
Abstract
The Plasmodium falciparum telomerase reverse transcriptase (PfTERT) is a ribonucleoprotein that assists the maintenance of the telomeric ends of chromosomes by reverse transcription of its own RNA subunit. It represents an attractive therapeutic target for eradication of the plasmodial parasite at the asexual liver stage. Automated modeling using MUSTER and knowledge-based techniques were used to obtain a three-dimensional model of the active site of reverse transcriptase domain of PfTERT, which is responsible for catalyzing the addition of incoming dNTPs to the growing DNA strand in presence of divalent magnesium ions. Further, the ternary complex of the active site of PfTERT bound to a DNA–RNA duplex was also modeled using Haddock server and represents the functional form of the enzyme. Initially, established nucleoside analog inhibitors of PfTERT, AZTTP, and ddGTP were docked in the modeled binding site of the PfTERT ternary complex using AutoDock v4.2. Subsequently, docking studies were carried out with 14 approved nucleoside analog inhibitors. Docking studies predicted that floxuridine, gemcitabine, stavudine, and vidarabine have high affinity for the PfTERT ternary complex. Further analysis on the basis of known side effects led us to propose repositioning of vidarabine as a suitable drug candidate for inhibition of PfTERT.
Introduction
Telomeres are repetitive G-rich regions that occur at the physical ends of chromosomes and prevent their nucleolytic degradation and physical fusion with other chromosomes. Protection and maintenance of the telomere length requires a specialized telomerase reverse transcriptase (TERT) that lengthens the telomeres. 1 Telomerase is a ribonucleoprotein that helps in maintaining the terminal repeats of telomeres in most organisms by repeated reverse transcription of a short sequence using its own RNA subunit as a template 2 for synthesis of the dGT-rich strand of telomeres. 3 Lack of telomerase activity leads to shortening of the TERT and may result in replicative senescence of the cell. 4 At present, drugs targeting human TERT or its RNA subunit are in clinical trials for treatment of cancer. 5
Malaria is a worldwide serious health problem that affects almost 207 million patients globally and causing an estimated 627,000 deaths. The poor populations of sub-Saharan Africa are at greatest risk as these patients receive the least care and account for 90% of the mortality burden. Four species of Palsmodium parasites cause malaria, namely: P. falciparum, P. vivax, P. malariae, and P. ovale. Malaria is spread by the bite of the female Anopheles mosquito (or malaria vectors) active between dusk and dawn. P. falciparum is the deadliest of the malarial parasites and is the causative organism 6 for almost 50% of the cases reported from India. 7
The intensity of the malaria transmission depends mainly on the vector, for example, vector lifespan, human biting habit, and climatic conditions affecting the vector number and survival. It is also modulated by the immunity of the human host. Long-lasting insecticidal nets, indoor spraying, access to rapid diagnostic tests, and artemisinin-based combination therapy (ACT) for malaria are the mainstays of malaria control and intervention. The factors confounding efficacious control of malaria include emergence of resistance of the mosquito to the insecticide used for control and recurring resistance of the parasite to the drugs used for treatment. The resistance of P. falciparum to chloroquine and primaquine became rampant between 1970 and 1980. As per WHO recommendations, P. vivax can still be effectively treated with chloroquine, while P. falciparum cases are treated with ACT due to development of drug resistance. However, resistance to artemisinin has recently been reported from four countries of Southeast Asia. 8
The genome of P. falciparum, the malaria-causing parasite, is 22.8 Mb long. The genome is arranged in 14 chromosomes of sizes ranging from 0.7 to 3.4 Mb. 9 Chromosome-mapping studies have revealed that the chromosomes are compartmentalized, containing conserved regions at their central domains and polymorphic regions at their terminal domains. In view of the requirement of the pathogen for maintaining chromosomal integrity, P. falciparum TERT (PfTERT) represents a potent therapeutic target for development of drugs for combating malaria. 10 The chromosomal ends of P. falciparum are consisted of 5′-GGGTT[T/C]A-3′ present as tandem repeats, and preceded by a set of noncoding DNA elements at the subtelomeric regions. 11,12 In P. falciparum, the highly variable var family of genes, present adjacent to the telomere-associated repetitive elements, is responsible for the antigenic diversity of the parasite.
It has been proposed that the heterochromatic structure at the telomeres is responsible for the silencing of the var genes. 13,14 Therefore, apart from the usual function of chromosome maintenance during the highly replicative stages of the life cycle of the parasite, PfTERT may also contribute indirectly to virulence and pathogenesis. Attempts to prepare viable knockouts of PfTERT were not successful, indicating that the enzyme was necessary for the proliferation of the parasite. 15 Immunofluorescence analysis revealed that PfTERT is expressed in the asexual liver- and blood-stage parasites that have begun DNA synthesis. 16 As the liver-stage parasites are present in smaller numbers, targeting the pathogen at this stage of the life cycle is likely to lead to effective eradication. Berberine, an alkaloid present in plant extracts, is shown to inhibit PfTERT in a dose-dependent manner. 16 Two nucleoside analog drugs targeted against HIV-1 reverse transcriptase (RT) have previously shown inhibition of PfTERT. 12 Antimalarial activity of HIV-1 protease inhibitor drugs lopinavir and saquinavir, as non-nucleoside reverse transcriptase inhibitors (non-NRTIs) drug nevirapine has also been determined against liver-stage P. falciparum and is in the micromolar range. Protease inhibitor drugs of aspartyl protease of HIV-1 are expected to inhibit P. falciparum liver-stage aspartyl proteases, while non-NRTI drugs are proposed to possess antiplasmodial activity due to increase in oxidative stress on the parasite. 17
PfTERT is an unusually large (∼2500 amino acid) basic protein, approximately three times the size of TERT from other organisms. It shares specific motifs with telomerases of other organisms. This plasmodial protein contains all of the canonical motifs of RTs as well as conserved amino acids known to be critical for RT activity, but contains insertions of basic amino acids between these conserved motifs. 15 In view of the potential of PfTERT to act as a therapeutic target for malaria, there is keen interest in its three-dimensional structure. In the present work, we report the modeling of the catalytic site of RT domain of PfTERT. Additionally, we modeled the ternary complex of the active site of the protein bound to a RNA–DNA hybrid strand by similarity to the ternary complex of HIV-1 RT. 18 This consists of the catalytic domain of PfTERT in complex with two bases of its own RNA and the complementary telomeric DNA to be elongated and represents the functional form of the enzyme.
It has been shown that nucleoside analogs ddGTP and AZT can inhibit the telomerase activity of PfTERT. 12 In view of the similarity of the PfTERT catalytic site to HIV-1 RT, it was hypothesized that the analog NRTIs of HIV-1 RT are likely to inhibit telomerase activity of PfTERT by a similar mechanism. Docking of the ternary complex with AZTTP, ddGTP, and 14 commercially available nucleoside analog drugs was carried out in order to explore their possible repositioning for treatment of malaria. Docking studies showed that floxuridine, gemcitabine, stavudine, and vidarabine are likely to have the highest efficacy for inhibition of PfTERT. Of these, vidarabine has fewer toxic side effects and is already in use as an antiviral agent. Our study has implications for the potential reuse of vidarabine and three other nucleoside analogs as antimalarial agents.
Methods
Molecular Modeling
The amino acid sequence of PfTERT was retrieved from GenBank sequence database of NCBI (GI: 23615295). The PDB was searched for sequence similarity with the query protein sequence using DELTA-BLAST algorithm for sensitive similarity detection. 19 Triboleum castaneum TERT (PDB ID: 3DU6) was selected as the template due to high query coverage of the PfTERT RT domain (78%) combined with low e-value of 8×10−7. This is also a functional telomerase containing the RT domain. Functional annotation of conserved domains in the protein and secondary structure prediction was carried out using CD Search and PSIPRED, respectively. 19 –23 The model of the catalytic site of PfTERT model was generated for residues 1881–2040 with 3DU6 structure as template using MUSTER, a MUlti-Source ThreadER program. MUSTER incorporates six additional sequence and structure parameters for improvement of the dynamic programming algorithm and sequence alignment against various structural templates. 24 The residues K1425 and R1433 of PfTERT were modeled by similarity to 3DU6 residues K189 and R194.
Structure Validation
WHATIF, Procheck, and QMEAN server were used to analyze geometrical and stereochemical quality of the modeled PfTERT. 25 Structural comparisons of the modeled structure with the template and HIV-1 RT were made using SwissPDB Viewer 26 and Dali server. 27
Placement of Magnesium Ions in the Modeled Active Site of PfTERT
P. falciparum telomerase helps in maintaining the length of chromosome and de novo telomere formation on broken chromosomes. This chemical reaction requires the presence of divalent metal ions in the active site. 28 By similarity with the RT domain of HIV-1 (PDB ID: 1RTD), the catalytic triad required for forming a coordinate covalent complex with the incoming dNTP and directing it to the nucleotide-binding site of the enzyme was inferred. 29 Two magnesium ions stabilize the interaction of the catalytic aspartates with the incoming dNTP. In view of the importance of Mg2+, two divalent magnesium ions were initially placed in the modeled active site of PfTERT by similarity with 1RTD. Average distances of the Mg2+ ions from the coordinately bound aspartate residues in the catalytic site were derived from the structure of 1RTD. Subsequent constrained minimization of the Mg2+-incorporated structure was carried out in Sybyl6.9 using the Tripos forcefield 30 till the distance constraints were satisfied.
Modeling of the Ternary DNA–RNA Complex with Catalytic Domain of PfTERT
The active form of the PfTERT catalytic site is represented by the complex of the enzyme with its own subunit RNA and the complementary growing DNA strand being synthesized. Once divalent metal ions were fixed in the active site of PfTERT, the RNA–DNA complex was placed in the TERT active site. The structure of the DNA–RNA dinucleotide base pair was obtained from the crystal structure of the HIV-1 RT complex (PDB ID: 1T03). The ternary complex of the DNA–RNA hybrid with the active site of PfTERT was modeled using HADDock server for docking of biomolecular complexes. 31,32 After docking, HADDock also performs rigid-body energy minimization followed by a semiflexible refinement in torsion angle space and a final refinement in explicit solvent. 31 The conformation with the best docked score and similarity with the superimposed crystal structure of the RT-bound ternary complex was chosen using SwissPDB viewer and Pymol. 33,34 The N-terminal loop residues 1–35 were remodeled using Modeller loop remodeling protocol of Chimera 35 interface as they were found to sterically hinder the PfTERT interaction with the DNA–RNA complex. This ternary complex of the PfTERT active site was then used for further docking studies with nucleoside analogs.
Standardization of Docking Studies
In order to standardize the docking protocol, AZTTP and ddGTP (known inhibitors of PfTERT) were first docked with the modeled active site using AutoDock v4.2. The AutoDock GUI was utilized to generate grids, calculate dock score, and evaluate conformers. Polar hydrogens and Gasteiger charges were added to both receptor and ligand with eight allowed active torsions. Mg2+ ions were incorporated into the receptor pdbqt files with a charge of 2+. Chemical affinity and electrostatics maps were computed and centered near the metal ion (MGI) with 56×56×56 grid points covering the putative binding cavity of the receptor with a spacing of 0.347 Å (Supplementary Table S1; Supplementary Data are available online at
Inhibitor Docking and Analysis
After standardization of docking protocol by docking of AZTTP and ddGTP, 14 approved nucleoside analogs were docked with the ternary complex of PfTERT active site using the same parameters. The 3D conformation of seven FDA-approved nucleoside analogs was utilized: zidovudine, stavudine, tenofovir, and abacavir were taken from Protein data bank (3V4I, 1RTD, 1T03), while lamuvidine, zalcitabine, and didanosine coordinates were retrieved from Pubchem Compounds (CID: 454110, 119119, and 129711). In order to search the entire chemical space of NRTIs, we systematically searched all the approved drugs in Drug Bank v3.0 36 with Tanimoto similarity to AZTTP with threshold 0.6 and found nine more nucleoside analog drugs to be assessed for antimalarial activity that are listed in Supplementary Table S2 and were also docked with the modeled ternary complex of active site of RT domain of PfTERT in order to predict their inhibitory potential. In all cases, the active phosphorylated form of the drug was used for docking studies.
Results
Sequence and Structural Features of PfTERT
The PfTERT sequence contains a conserved telomerase/RNA binding domain in the N-terminal half and an RT-like domain in the C-terminal half of the protein. The telomerase domain supports efficient binding to the RNA template, while the RT domain carries out the synthesis of DNA complementary to the RNA template. 15 Accordingly, a conserved telomerase domain and an RT-like domain was detected by CD search in the N-terminal region of the protein. An RT-like domain was present in the C-terminal region (residues 1881–2040). The RT domain of PfTERT has structural homology to the viral RTs and contains the finger, palm, and thumb subdomains. 37 It shares seven conserved motifs with the HIV-1 RT in the finger, palm, and thumb subdomains that lead to specific interactions with the RNA, DNA, and the incoming NTPs. 38 Mutational analysis has revealed that the residues of these motifs in TERT perform nearly identical functions in RT. The catalytic aspartates are D1885, D1982, and D1983. 29 Specifically, D1885 of PfTERT corresponds to D115 of HIV-1 RT, while the FIDD loop (residues 1980–1983) corresponds to the YMDD loop (residues 183–186) in HIV-RT. Together, these conserved residues are responsible for nucleotide addition in both RT and TERT. 39 –42
The PfTERT RT domain sequence (residues 1881–2040) showed 45% similarity with the TERT from T. castaneum (PDB ID: 3DU6). Figure 1 shows the alignment of RT domain of PfTERT with T. castaneum TERT. Using this alignment, a model could be generated for the PfTERT RT domain using MUSTER. The modeled PfTERT could be superimposed with T. castaneum TERT with root mean square deviation (rmsd) of 1.50 Å between 300 Cα atoms (Fig. 2A). The modeled structure spans residues of the finger, palm, and thumb subdomains and contains the conserved motifs A, IFD, B′, C, and D responsible for catalytic activity. The PfTERT model has good structural alignment with T. castaneum TERT and HIV-1 RT along with the presence of all the conserved motifs and residues that are important for the structure and function (Fig. 1A, B). The catalytic residues D1885, D1982, and D1983 in the model of PfTERT were found to be in structurally equivalent positions with the corresponding catalytic residues in T. castaneum TERT and HIV-1 RT.

Target-template alignment of PfTERT and 3 DU6 sequence used for homology modeling. Dashed arrows of different styles have been used to show the finger domain (), insertion in the finger domain (IFD  ), the palm (
), the palm ( ), and the thumb part (
), and the thumb part (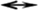 ) of the sequences. Start identify putative catalytic aspartates residues, while triangles represent other important residues that form the polymerase binding site. Secondary structural elements were derived from crystal structure of 3DU6 and are shown as thick arrows for β-sheets and helices for α-helices. PfTERT, Plasmodium falciparum telomerase reverse transcriptase.
) of the sequences. Start identify putative catalytic aspartates residues, while triangles represent other important residues that form the polymerase binding site. Secondary structural elements were derived from crystal structure of 3DU6 and are shown as thick arrows for β-sheets and helices for α-helices. PfTERT, Plasmodium falciparum telomerase reverse transcriptase.
As inferred by analogy with the structure of TERT from T. castaneum, the nucleotide binding pocket of PfTERT lying at the interface of the fingers and palm subdomains contains conserved residues that are responsible for template and nucleotide binding. A comparison of the conserved motifs of PfTERT present in the nucleotide binding domain, that is, motifs 1, 2, A, C, B′, and D with the corresponding motifs in HIV-1 RT, is shown in Table 1. Of these, only motifs 1 and 2 of the finger domain were not present in the homology model obtained. Due to low sequence similarity in the finger domain, modeling of the entire finger domain segment of PfTERT could not be carried out. However, residues corresponding to K1425 and R1433 in motifs 1 and 2 of PfTERT are conserved in HIV-1 RT, and T. castaneum TERT. Structural as well as mutational studies on finger domain of HIV-1 RT have confirmed the importance of the corresponding residues, namely, K65 and R72, for binding to the incoming dNTP/nucleoside analog (PDB ID: 3V4I) 43 and are also structurally conserved with T. castaneum TERT residues K189 and R194. Therefore, in order to achieve completeness of the PfTERT active site, these conserved residues were placed in the active site of PfTERT by similarity to the crystal structure of T. castaneum TERT. 37
Comparison of the Domains, Subdomains, Motifs, and Residues of PfTERT with HIV-1 RT
NRTI, nucleoside reverse transcriptase inhibitor; PfTERT, Plasmodium falciparum telomerase reverse transcriptase; RT, reverse transcriptase; TERT, telomerase reverse transcriptase.
Structure validation report of the modeled active site of PfTERT showed no anomalous bond lengths/angles, and revealed no bad contacts. The structure possesed a good QMEAN Z-score. The modeled palm subdomain of PfTERT also showed good structural alignment with the HIV-1 RT (PDB ID: 1RTD). While the palm subdomains of both proteins aligned with rmsd of 1.77 Å for 204 Cα atoms, considerable deviations are observed in loop regions (Fig. 2B, inset).
Active Site of PfTERT
The catalytic aspartate residues of HIV-1 RT impart a predominantly negative charge to the active site. Thus, the presence of metal ions is essentially required to mediate the binding of the negatively charged phosphate groups of the incoming dNTP. 29 Thus, binding/catalysis in HIV-1 RT involves the presence of and coordination with positively charged Mg2+ ions. In HIV-1 RT, the carboxylate groups of D110 and D185 are at coordinating distances of both metal ions. 28 While the native uncomplexed structure of 3DU6 does not show the presence of bound Mg2+ ions, the biological assay procedure for PfTERT lists the use of Mg2+ ions for activity testing. 37 Further, the active-site information in PDBSum for the available HIV-RT structures bound to nucleoside analogs was collated to reveal that the nucleotide binding site is composed of residues K65, R72, D110, V111, G112, D113, A114, Y115, F116, Q151, M184, D185, D186, and K219. In T. castaneum TERT, the corresponding structurally equivalent residues are K189, R194, D251, I252, R253, D254, A255, Y256, G257, Q308, V342, D343, D344, and K372. 37
By alignment and structural analogy to T. castaneum TERT, the equivalent residues of PfTERT were inferred to be K1425, R1433, D1885, I1886, K1888, Y1889, H1890, K1891, G1920, Q1925, I1981, D1982, D1983, and K2015, respectively. These residues were found to be in structurally equivalent positions. Of these, conserved PfTERT residues D1885 from the finger domain and residue D1982 from the palm subdomain of PfTERT are the probable catalytic site residues that are required for DNA polymerization by analogy with HIV-1 RT.
D110 in the finger domain as well as D185 in the palm subdomain of HIV-1 RT coordinate one Mg2+ ion with the triphosphate moiety of the incoming dNTP to form a pentacovalent transition state, leading to its incorporation in the growing DNA strand. The second Mg2+ ion is also coordinated by the carboxylate groups of D110 and D185, and is required for the proper placement of the substrate in the nucleotide binding site. 28 The flexibility of the YMDD loop (residues 183–186) is inherently required for the formation of the pentacovalent transition state with both the Mg2+ ions in the active site. Subsequently, hydrolysis takes place and the pyrophosphate tail of the dNTP along with one of the bound Mg2+ dissociates from the active site. The other Mg2+ ion remains bound in the active site ∼3.6 Å away from the first and presumably provides a positively charged atmosphere favoring the binding of another dNTP. 29,35,44 In the final metal-bound model of the active site of PfTERT, both Mg2+ ions were coordinated by D1885 and D1982. The first Mg2+ ion (MGI) is placed for coordination of D1885 and D1982 with the triphosphate moiety of the incoming dNTP substrate. The second metal ion (MGII) is also coordinated by D1885, by D1982, and by the carboxylate group of D1983 to complete the pentacovalent coordination. The final coordinating distances of the Mg2+ ions obtained with catalytic aspartates and their relative positions in the active site of PfTERT are shown in Figure 2B. The coordinating distances of both Mg2+ ions were compared with HIV-1 RT and are shown in Supplementary Table S3.
Structure of Ternary Complex of PfTERT
The main function of the RT domain of PfTERT is to add dNTPs to the growing strand of DNA by making necessary phosphodiester bonds between the new incoming dNTP and the 3′-hydroxyl group of the growing strand of DNA. The incoming dNTP substrate is positioned such that it forms a base pair with the template RNA and stacks with the existing DNA base at the 3′ end. The active form of the catalytic domain is therefore represented by an RNA–DNA duplex. In order to correctly model the relevant active-site conformation of PfTERT in complex with the RNA–DNA duplex, the nucleotide bases in contact with the nucleotide binding site were taken from the crystal structure of HIV-1 RT crosslinked to tenofovir-terminated template–primer complex (PDB ID: 1T03). Docking of this two-base DNA–RNA duplex with the metal-bound active site of PfTERT was carried out to obtain the ternary complex.
The total energy of −4506 kJ/mol indicated the stability of the ternary complex. Structural superposition of the modeled PfTERT ternary complex with crystal structure of HIV-1 RT ternary complex showed rmsd of 1.78 Å for 154 atoms and is shown in Supplementary Figure S1. The ternary complex thus obtained provides the active site of PfTERT, the coordinating metal ions, the RNA template, and the stacking DNA base, thus defining a complete active site as required for the binding of a nucleotide substrate or analog. However, in our model, the IFD residues were not found to be in contact with the primer or the template. It is possible that a structural change may bring about its contact with the DNA–RNA duplex and further modulation of telomerase activity by IFD residues.
Binding Mode and Affinity of ddGTP and AZT with PfTERT Ternary Complex
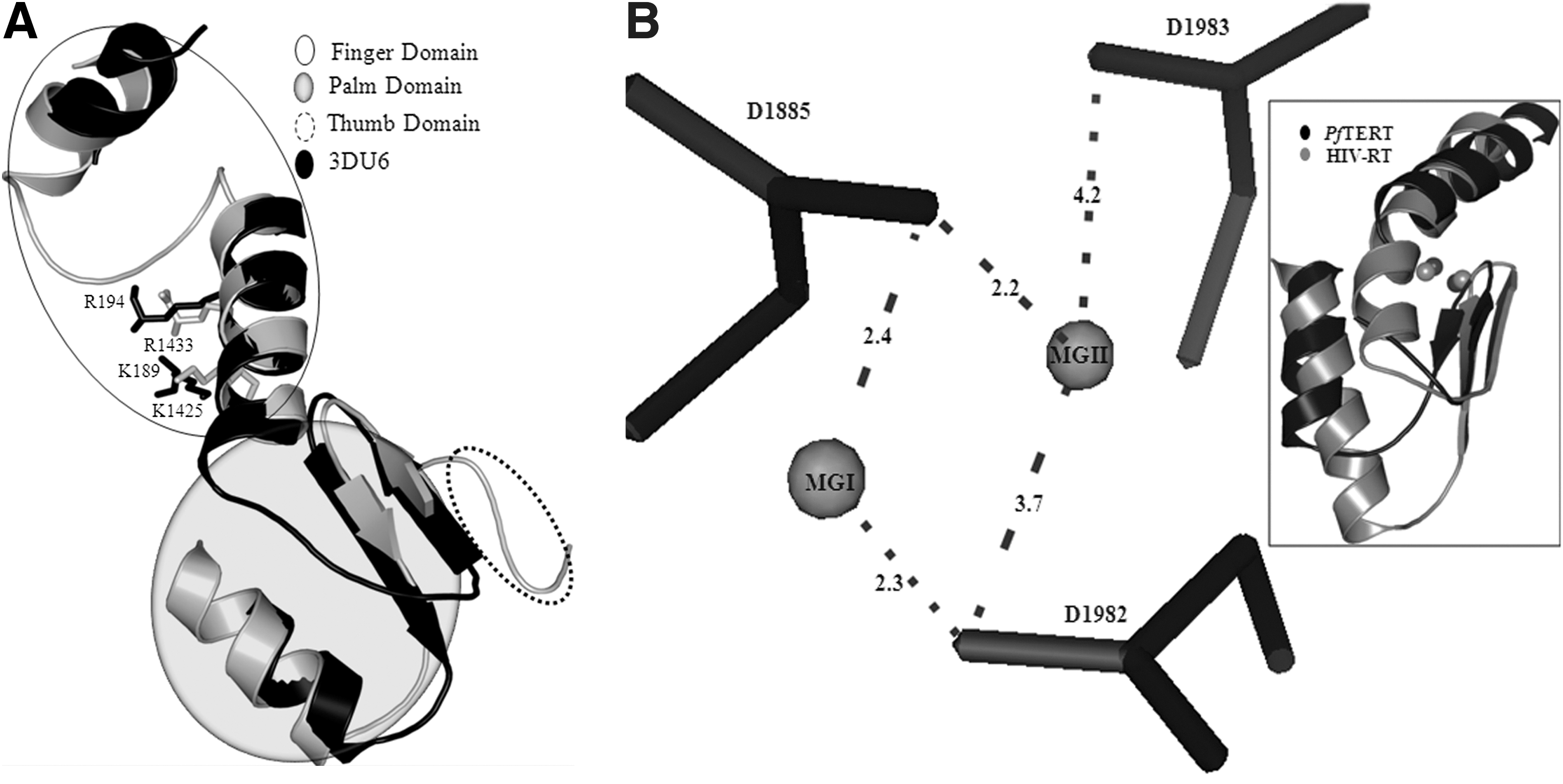
As ddGTP and AZTTP have been demonstrated to inhibit RT activity of PfTERT, 12 we first docked the two inhibitors with the ternary complex obtained according to the docking methodology described. In view of the sequence and structural similarity of the PfTERT RT domain with HIV-1 RT, it is expected that the mode of binding of nucleoside analog inhibitors will be the same between the two proteins. Therefore, as no inhibitor-bound structure is available for TERT, the docked pose of ddGTP and AZTTP was validated by superposition with HIV-1 RT bound to ddGTP (PDB ID: 3JYT) and AZTTP (PDB ID: 3V4I). The rmsd between the docked positions of ddGTP and AZTTP as compared with the crystal structure was 1.78 and 1.66 Å, respectively. The docked positions of ddGTP and AZTTP are shown in Figure 3A and B, respectively.
Docked positions of
Hydrogen-bond distances of the docked nucleoside analogs with the active-site residues were comparable to those observed experimentally. The interaction of both inhibitors with the critical PfTERT residues takes place at the ribose moiety and the pyrophosphate tail, whereas the nucleoside base is stabilized by stacking interactions with the neighboring base, hydrogen bond interactions with the complementary base, and hydrophobic interactions with selected side chains of PfTERT. Significantly, the pyrophosphate moiety is placed in a suitable position to form coordinate covalent bonds with the Mg2+ ion (MGI). Placement of MGI near the catalytic aspartates points to the formation of a pentacovalent complex as seen for AZTTP with HIV-1RT. The phosphate group also makes hydrogen bonds with K1425 and R1433, similar to that of AZTTP with residues K65 and R72 in HIV-1 RT as shown in Supplementary Table S5. The superposition of docked position with AZTTP bound to HIV-1 RT is shown in Supplementary Figure S2. Additionally, the AutoDock-estimated Ki values are similar to the experimentally determined Ki values obtained from the literature. 12 Thus, the same docking protocol was repeated for 14 commercially available approved nucleoside analogs.
Virtual Screening of Approved Nucleoside Analog Drugs
The efficacy of NRTI binding and inhibition of PfTERT was predicted by carrying out the in silico docking studies with the commercially available nucleoside analogs. The docking results for all the 16 Nucleoside analogs used in this study are summarized in Supplementary Table S2. Docking results reveal that floxuridine, gemcitabine, stavudine, and vidarabine satisfy all the important interactions needed for the inhibitor to form stable complex in the nucleotide binding site. The AutoDock lowest binding free energy were found to be −10.2, −8.8, −9.98, and −8.0 kJ/mol, respectively. These four nucleoside analogs were predicted to show low Ki for PfTERT with floxuridine being the most effective. The final docked position of vidarabine, gemcitabine, stavudine, and floxuridine in the modeled ternary complex of PfTERT is shown in Supplementary Figure S3A, B, C, and D, respectively.
In floxuridine, MGI helps bind all the three catalytic aspartates, D1885, D1982, and D1983, with the triphosphate tail. The three aspartate residues and MGI define a platform for binding the triphosphate in the polymerase active site. The high binding affinity of floxuridine to PfTERT could be attributed to the presence of negatively charged fluorine substituent in the ribose group of floxuridine near the second Mg2+ ion (MGII) and positively charged residues of the finger domain, thus providing additional interactions for its stabilization as shown in Supplementary Tables S4 and S5. The final docked positions of the nine other commercially available nucleoside analogs are shown in Supplementary Figure S4A–I. A comparison of the interactions and hydrogen bonds obtained between the different docked nucleoside analogs and the active site of PfTERT is shown in Supplementary Tables S4 and S5.
Discussion
PfTERT is a potential drug target for development of therapeutic compounds for treatment of malaria. However, it is an atypically large sequence of undetermined structure and limited sequence similarity to other TERTs with experimentally determined structure, due to which modeling efforts have not been successful. In this work, we modeled the active site of the RT domain of PfTERT based on sequence comparison, motif identification, and TERT structure information. Information about the active-site residues was used in conjunction with knowledge derived from site-directed mutagenesis to develop a complete model of the nucleotide binding site of PfTERT. A ternary complex of the active site was modeled by comparison with the experimentally determined structures of HIV-1 RT in complex with a DNA–RNA hybrid and was used to probe the affinity of binding with various known nucleoside analog inhibitors. These nucleoside analogs are incorporated into the viral DNA, and act as DNA chain terminators due to the lack of a 3′-OH group. Thus, they act as PfTERT inhibitors with a binding mode analogous to HIV-1 RT-bound nucleoside analogs.
After standardization of docking with known PfTERT NRTI inhibitors, 14 commercially available NRTI drugs were docked into the modeled PfTERT binding site in order to explore the possibility of repositioning them for antimalarial therapy. It was found that floxuridine had the highest predicted affinity for binding with PfTERT, while gemcitabine, stavudine, and vidarabine also showed submicromolar affinity. Due to their interference with DNA synthesis in rapidly proliferating cancerous cells, floxuridine and gemcitabine are FDA-approved anticancer drugs against ovarian, breast, and pancreatic cancer, whereas the HIV-1 RT inhibitor stavudine is used as an antiretroviral agent in pediatric HIV infection. While these three drugs are FDA approved, however, they show high mitochondrial toxicity and LD50 values. 45,46
Nucleoside analogs are shown to be effective inhibitors of telomerase from human, Tetrahymena, as well as P. falciparum. 12,47,48 While some nucleoside analogs have demonstrated antitelomerase activity, in vitro testing of these drugs as antimalarials has been reported to be efficacious at micromolar range in killing P. falciparum after 3–5 blood cycles. 15 Antiretroviral therapy with nucleoside analog NRTIs has been reported to cause an improvement in the clinical presentation of malaria in HIV-1 patients. However, this has been attributed to the improvement of immune function. 49 Thus, the cause for the effectiveness of antiretroviral treatment against P. falciparum has remained obscure. 50 In view of the action of nucleoside analogs as telomerase inhibitors, this may be one of the causative factors underlying the observed effect of nucleoside analogs as antimalarial drugs. In vitro drug susceptibility assay against two strains of P. falciparum has previously shown low efficacy (IC50 >50 μM) of five NRTIs. 51 However, the effect of vidarabine, gemcitabine, or floxuridine, that is, the compounds predicted to have highest affinity as per this study, has not been reported in the literature and is required to validate the accuracy of the in silico predictions. Also, the reported antiplasmodial activity of non-NRTIs may be caused due to inhibition of telomerase by these compounds. Elucidation of the structure of PfTERT will aid in exploring the structure–activity relationships of both the nucleoside analog and non-NRTI drugs at their respective binding sites in the RT domain of PfTERT.
Despite their extensive use for treatment, NRTIs exert a large spectrum of long-term side effects like bone marrow suppression, lactic acidosis, nerve disorders, and pancreatitis. 52 In vitro studies show that stavudine, a pyrimidine analog RT inhibitor, causes the lowering of intracellular pyrimidine pools, thus leading to mitochondrial DNA depletion. 53 Stavudine-induced mitochondrial toxicity can be reversed by supplementation of uridine analogs. 54 Similarly, the toxic effects of nucleoside analogs may be attributed to their affinity with the host mitochondrial DNA polymerase. The order of the potency of nucleoside analog NRTIs for inducing mitochondrial dysfunction has been reported to be ddC=ddI=FIAU=D4T ≫ 3TC>tenofovir>AZT>carbovir. It has also been observed that pyrimidines are more toxic to mitochondrial DNA than purines. 55 –57 In view of this, floxuridine (Drugbank ID: DB00322) and gemcitabine (Drugbank ID:DB00441) may be rejected due to a larger number of expected side effects.
On the basis of our study and analysis, vidarabine is proposed as a suitable candidate antimalarial drug. Vidarabine is an adenosine analog and does not show significant mitochondrial toxicity. Accordingly, the Drugbank LD50 value for floxuridine is 215 mg/kg, while that for vidarabine (Drugbank ID: DB00194) is 5020 mg/kg. Also, vidarabine is an approved drug currently already in use as an antiviral agent for treatment of herpes simplex encephalitis virus. 58 Therefore, it is a promising antiplasmodial drug candidate. In support of this, in vivo exposure to as well as in vitro incubation with vidarabine has been reported to markedly inhibit protein synthesis and cause distinct changes in gene expression in P. Burghei. 59 Further testing of vidarabine and the other inhibitors as identified in our study will help to determine their potential for repositioning and will validate our work.
Conclusions
Drug repositioning can help overcome the high rate of attrition in drug discovery. In this study, we report the knowledge-based modeling and docking of approved nucleoside analog drugs at the active site of RT domain of PfTERT in order to explore the possible use of nucleoside analogs as antimalarial agents. Our study shows that floxuridine, gemcitabine, stavudine, and vidarabine are promising high-affinity compounds that may act as antimalarials by binding to PfTERT. Based on its low mitochondrial toxicity and few side effects, vidarabine (an approved antiviral drug) can be selected as the candidate drug of choice. In view of this, extensive testing of these inhibitors against P. falciparum may allow their repositioning as antimalarial agents.
Footnotes
Acknowledgment
The authors would like to acknowledge Mayank Chhavri for technical assistance.
Disclosure Statement
The authors confirm that this article content has no conflicts of interest.