decades. However, the systematic development of TCOs is still challenging because of the difficulties with characterizing protonic behaviour in TCOs. A quantitative property assessment and prediction are still not available for ternary oxides. As the operation of PCECs is being pushed to lower temperatures (≤ 500°C), surface modification and decoration may be effective strategies to compensate for the catalytic activity loss from reduced thermal activation
The Ni-based fuel electrode is and still will be the best fuel electrode in the near future due to their low cost, high electronic conductivity, and excellent thermal/chemical compatibility. The introduction of nano-catalysts via various strategies has dramatically enhanced its resistance to coke deposition and sulphur poisoning at moderate temperatures. However, more rigorous challenges rise at lower temperatures.
Advanced materials optimization and architecture innovation are carrying the development of PCECs from laboratory exploration to the next phase. Significant breakthroughs are expected in promising protonic energy conversion and storage applications beyond the current technologies. Scaling up the footprint of the cells and readying them for in-field demonstration will be needed to demonstrate the advantages of PCECs in the future.
Footnotes
Disclosure statement
No potential conflict of interest was reported by the author(s).