
Abstract


John W. Kozarich, PhD, is Chairman and President of ActivX Biosciences, Inc. He received his BS degree in chemistry from Boston College and his PhD from the Massachusetts Institute of Technology. Dr. Kozarich completed a National Institutes of Health (NIH) postdoctoral fellowship in biochemistry at Harvard University. After an early career in academia, with tenured full professorships at Yale University School of Medicine and the University of Maryland, he moved to industry and while on sabbatical initially served as chief scientific officer of the start-up company Alkermes in Cambridge, Massachusetts. During his tenure as Vice President of Biochemistry at Merck Research Laboratories in Rahway, New Jersey, he led the development of Merck's Boston Research Center. In 2001 he moved to San Diego to head ActivX, which has pioneered the development of activity-based proteomics and its application to drug discovery and development. Tokyo-based Kyorin Pharmaceuticals, Inc., acquired the company in 2004.
Dr. Kozarich, your career has taken you from the halls of academia as a professor in a medical school and a university setting, to being a director in one of the world's largest pharmaceutical companies and currently president of a biotechnology company. Drawing on these experiences, how do these three professions complement each other to enable drug discovery?
Looking back on my career path, I think my own evolution has been a reflection of my research interests and how I could optimize the impact I could have on biochemistry, biotechnology, and drug discovery. I started out as a pure academician. I got my Bachelor's degree from Boston College in Chemistry, went to MIT, and worked more in the area of biological chemistry—at the interface of chemistry and biochemistry. I was then doing a postdoc in biochemistry at Harvard in Jack Strominger's lab, when I began to look for a job. I had a lot of great interviews at various chemistry and biochemistry departments, and in those days they were generally separate entities; very few departments called themselves departments of chemistry and biochemistry, as you often find now. When I would interview at a chemistry department, I was viewed as more of a biochemist; whereas, in a biochemistry department they tended to view me more as an organic chemist.
I went to Jack Strominger and told him I felt like I was falling through the cracks. He suggested that I apply to a pharmacology department. The subtext of that comment is that before Jack went to Harvard to teach biochemistry, he was chairman of pharmacology at the University of Wisconsin, and before that at Washington University. He was well known for his work in understanding the mechanism of penicillin action in bacteria. I asked him half-jokingly, “What is pharmacology?” But I took his advice, applied to the departments of pharmacology at Harvard and Yale and was offered jobs at both. In pharmacology you are looking at the interface of chemistry (small molecules/drugs) and how they interact with biological systems. I decided to go to Yale, learned a lot of pharmacology there, and began to think about drug discovery.
I had a tenured professorship at Yale, but I decided that I wanted to go back to a chemistry department, and that is when I moved to the University of Maryland at College Park. Compared to being in a pharmacology department, that gave me more access to graduate students and I was able to increase the size of my lab. Together with several of my colleagues at the University of Maryland, we succeeded in having the name of the chemistry department changed to the Department of Chemistry and Biochemistry.
I had become interested in the biotech sector over time, and while I was a professor at Maryland, one of my former mentors at MIT, Paul Schimmel, who is now at The Scripps Research Institute in La Jolla, was instrumental in starting a lot of early biotech companies. He was a cofounder of Repligen, Alkermes, Cubist, and Alnylam, for example. Paul called me about the new company he was starting then, Alkermes, and asked if I would be interested in heading R&D at the company. In 1990, I took a year's sabbatical from Maryland and went to Alkermes. Within about 10 months we were convinced that we could file an investigational new drug application (IND) on a drug delivery method. I hired a head of clinical development and we began the work to file an IND. I was literally learning this stuff de novo. I extended my sabbatical for about another 6 months. We hired a talented, young CEO, Rich Pops, in February of 1991, took the company public 2 months later, and filed an IND that summer. It was an incredible ride.
As the company was becoming increasingly developmentally focused, and I was also still running my lab at the University of Maryland, I decided to go back to Maryland. Alkermes kept me on for another 18 months as a 1-day-a week CSO. I had a great experience with Alkermes, have fond memories of my experience there, and learned a lot. Alkermes is now a $3+ billion company.
The next summer, in 1992, Merck contacted me. They were looking for someone to take a leadership role in biochemistry at the Rahway site. I think they were intrigued that I had been involved in a biotechnology company. Merck has always liked to bring in people from academia. When I joined Merck, as an executive director initially and then as a vice president, I was one of the only people in management who had experience in a biotechnology company; most came from academia or moved up through Merck. As a result, I did a lot of biotech-related work for Merck, and that is how I became involved with the Aurora collaboration, for example.
I worked at Merck until early 2001. I had been doing a lot of work in San Diego and was involved in Merck's acquisition of SIBIA Neurosciences, which became the Merck San Diego site, but subsequently closed. In late 2000, Paul Schimmel, then at Scripps, called me about the new company a young colleague of his, Ben Cravatt, was starting called ActivX Biosciences. “It's right up your alley,” he said. And I joined ActivX.
How did each previous experience inform the next? What have you taken away from each on a broad scale that you have been able to apply to subsequent challenges or opportunities?
While I was at Merck I learned a lot about big pharma, drug discovery, clinical trials, and all of the issues involved in what projects to work on—not only from a scientific standpoint but also from the perspective of market potential. I was involved in the early work on Januvia, for example, and I have done a lot of work in the areas of antibiotics, statins, and steroid 5-alpha-reductase for prostate cancer and male pattern baldness.
I have been at ActivX for 12 years now, which is the longest I have been anywhere, and I am having so much fun!
Do you see opportunities for industry and academia to work together more closely to stimulate and accelerate drug discovery and development research? Particularly in light of recent questions related to the future of publicly funded academic research, do you envision new models in which industry would increasingly help to fund and partner with academic research institutions? Are these types of collaborations increasing, and what are the main benefits and barriers at present?
Since we started ActivX in late 2000, the venture capital models have changed. It was relatively easy to raise $20–$30 million or more upfront in those days. We did all of our early financing for ActivX before 9/11. After 9/11 and the dot.com bust, things changed fairly dramatically. We raised about $26 million for ActivX. In fact, when we were looking for a Series C financing in 2003–2004, the venture funding models had changed. We started out as a second-generation proteomics company developing activity-based proteomics for drug discovery and development. If you were an “omics” play in the early 2000s, you could raise money pretty easily. Later, having an omics-based technology wasn't enough; if you weren't in clinical trials, and preferably in phase 2 clinical trials, the venture companies didn't even want to talk to you.
When I was at Merck and headed antimicrobial drug discovery, I had developed a close relationship with a Japanese pharmaceutical company called Kyorin. Merck had a long-term relationship with Kyorin, a relatively small company with about 1800 employees and its own sales force that has been around for more than 90 years. It is a public company in Japan and its relationship with Merck was very interesting. In the early 1980s, Kyorin discovered the first fluoro-quinolone antibiotic, norfloxacin, which it licensed to Merck for global marketing.
So many things that happen are totally fortuitous. When I left Merck for ActivX, one of the heads of research at Kyorin sent me an e-mail saying how much they had enjoyed working with me and that they had heard about my move to ActivX and the proteomics technology it was developing, and expressing an interest in a collaboration. We were looking for companies to support our work, and ActivX's first corporate collaboration was with Kyorin. It turned out to be a very successful collaboration. When ActivX later ran into financial trouble, I contacted Kyorin, and they stepped in and bought ActivX in 2004. They asked me to stay and run the company.
At about the same time, Takeda bought the San Diego–based company Syrrx, and the next day Syrrx became Takeda-San Diego. Often when a company is bought, many of its leaders will leave, because the company's model changes. In contrast, when Kyorin bought ActivX, I agreed to Kyorin's request that I stay on and run the company with a couple of conditions. First, that ActivX remained a U.S. corporation, even as we were becoming a wholly owned subsidiary of Kyorin. Second, the activity-based proteomics technology ActivX was developing—that led to the KiNativ platform—was just starting to take off, and I suggested that Kyorin leverage us as a biotech company and take advantage of the access it would have to our established network with pharma and other biotech companies. They agreed, and Kyorin allowed ActivX to continue its third-party collaborations, while we also pursued R&D projects for them. At that time, that was a fairly avant-garde strategy for a Japanese company. Nowadays, it is not as unusual; for example, Takeda bought Millennium Pharmaceuticals, which remained Millennium.
We are seeing many of the big pharma companies downsizing their R&D groups now, largely because they cannot compete internally with the technology and drug development taking place in biotech and being spun out of academic institutions. The most cost-effective strategy is to let the small young companies share in the risk/benefit. At some point, their goal is to get bought by a company with money to develop the technology further. So we are seeing the pharma companies sit back and wait, then cherry-pick and buy companies at a later stage. Some pharma companies, such as Johnson & Johnson and Pfizer here in San Diego, are setting up biotech incubators within their facilities, giving them an inside track to acquire the more successful technologies down the road.
For younger scientists contemplating careers in drug discovery, how would you compare and contrast the scientific environment in a large pharmaceutical company versus a smaller biotech firm? What advice would you offer them?
Much of it depends on temperament. There are pluses and minuses to both. At a small company there is an intrinsic instability. Biotech has more of the elements and flavor of an academic institution, if that is an appealing environment for a young scientist. In big pharma, you are in a much more hierarchical organization. Whereas pharma companies used to be in more isolated locations, like Merck in Rahway, New Jersey, Pfizer in Groton, Connecticut, and Upjohn, in Kalamazoo, Michigan, now we see much more of an interface between academia, big pharma, and biotech. For the scientists it depends on where they will feel more comfortable.
From the perspective of training, learning more chemistry is always more beneficial, especially for small molecule drug discovery. Nowadays, biologics are coming more into play, so molecular biology is increasingly important. The really hard-core chemists become medicinal chemists or process chemists, and that is a unique kind of space. They come from select academic organic chemistry labs where they learn new reaction methodologies for both medicinal and process chemistry. The people who ultimately rise to leadership positions in pharma and biotech are generally those who have good chemistry backgrounds and are also strong on the biology side.
What type of training is required for an enzymologist working in drug discovery at a pharmaceutical or biotech company, and do you find that new employees have been appropriately trained in this area?
I like to hire the best possible candidates in a certain space, even if most of them may not be that familiar with drug discovery and development issues. Those are the kinds of things that become part of your training in a company. When I have a job opening I can easily get hundreds of applications and many good candidates. Overall, I am impressed with students who have good biochemistry backgrounds, and many now have good technology training as well, such as in proteomics, molecular biology, and crystallography methods. Many have experience working at the interface of scientific disciplines. The interfaces are much more well-populated than they were when I was a student.
For our readers who might not be familiar with your company, ActivX Biosciences, how would you summarize its mission and accomplishments to date? Where does the technology you provide fit into the overall drug discovery and development pipeline?
When we started the company we focused on activity-based proteomics, which is based on old biochemical concepts. First-generation proteomics companies focused mainly on the hardware, on methodologies for separating proteins and characterizing the proteome using mainly two-dimensional gels and mass spectrometry. In many ways, the proteome is much more complicated than the genome, and we focused on developing a chemical toolbox you could use to interrogate a proteome. Say, for example, that you had a chemical that could recognize all of the serine hydrolases in the body. Serine hydrolases—of which there are about 250 in the human genome—include proteases, lipases, and esterases, and they share the characteristic feature that they have a serine in their active site that forms a covalent intermediate with the substrate. They all use a similar ensemble of active site residues to activate that serine. By labeling a halo-phosphonate—which covalently binds to serine—with biotin, for example, you essentially have a probe you can use to pull out of a complex proteome a unique set of proteins that are likely to be serine hydrolases. If you have inhibitors of serine hydrolases, which tend to bind to the active site of the enzymes, you could see which ones a drug affects by determining which signals disappear when you add the drug (Fig. 1). This was the original technology from Ben Cravatt's lab at Scripps that got ActivX up and running as a start-up. That is what we started with at ActivX. It is an old, simple idea that we were applying to a whole proteome, instead of to a single isolated protein. We are able to do a backend analysis from a complex proteome. You can use gels or mass spectrometry to interrogate what proteins you pulled out and identify them en masse. That is very powerful.
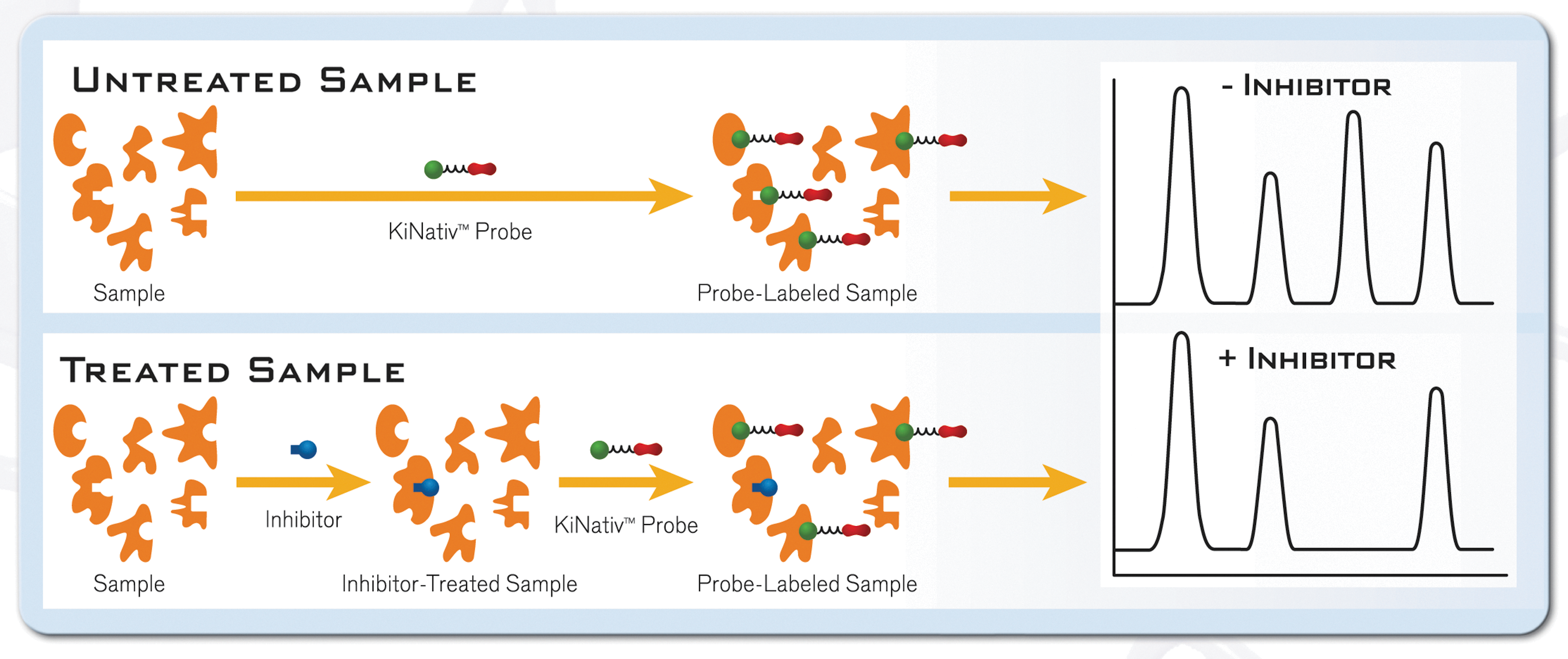
Activity-based proteomics used at ActivX.
Can you please go into some detail in describing the KiNativ platform of ActivX Biosciences, which offers an in situ approach for generating a quantitative binding profile of protein and lipid kinases in cell or tissue lysates?
We also wanted to look at other families of proteins, such as protein kinases. There are 518 predicted protein kinases in the human genome (Fig. 2). Do they have a conserved family trait you could use to define a specific chemical tool for proteome interrogation? The trait we focused on is a group of lysine residues, found very discretely and with high conservation in the active sites of protein kinases where the ATP binds. We are able to use these lysine residues to interrogate protein kinases in complex proteomes. You can put an acyl group on the terminal phosphate of an ADP or ATP to make an acyl-phosphate. When the ATP binds to the active site of the enzyme, the acyl-phosphate will be very near the lysine residues, and the acyl group will be transferred to the ɛ-amino group of lysine, resulting in a stable amide bond that we can use as a site of interrogation (Fig. 3).

The Kinome. The KiNativ™ Platform has identified more than 400 of the 500 protein kinases (shown in red).

ADP or ATP acyl-phosphate KiNativ™ probes and their reaction with active site lysine side chains.
So we use ATP- and ADP-based acyl-phosphate probes to interrogate the kinome. If you are developing an inhibitor, how do you know if it is hitting only one protein kinase, and the one you want to target, or several different ones? We can put a compound into our proteome and see which signal is affected, producing a profile in the context of a particular cell or tissue. In contrast, a typical single-well recombinant assay can screen your compound against say 300 protein kinases, using a recombinant enzyme for each protein kinase. The assay results will tell you that your inhibitor hits the kinase target you want, but, for example, it also hits five other kinases with varying potency. That is very useful information.
We can do the same sort of experiment, but not in single assays. We can tell you what your compound does in a particular cell line or tissue. We put your compound into a proteome and then use our probes to see which kinase signals disappear. Your drug will block the ATP binding site and prevent our probe from binding. If you compare the profiles obtained from single-well recombinant assays with those obtained using the KiNativ assay platform in cell lines or tissues, the results can be dramatically different.
Another important difference in our technology relates to its use in drug testing in animal models. I mentioned that there are 518 human protein kinases; about eight of which are unique to humans. There are 540 protein kinases in the mouse, and about 30–40 are unique to mouse. The last time I checked the dog kinome, it contained more than 600 protein kinases, and the chimpanzee has about 580. This is important, because before a kinase inhibitor in development as a therapeutic ever gets tested in a human being, it has to go through preclinical testing in animals, such as mouse, dog, and monkey. If they have a different number of protein kinases and you see toxicity, how do you know what the toxicity is due to? The beauty of the KiNativ platform is that you can use it in any species. We analyze the protein fragments by LC/MS-MS to get an amino acid sequence, which we then convert to a genome sequence, which tells us the protein kinase gene. As long as we have good genome information for a species, we have an assay for that species.
How do you achieve sensitivity for low abundance kinases?
What ActivX is doing today we could not have even conceived of doing when we started the company. The mass spectrometry technology has become so powerful, efficient, and sensitive that we can take 107 cells and have enough protein to achieve basically single molecule per cell detection. We are certainly down to the single numbers per cell.
When someone sends us a new cell line or tissue and wants to profile its protein kinase inhibitors, first we determine how many protein kinases are in that cell line or tissue. A rule of thumb is that of the 518 protein kinases in humans, any cell line expresses about 40% of the kinome, so about 200–225 protein kinases. We initially use data-dependent mode, telling the mass spectrometer to scan the LC readout and, when it sees peaks, to figure out what they are as quickly as possible. That may yield about 150 protein kinases in the first pass. So many proteins are in a complex sample that you may have to do about a dozen runs until you are no longer getting new peaks.
Then we are ready to switch to targeted analysis mode. Because we are doing MS-MS we will often use a daughter ion—a fragment of the parent ion, because it comes out very clean in the LC analysis and yields good signal-to-noise. We know that at certain time lines on the LC spectra, specific kinases will come out, and we target the mass spec to analyze the ions that come out during a specific window of time with a high degree of mass precision. When you do that you get very high quality and good signal-to-noise that allows you to do quantitation and generate an IC50 curve. Targeted analysis is where you go once you know what you have in the sample, and we can do that across all the kinases in a sample.
How does the KiNativ method allow for an understanding of kinase target engagement with an inhibitor versus efficacy for a cellular process? How do you define target engagement and why is it important?
Since protein kinases are signaling molecules, their signal gets amplified in a cascade. Suppose you have a drug that inhibits a protein kinase by about 50% in a cell. You might not see any biological effects from this inhibition because the signal amplification is so strong that 50% inhibition just doesn't cut it. To see a biological effect you might need to inhibit the enzyme more than 90%. That is a target engagement factor. This is a phenomenon you see with receptors too.
This is important to know, because let's say you are designing a kinase inhibitor to prevent hepatotoxicity. You might find that you need high concentrations of the drug to get the desired biological effect, not because the drug has an issue in terms of cell permeability, but because you need very high target engagement. While the drug may have an IC50 on the target of 10 nM, to get 90% inhibition you might need 500 nM. The scientists want this information so they can be convinced that the drug is getting where it needs to get at the correct level of target engagement, so they know they should be seeing the biological effect.
How is the KiNativ platform being used in drug discovery?
We are using it at almost all levels of drug discovery, beginning in early target identification, for profiling human tissue and tumor samples, for example. We also do preclinical work, looking at different animal species. Our business spans from early target and drug discovery to early clinical sample analysis.
What are the main challenges in applying the ActivX technology to new target classes, such as epigenetic enzymes? What strategies can help overcome those challenges?
Epigenetics normally refers to gene expression of proteins, and there has been a lot of work on doing very expensive gene expression profiling. Gene expression is really telling you the amount of mRNA being produced for a particular protein, but it does not necessarily reflect protein expression levels. We have done experiments comparing gene expression profiles and ActivX KiNativ profiles and have seen quite dramatic differences. Just because a gene might be overexpressed does not tell you very much about how much protein is being produced at any particular point in time. Our technology gives you a correlative readout that you can use with gene expression data to get a more accurate picture of the protein cycle.
Finally, it is clear you have had a lifelong passion for science and technology and have pursued varied avenues for fulfilling that passion. Did you ever envision that your career would follow the type of path it has and that one day you would be working in the biotech arena developing technology to drive drug discovery?
Right now I am having as much fun as I have ever had in my career. I have always been unafraid to do things that I thought were interesting. Leaving academia and giving up tenure wasn't an easy decision, but there were other things I wanted to do at the time. Could I have predicted that I would be doing what I am now? No. On the other hand, it all makes perfectly logical sense that I am where I am today! The combination of experiences has given me the ability to do a number of other things as well, such as serve on the Board of Directors of Ligand Pharmaceutical, a publicly traded company, and I have been involved on other boards as well. I have extracted good lessons from all of these experiences and appreciate the opportunities to continue applying them to what I enjoy doing.