
Abstract
In the present study, mupirocin (MP), an antimicrobial agent, was formulated as a nanostructured lipid carrier (NLC) by using a novel method named as melt emulsion ultrafiltration method. For the formulation of NLC, glyceryl monostearate and watermelon seed oil were used as solid and liquid lipids, respectively. The method was optimized for various parameters by Taguchi design of experiment and prepared NLCs were characterized for particle size, polydispersity index (PDI), shape, zeta potential, % drug loading, and in vitro release profile. The optimized NLCs were found to be smooth, monodisperse with PDI 0.229 ± 0.093. NLCs were found to have an average particle size of 139 ± 0.75 nm and +21.9 ± 0.98 mV as zeta potential. The % drug loading of optimized NLCs was found to be 59% ± 0.13%. The optimized NLCs were able to release the drug up to 24 h. The release kinetic study revealed mixed-order kinetics. Hence, it was concluded that the novel method is simple and able to fabricate MP-loaded NLCs with sustained release property and being stable in terms of particle size, PDI, and % drug loading.
Introduction
The antimicrobial agents (AMA) are a broad chemical class of therapeutic agents obtained from natural sources (molds, bacteria, etc.), considered a milestone among the greatest discoveries of science and technology. Antibiotics are the molecules made by living cells to battle against the infections and have been adapted by man to combat unicellular predators. 1
It is the need of the hour to chemically modify these natural molecules to broaden the activity spectrum, reduce toxicity, with enhanced ability to check the growth of invader microbes.
The use of AMA is quite challenging due to antimicrobial resistance, resulting in treatment failure, and hence, the area of research in AMA formulation is declining. Systemic infections and their resistance are major contributing factors to this decline. High cost and time taken for new drug discovery also make the area of AMA research more uninteresting. 2
One of the alternatives is the reformulation of AMA to attain flexibility in application. In 1971, a researcher had isolated a novel antibacterial agent mupirocin (MP) (9-[(E)-4-[(2S,3R,4R,5S)-3,4-dihydroxy-5-[[(2S,3S)-3-[(2S,3S)-3-hydroxybutan-2-yl]oxiran-2-yl]methyl]oxan-2-yl]-3 methylbut-2-enoyl] oxynonanoic acid) from Pseudomonas fluorescens with a peculiar chemical structure (molecular formula∼C26H44O9) (Fig. 1) and mode of action. MP is highly effective against aerobic gram-positive bacteria, such as Staphylococcus aureus, Staphylococcus epidermidis, and Streptococcus pyogenes, and other gram-negative bacteria such as Haemophilus influenzae, Neisseria gonorrhea, Branhamella catarrhalis, and Pasteurella multocida. 3
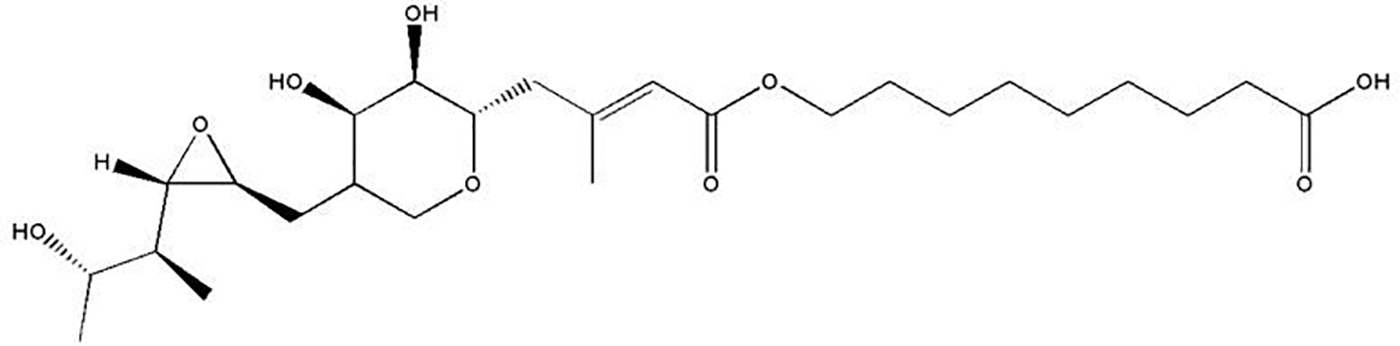
Molecular structure of mupirocin.
It acts by inhibiting the synthesis of bacterial RNA and protein synthesis without causing substantial toxicity to the human cell. 4
MP shows very less interference with mammalian isoleucyl-transfer-RNA synthetase.
3
The use of MP is limited to conventional topical marketed products such as cream and ointment (2% MP) for the treatment of skin ailments. The oral or intravenous (i.v.) route poses a challenge for formulators because of its rapid metabolism to inactive form monic acid with no antibacterial activity after reaching the systemic circulation. Moreover, i.v. route is invasive and needs assistance for administering the dose. The serum protein binding of MP was found to be high (96.5%) leading to a 10–12
The noninvasive transdermal route may be an alternative for delivering MP for improved therapeutic efficacy. The route also provides ease of self-administration and removal of therapy at any time course of treatment.
Hence, to deliver the drug via skin and also to protect it from getting metabolized, there exists a need for a drug carrier. Drug delivery carriers play a key role in improving the therapeutic efficacy of active moiety. The carrier must be able to protect the active from various degrading factors (e.g., water, various enzymes, and plasma proteins) during its course of movement from the site of application to the site of action. 6
In the present study, the approach to entrap an active drug into a particulate carrier system was studied to overcome this problem. Incorporation into a carrier system may achieve drug targeting and will protect from degradation in vitro as well as in vivo conditions.
Polymeric nanoparticles can deal with drug degradation and targeting, but regulatory issues, the feasibility of scale-up, fewer production yields, and toxicity are some of the major obstacles. A biocompatible nanocarrier such as liposome was also not chosen because of drug degradation and less stability. Solid lipid nanoparticles (SLNs) had emerged as an alternative delivery system with a lack of the abovementioned problems of liposomes, but drug expulsion during the storage of SLNs led to poor drug loading. 2,6 Consequently, the development of nanostructured lipid carriers (NLCs) seems to be an attractive approach system to control the problem of drug expulsion. NLCs are composed of physiological and biodegradable solid lipids in which the core is dispersed with liquid lipids to minimize the possibility of polymorphic changes and recrystallization. 7 The small size ensures close contact with the stratum corneum and can increase the amount of drug penetrated in the skin. Due to the occlusive properties of lipid nanoparticles, enhanced skin hydration is also observed. Furthermore, lipid nanoparticles can enhance the chemical stability of compounds sensitive to light, oxidation, and hydrolysis. 6 Skin hydration after applying NLC leads to the reduction of coenocyte packing and an increase in the size of the coenocyte gaps. This will facilitate the percutaneous absorption and drug penetration to the deeper skin layers. 8,9 Therefore, in the current study, NLCs were selected as a choice of carrier for the transdermal delivery of MP.
Materials and Methods
Reagents and Chemicals
MP I.P (>99%) was obtained as a gift sample from Kawman Pharma (Cuddalore, Tamilnadu, India). Glyceryl monostearate, stearic acid, Pluronic F-68, sodium chloride, and potassium bromide were purchased from Hi Media Laboratories Pvt. Ltd. Mumbai India. Methanol, ethanol chloroform, acetone, and isopropyl alcohol were purchased from Qualigens Fine Chemicals—A division of Glaxo India Limited (Mumbai).
Preliminary Study of Drug
The preliminary studies such as infrared (IR) spectroscopy and melting point analysis were performed in the laboratory.
IR spectrum
The IR spectrum of MP was obtained using Fourier transform infrared spectroscopy (FTIR) (Jasco FTIR-4100, Japan). One milligram of the drug was mixed with 100 mg of potassium bromide in a mortar by trituration and the mixture was compressed into a pellet at 10 ton/cm2 in a pellet maker. The sample was scanned at 4,000–400 cm−1(Fig. 2).
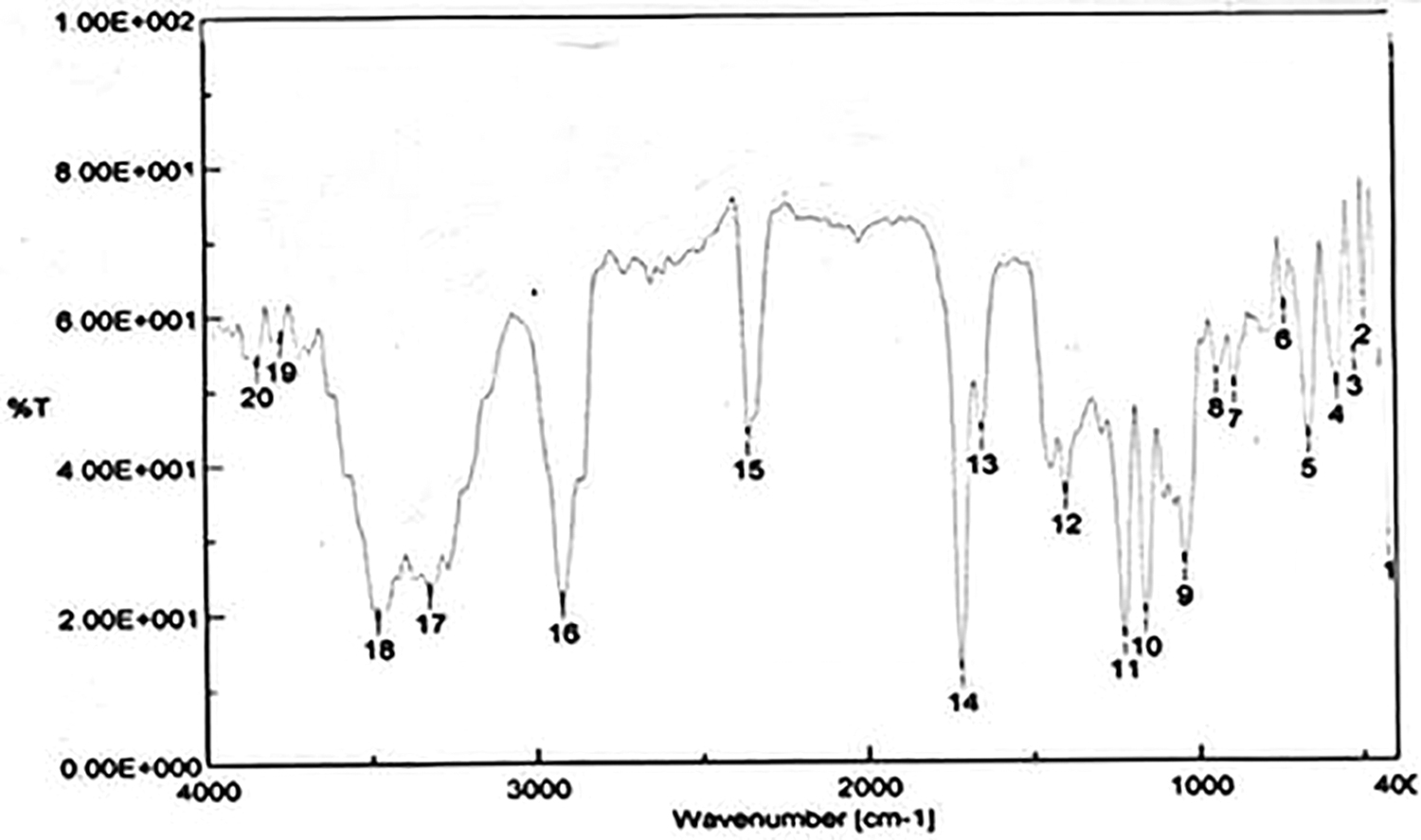
FTIR spectrum of mupirocin. FTIR, Fourier transform infrared spectroscopy.
Melting point
The melting point of MP was determined by using the capillary melt method. In this method, the glass capillary was sealed at one end and a very small amount of drug sample was filled through the open end by gentle tapping on a soft surfaced table top. After filling, the capillary was placed in the melting point apparatus (PERFIT, India), and the temperature at which melting occurred was noted as melting point. 10
Screening of Solid Lipid
Solid lipid was selected based on drug solubility and miscibility in melted lipid as the solubility of the drug in the lipid plays an important role in drug loading. The lipid for the current study was selected on the basis of solubility of MP in three lipids, glyceryl monostearate (GMS), Gelucire 50/13, and Compritol 888 ATO (Table 1).
Solubility of Mupirocin in Solid Lipid
Data represented as mean ± SD, n = 3.
GMS, glyceryl monostearate.
The solubility of MP in different lipids was determined by the method reported earlier. Ten milligrams of MP was taken in a wide mouth screw-capped bottle. The solid lipid was separately heated above its melting point and was gradually added in portions to MP with continuous stirring using a vortex mixer. The amount of molten lipid required to solubilize the drug was calculated in triplicate (n = 3). The endpoint of the solubility study was the formation of a clear, pale yellow solution of molten lipid. 11
Screening of Liquid Lipid/Oil
The solubilizing power of liquid lipid for the drug and miscibility of solid lipid with liquid lipid were the criteria for selecting liquid lipid. Watermelon seed oil (WSO) and almond oil (AO) were selected for the present study.
Solubility of the drug in liquid lipid
MP (from 1 to 200 mg) was added in incremental parts and dissolved in 5 mL of each liquid lipid under constant stirring, thermostatic at 70°C to simulate conditions for NLC production, and was allowed to equilibrate for 24 h. The maximum amount of drug dissolved was noted in triplicate (Table 2). MP solubility in each system was evaluated by visual observation of the disappearance of drug crystals and the formation of a transparent homogeneous system. 12
Solubility of Mupirocin in Liquid Lipid
Data represented as mean ± SD, n = 3.
Miscibility of solid and liquid lipid
The solid lipid GMS was mixed with each liquid lipid (WSO and AO) in the 90:10, 80:20, and 70:30 w/w ratio, to evaluate the miscibility of the two lipids. The mixtures were heated for 30 min at 60°C and then allowed to cool at room temperature. After cooling, the mixture was placed on filter paper and visually observed for any possible sign of turbidity or phase separation as the droplet. 13
Drug-Excipient Compatibility
The compatibility study of drug and excipients (lipid, oil, and surfactant) was done to ensure the stability of the final formulation. A physical mixture of drug and lipid in the ratio of 1:1 with a small amount of Pluronic F-68 was placed in glass vials, sealed, and kept at room temperature. The sample was examined for the physical and chemical integrity of drug and excipients. Parameters such as color change, odor, or gas formation were examined. Compatibility was further confirmed by taking the FTIR spectrum (Jasco FTIR-4100, Japan) of drug, lipid, and physical mixture (Fig. 3).
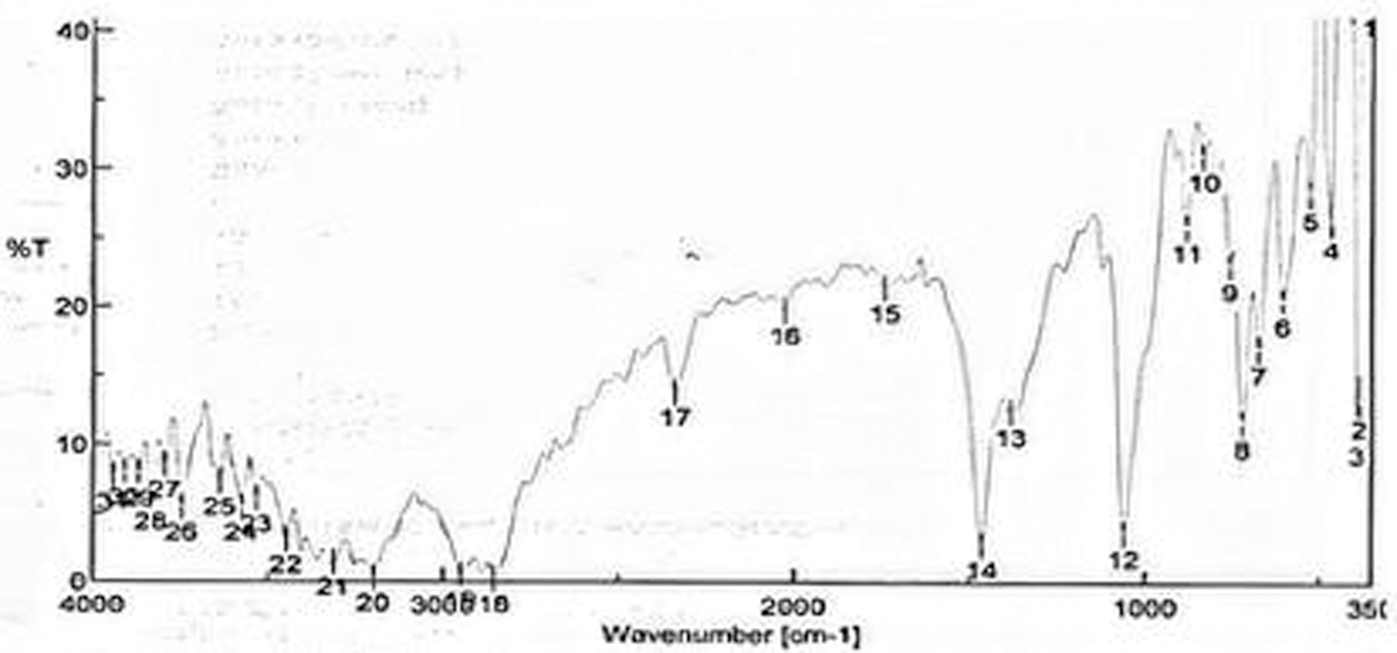
FTIR spectrum of physical mixture.
Preoptimization of Various Processing Variables
Various literatures have reported the effect of various processing parameters (such as a drug-to-lipid ratio, surfactant concentration, and sonication time) on the characteristic of NLCs. In this study, we have primarily optimized the concentration of surfactant, and drug-to-lipid (mix) ratio and sonication time.
Optimization of surfactant concentration
For selecting surfactant, different concentrations of surfactant from 1% to 3% (w/v) were used based on literature by keeping other parameters (a drug-to-lipid mix ratio and sonication time) constant.
Optimization of drug-to-lipid mix ratio
Different ratios of drug-to-lipid mix ranging from 1:1 to 1:5 were used by keeping rest parameters (surfactant concentration and sonication time) constant.
Optimization of sonication time
Time for sonication was kept 5, 10, and 15 min as per previous literature keeping rest parameters constant.
Optimization of Parameters by Design of Experiment
Taguchi design of experiment 14 was used to optimize the formulation parameters and study the effect of processing variables selected above, that is, the concentration of the surfactant, drug-to-lipid mix ratio, and sonication time. Each of the above-selected values of variables was taken as medium and two more variables based on the medium were taken as low and high, that is, low (1), medium (2), and high (3) (Table 3). A design matrix consisting of nine experimental runs was constructed (L9array). The respective responses of the dependent variables of particle size, polydispersity index, zeta potential, and % drug loading were observed and recorded.
Variables in Taguchi Design L9 Array
Preparation of MP-loaded NLCs
MP-loaded NLCs (MNLCs) were prepared by a novel melt emulsion ultrafiltration technique. Glyceryl monostearate and WSO were taken as solid and liquid lipids, respectively, in a ratio of 7:3. A total of nine formulations were prepared as per the composition given by the L9 array (Table 4). For 100 mg MP, 100 to 300 mg (1:1, 1:2, 1:3; drug-to-lipid ratio) of lipid mix (in a ratio of 7:3) was weighed and melted on a thermostat water bath at 60°C. Accurately weighed 100 mg drug was mixed in the above lipid melt. This lipid phase containing the drug was sonicated using a bath sonicator (ULTRASONIC CLEANER CD-4800, 70 W) for 2 min at the same temperature. Ten milliliters of aqueous Pluronic F-68 solution [in a concentration ranging from 2.5% to 3.5% (w/v) for all batches] was prepared by using distilled water and heated on a water bath to the same temperature as that of lipid mix melt (i.e., 60°C). Both the phases were mixed and homogenized (HICON, India) for 30 min at 3,000 rpm at 60°C to obtain a primary emulsion. The primary emulsion was then sonicated for 4 to 6 min for all batches; the primary emulsion was passed through a membrane made up of mixed cellulose ester (pore size 0.7 μ, MICROFIL FILTER; MERCK MILLIPORE, India) and was allowed to cool quickly in the ice bath to get MNLCs.
Composition as per L9 Orthogonal Array
MNLC, mupirocin-loaded NLCs.
Characterization of MNLCs
Particle size and polydispersity index
The particle size and polydispersity index (PDI) of prepared MNLCs were measured by dynamic light scattering using ZetasizerNano ZS (Malvern Instruments, United Kingdom). MNLC suspension, 1.0 mL, was diluted 10 times (10 mL) with distilled water and the average particle size and PDI were measured in triplicate (n = 3) using photon correlation spectroscopy. 15
Surface morphology
The surface morphology of MNLCs was determined by transmission electron microscopy (Hitachi, Japan). A drop of NLC dispersion was spread on a 200-mesh copper grid and negatively stained with 2% phosphotungstic acid for 30 s. The grid was dried at room temperature and then observed by transmission electron microscopy.
Zeta potential
The surface charges of MNLCs, denoted as zeta potential, were determined by the electrophoretic mobility of MNLCs in a disposable zeta cell at 25°C using a Zetasizer (Malvern, United Kingdom).
% Drug loading
Estimation of % drug loading of all formulation was done by using the centrifugation method.
16
Ten milliliters of MNLC suspension was taken into a centrifuge tube with a pipette (10 mL, Borosil), and centrifuged at 18,000 rpm for 30 min at 10°C temperature (R-8C Remi Cooling centrifuge, India). The lipid portion was isolated from the supernatant and the amount of drug present in the supernatant was determined using UV-Visible spectrophotometer (UV1800 Shimadzu, Japan.) at 220 nm in a solvent system of acetonitrile and phosphate buffer (pH 6.4) in a ratio of 50:50 v/v.
17
The % drug loading was calculated in triplicate (n = 3) by the following equation.
18
where W1 is the weight of MP added to the system, WS is the weight of the drug in the supernatant after centrifugation, and WL is the weight of lipid used for MNLC preparation.
In Vitro Release
The in vitro drug release studies of all nine formulations were carried out using Franz diffusion cell in triplicate (n = 3). Dialysis membrane (Hi-Media, Mumbai, India) was soaked in double-distilled water for 12 h before mounting in Franz diffusion cell. MNLC suspension was placed in the donor compartment and phosphate buffer (pH 7.4), as a dissolution medium, was placed in the receptor compartment. The temperature of the receptor compartment was maintained at 37°C ± 0.5°C with the help of a circulating water bath. The solution in the receptor compartment was stirred continuously (100 rpm) using a magnetic bead. The sample (1.0 mL) was withdrawn at predetermined time intervals and replaced with an equal amount of fresh dissolution media to maintain the sink condition. The samples were analyzed spectrophotometrically (UV-1800, Shimadzu, Japan) at 220 nm. The cumulative % drug release was calculated in triplicate. 18
Release kinetic study
In vitro release data of optimized formulation MSLN3 were analyzed by various kinetic models such as zero order (C = k 0 t), first order (Log C = LogC 0 − k f t/2.303), Higuchi's model (Q = Kt 1/2), and Korsmeyer–Peppas model (Mt /M ∞ = Ktn ) to investigate the release pattern and mechanism of drug release. 19
Stability Study
The stability of the optimized formulation MNLC4 was evaluated using stability chamber (Stericox, India). The stability studies were undertaken at 25°C/60% relative humidity (RH) and 40°C/75% RH in triplicate (n = 3). Sampling was done at 0, 15, 30, and 60 days following storage. At each sampling time, the formulation was evaluated for any physical changes, particle size, PDI, and % drug loading. 20
Result and Discussion
Preliminary Study of Drug
FTIR spectra of MP (Fig. 2) showed characteristic peaks corresponding to the major functional group between 1720.19 cm−1, which is due to carbonyl stretching (C = O) of the ester group present in the molecule. The peak corresponding to 2923.56 cm−1 indicates the acidic hydroxyl group (R-COOH). Another peak at 3772.08 cm−1 may be due to the free hydroxyl (R-OH) group present terminally in the molecule (Fig. 2). Hence, the molecule is interpreted as MP due to the presence of the above-stated peaks.
Melting of the drug was initiated at 76°C and completed at 78°C so instead of giving a definite melting point, the author suggested a melting range from 76°C to 79°C, which is the reported melting point (77°C–78°C). 21
Screening of Solid Lipid
Solid lipid was selected based on the solubility of the drug in the lipid as drug loading depends on the solubility of the drug in the lipid. The significantly high solubility of MP, that is, 73 ± 0.84 mg per 1,000 mg of lipid, was observed for GMS compared with Compritol 888 (65 ± 0.92 mg/1,000 mg of lipid) and Gelucire (60 ± 0.43 mg/1,000 mg of lipid). Hence, GMS was selected as solid lipid. The high solubility of the drug in GMS could be due to the high hydrophobicity of glycerides due to the esterification of glycerol by long-chain fatty acids in GMS, and having a higher proportion of mono- and diglycerides compared with Gelucire and Compritol 888. 22
Screening of Liquid Lipid/Oil
The selection was based on the solubilizing power of liquid lipid for MP as it affects drug loading and entrapment. 23 It was observed that the solubilizing power of WSO was more (78 ± 0.34 mg) compared with AO (70 ± 0.97 mg) (Table 2). Miscibility of solid lipid with liquid lipid is also a very important criterion as it reduces the recrystallization of lipids. 24 The optimized ratio of WSO and GMS was found to be 2:8, as a higher amount of WSO leads to phase separation on visual observation of filter paper smear test. AO in the same ratio tends to reduce the melting point of GMS (from 71°C to 48°C). There may be a chance of the formation of the polymorphic form of GMS (recrystallization) with a decreased melting point. 25
Drug-Excipient Compatibility Study
As FTIR spectra cannot be identical for different molecules, the IR spectroscopy was used to study the compatibility of the drug with the excipients. It was observed that the physical mixture characteristic peaks (obtained for different functional groups) were found in the same position and have an almost equal intensity (Fig. 3). It was concluded that there is no interaction between the drug and excipients used as there is no change in the position and intensity of spectra of the functional groups of MP.
Preoptimization of Various Processing Variables
In the preparation of NLCs, there exist variables that can affect the particle size, PDI, drug loading, and in vitro drug release. Based on the literature, processing variables such as the concentration of surfactants, the drug-to-lipid ratio, and the time of sonication were priorly optimized. Preoptimization is necessary for screening out a range of variables according to desired outcomes. In the preoptimization, the concentration of the surfactant was selected as 3% w/v. Surfactant concentration of 1% w/v was too less to prepare the MNLC (lipid get separated as a precipitate) and for 2% surfactant concentration, the prepared MNLC gets milky after 24 h. The milky appearance may be due to aggregation of prepared MNLCs, but a 3% surfactant concentration gave stable results. Similarly, the drug-to-lipid mix ratio also experimented from 1:1 to 1:5. The best result with respect to particle size and drug loading was found for the drug-to-lipid ratio 1:2. At 1:1 drug-to-lipid ratio, NLC was not fabricated, while the size of the particle was increased for 1:3 and 1:4. This is in agreement with previous work that reported an increase in size with an increased drug-to-lipid ratio. 17 The sonication time was chosen as 5 min because sonication for 10 and 15 min was not sufficient enough to show any change in particle size of MNLC.
Optimization of Parameters by Design of Experiment
To optimize processing parameters simultaneously, at three different levels namely, medium (2) was taken from a preoptimized value, a fixed amount is reduced to get a lower level (1) and the same value is added in level (2) to get a higher level (3). For experimental optimization, the number of batches will be more, and so, to reduce the number of experiments, L9 array was used for three factors at three levels, where only nine sets of experiments (Table 4) were needed to completely optimize the effect of various parameters simultaneously.
Preparation of MNLCs
A novel melt emulsion ultrafiltration technique has been reported in this research work. The techniques reported earlier for the preparation of NLCs such as the solvent emulsification diffusion technique, 7 solvent-emulsification evaporation technique, 26 and solvent injection techniques 27 used organic solvents, and pose a risk of contamination of prepared NLCs with residual solvents. Other techniques such as high-pressure homogenization 28 and membrane contractor 29 require specialized equipment. The novel melt emulsion ultrafiltration method does not make use of organic solvents and can be easily operated with basic laboratory equipment such as bath sonicator and membrane filter. This technique could be a step forward in the green manufacturing of nanoparticles.
Characterization of MNLCs
Particle size and PDI
The particle size of prepared MNLCs was found in a range from 138 ± 0.67 nm for MNLC7 to 683 ± 1.98 nm for MNLC3. PDI value was found to be between 0.199 ± 0.045 for MNLC6 and 0.863 ± 0.079 for MNLC7. The particle size of MNLC7 was found to be 138 ± 0.67 nm, but the PDI for the same batch was found to be 0.863 ± 0.079 (Fig. 4 and Table 5). Particle size of MNLC4 was 139 ± 0.75 nm and PDI for the same was
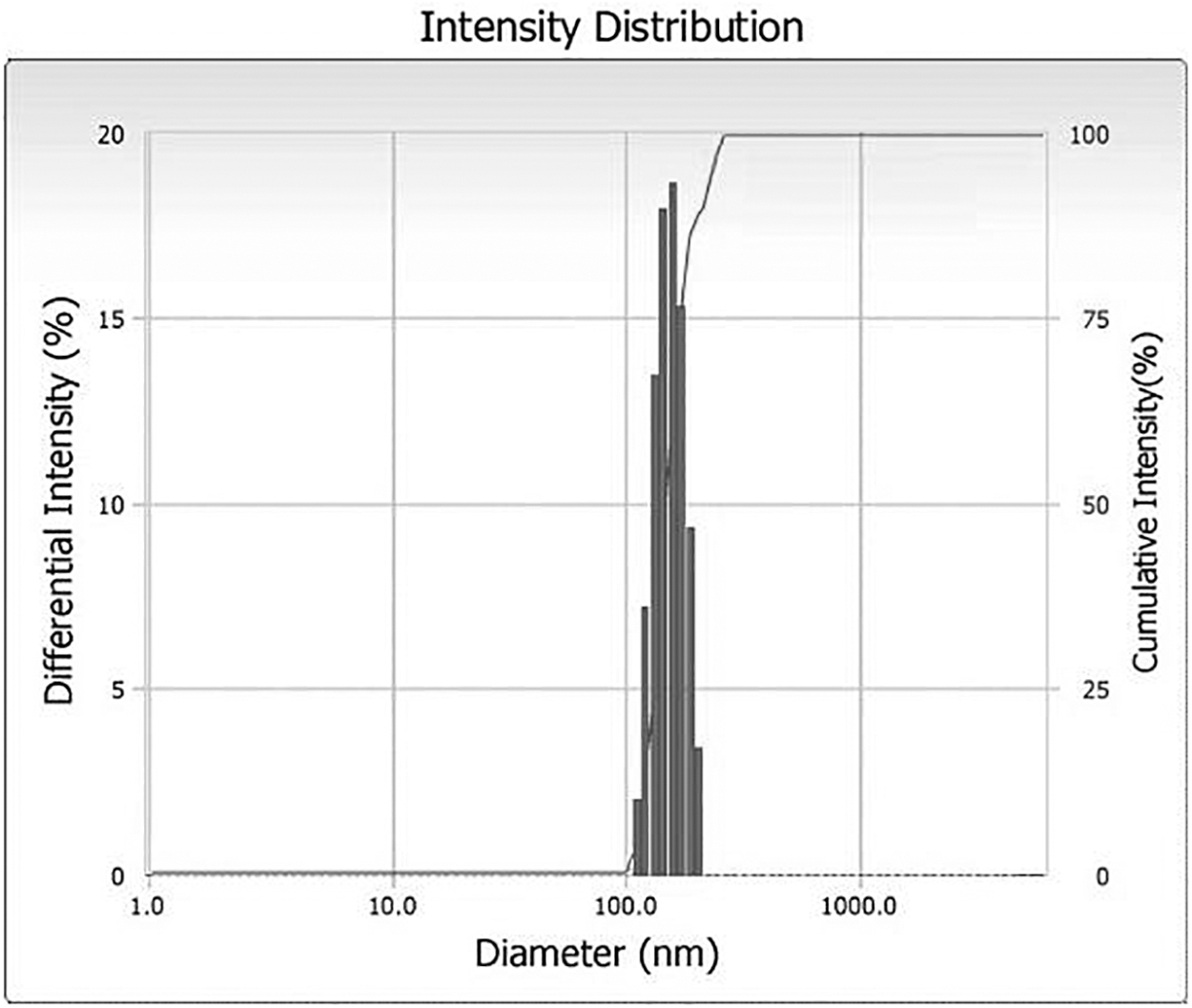
Particle size distributions of MNLC4. MNLC, mupirocin-loaded nanostructured lipid carrier.
Characterization of Various Formulations
Data represented as mean ± SD, n = 3.
The PDI is an indicator of the quality of dispersion and ensures the mono- or polydispersity of dispersions. The measurement of PDI plays an important role to predict the chance of agglomeration. The smaller value of PDI is considered ideal for nanoparticle homogeneity and to ward off aggregation. 31 It was also observed that the PDI was lower for the surfactant concentration up to a certain level, that is, 3%, but it increases when the concentration was raised to 3.5% in formulations MNLC7, MNLC8, and MNLC9. This may be due to the aggregation of particles by surface-bound surfactants at a higher concentration. 32 Although the lowest value is 0.199 ± 0.045 for MNLC6, so many formulations also have the value of PDI within an acceptable limit.
Surface morphology
Transmission electron micrograph showed that the prepared MNLC4 has a smooth surface and almost all particles were spherical (Fig. 5).
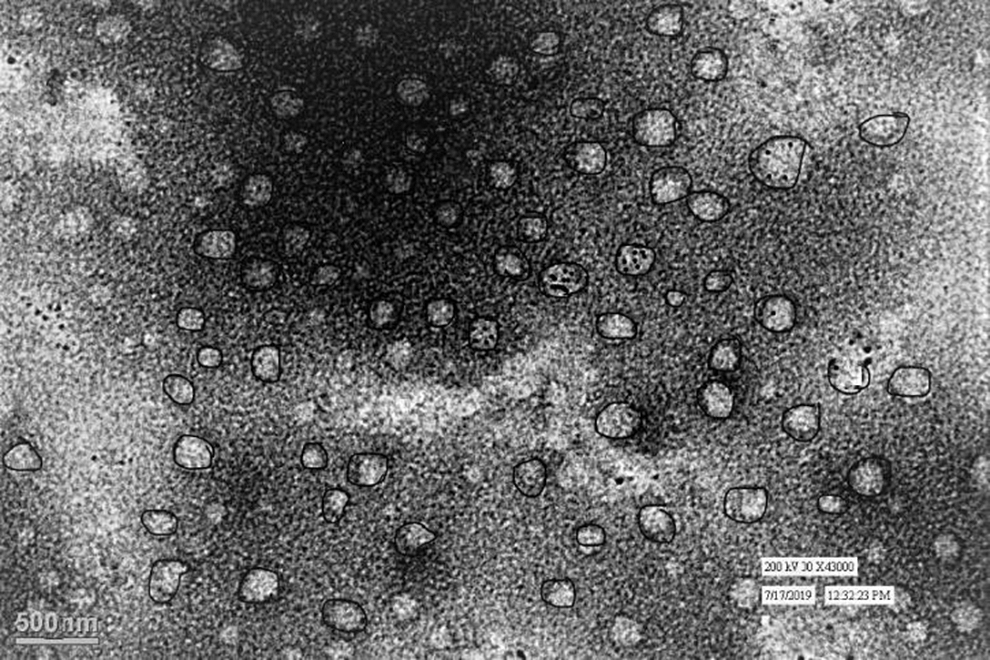
Transmission electron micrograph of MNLC4.
Zeta potential
Zeta potential is the measurement of the surface charge that particles gain in the dispersed state and gave an idea about the aggregation tendency of NLCs. The zeta potential of formulations was found in a range from +15.2 ± 0.91 to +28.1 ± 1.12 mV (Table 5). The positive value of zeta potential may be due to the surfactant that coated the surface of particles. It was observed that the sonication time and drug concentration affect the value of zeta potential. 33 A higher value of zeta potential with increasing sonication time may be due to an increase in surface area and the subsequent increase in surface energy followed by surfactant coverage. The value of zeta potential away from zero is considered to be stable. 34
% Drug loading
The % drug loading is the ratio between the entrapped drug and the total weight of the lipid mix used. It was found that the % drug loading was high and ranged from 50% ± 0.29% for MNLC1 to 62% ± 0.31% (Table 5). The drug-to-lipid ratio plays an important role in % drug loading; as the drug-to-lipid ratio increases, the % drug loading increases, it was observed due to disordered crystal matrix and consequently more space for the accommodation of drugs due to liquid lipid. 23
In Vitro Release
Percent cumulative drug release of MP for 24 h from all nine formulations was investigated (Fig. 6). Each sample was analyzed in triplicate. Four formulations MNLC1 (95.27% ± 1.12%), MNLC4 (98.76% ± 1.56%), MNLC5 (91.06% ± 1.29%), and MNLC7 (98.84% ± 1.87%) were able to release >90% of drug in 24 h among them. The release from MNLC4 and MNLC7 was found to be 98.76% ± 1.56% and 98.84% ± 1.87% after 24 h and only 72.76% ± 3.41% of drug release was obtained for MNLC3 after 24 h. It was observed that with increasing drug-to-lipid ratio, the particle size also increases. However, the rate and amount of drug release declined for a point of time. The author suggests that this decrease may be due to the increase in diffusional path length because of the large particle size. Higher concentrations of surfactant may also be a factor for maximum release of drug from MNLC4 and MNLC7, and the observation is in accordance with previous literature. 35
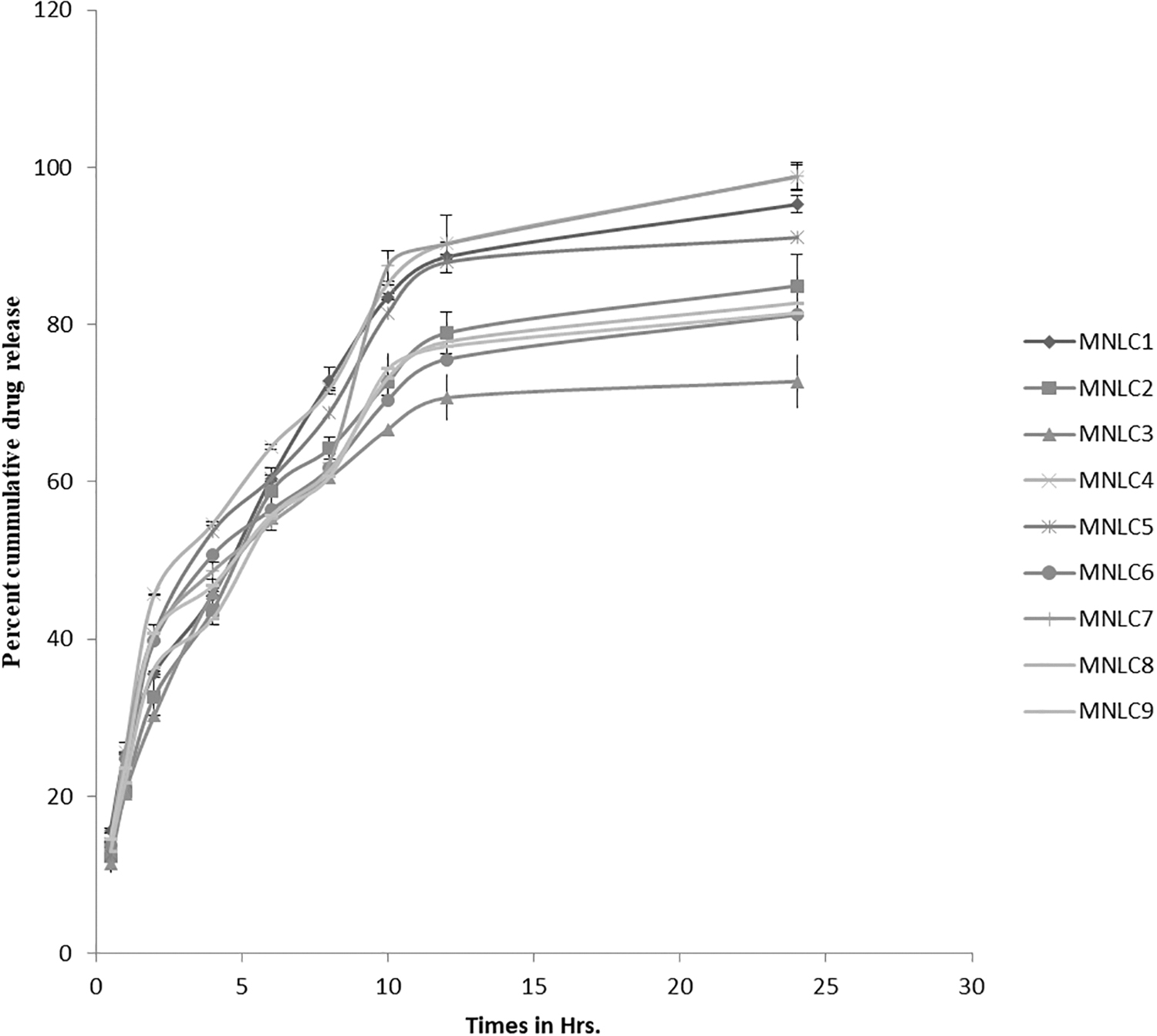
In vitro release profile of MNLCs.
Release kinetic study
After applying drug kinetics, the value of correlation (r 2 ) for the Higuchi equation was found to be 0.961 followed by 0.913 for the first-order and 0.892 for the zero-order model (Table 6). Since the highest r 2 demonstrates the highest correlation, the release profile of MNLC4 was found to follow Higuchi kinetics. The drug release from MNLC4 follows neither first- nor zero-, but mixed-order kinetics. To explain the mechanism of drug release (log % cumulative drug release vs. log time), the Korsmeyer–Peppas model 36 was studied. It showed good linearity (r 2 = 0.943). The release exponent “n” was found to be 0.439, indicating that the release of the drug was based on Fickian diffusion.
Release Kinetic Parameters of MNLC4
Stability Study
To perform stability studies, formulations were kept for 60 days at 25°C/60% RH and 40°C/75% RH. No changes were detected in visual appearance for MNLC4. The particle size showed changes after 15 days (139.4 ± 0.31 nm), 30 days (145.3 ± 0.65 nm), and 60 days (152.7 ± 0.28 nm) of storage; while the value of PDI increased slightly after 15 days (0.308 ± 0.27), 30 days (0.521 ± 0.48), and 60 days (0.714 ± 0.86), the % drug loading was found to decrease slightly after 30 days (58.2 ± 0.052) and 60 days (56.7 ± 0.73) at 25°C/60% RH (Table 7), but it was within range. At 40°C/75% RH, the particle size was 158.1 ± 0.25 nm after 15 days of storage, 172.7 ± 0.98 nm after 30 days, and 239.7 ± 0.76 nm after 60 days of storage, the PDI was 0.761 ± 0.765 after 15 days, 0.89 ± 0.853 after 30 days, and 0.94 ± 0.098 after 60 days of storage, and the % drug loading decreased (from 59 ± 0.13 to 39.9 ± 0.79) in 60 days at 40°C/75% RH. Particle size and PDI increased more when stored at 40°C/75% RH, compared with storing at 25°C/60% RH, the possible reason may be that an increase in the kinetic energy of particles at a higher temperature leads to particle aggregation, which consequently increases the particle size and PDI. The decrease in % drug loading at a higher temperature may be due to polymorphic changes in lipid at a higher temperature. 19 Hence, the prepared NLCs of MP were stable at 25°C.
Stability Studies of MNLC4
Data represented as mean ± SD, n = 3.
PDI, polydispersity index; RH, relative humidity.
Conclusion
In this study, a novel method, the melt emulsion ultrafiltration technique, was explored for the fabrication of NLC. The method furnishes the benefit of avoiding organic solvent, and hence, no risk of toxicity associated with residual solvent. The method can be easily performed without using sophisticated equipment. The method was found suitable for fabricating MNLC. The prepared formulations were nanoscale, monodisperse, and showed excellent drug loading. Formulation MNLC4 was the optimal formulation, although MNLC7 has a lesser particle size and a higher zeta potential compared with MNLC4, but sufficient to provide stability for MNLC4, also the PDI is high for
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.