
Abstract
HIV-1 BF intersubtype recombinants are frequent in Argentina, Uruguay, and Brazil, where among a high diversity of BF unique recombinant forms (URFs), eight circulating recombinant forms (CRFs) have been characterized. Here, we describe a new one, designated CRF44_BF, identified in HIV-1 samples from Chile. In a previous report, where partial pol sequences of 136 HIV-1 infections of Chilean subjects were analyzed, a phylogenetic cluster of HIV-1 recombinant BF viruses from 10 individuals, with coincident intersubtype recombination points, was detected. One virus of this cluster had been characterized along its near full-length genome. A second one, from an epidemiologically unlinked HIV-1-infected subject, is described here. Both genomes share identical mosaic structures, consisting of a predominantly subtype F1 genome with three fragments of subtype B. Coincident breakpoints and phylogenetic clustering of the newly identified CRF44_BF with CRF12_BF, CRF17_BF, and CRF38_BF support a common origin of different CRF_BFs identified in Argentina, Uruguay, and Chile.
I
BF intersubtype unique recombinant forms (URF BFs) probably represent the greatest variety of different mosaic patterns described for HIV-1, and the number of CRF_BFs characterized to date clearly surpasses any other subtype combination. Some of the CRF_BFs could have experienced successive rounds of recombination (mainly with subtype B), contributing to this wide genetic diversity, 14 which could be a consequence of epidemiological factors or biological features, such as a higher fitness, transmissibility, or replication capacity of the BF recombinant viruses compared to the parental subtypes. 15
In Chile, initial analysis by heteroduplex mobility assay (HMA) and sequencing of the HIV-1 env gene (a fragment comprising the C2-V3-C3 region) indicated the predominant circulation of HIV-1 variants of subtype B and the presence of two different clusters of Chilean subtype F variants detected in children born from HIV-1 heterosexually infected mothers. 16,17 Subsequently, we reported a study of protease-reverse transcriptase (PR-RT) sequences from 136 HIV-1-infected individuals. 18 Subtype B was predominant (85%), BF recombinants were detected in 15%, and one sequence was a nonrecombinant F1 subtype. Most Chilean BF recombinants clustered into two groups with a similar number of infections in each one. The nine viruses of one group clustered together with CRF12_BF. The second group, designated in that report as “Chilean BF cluster,” contained samples from 10 subjects infected with BF recombinant viruses sharing three breakpoints in PR-RT. All 10 infections had been diagnosed between 1997 and 2004 in north and central Chile. Six patients were children infected from their mothers, two were the mothers of two of the children, who had acquired the infection through heterosexual intercourse, and two were men who had sex with men. The near full-length genome characterization of one virus of this cluster (CH12) had been reported previously. 14 To determine whether these viruses represent a new CRF, 19 we proceeded to amplify and sequence the complete genome of a second virus of this cluster (CH80), which lacked epidemiological links to CH12. In the present report, we analyze its recombinant structure and phylogenetic relations with other BF recombinant forms of South America.
The near full-length genome of CH80 was amplified by nested polymerase chain reaction (PCR) in four overlapping fragments of 1.8–3 kb each from the patient's peripheral blood mononuclear cells (PBMCs) DNA. 6 Direct sequencing was performed with the ABI Prism BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA). Neighbor-joining phylogenetic trees were constructed using MEGA v. 3.1. 20 Recombinant patterns were analyzed by bootscanning with SimPlot v. 3.5.1, 21 and phylogenetic analyses of small fragments were performed with maximum likelihood using PhyML. 22
In a phylogenetic tree of full-length genomes, CH12 and CH80 clustered together, and both grouped in a cluster with CRF12_BF, CRF17_BF, and CRF38_BF (Fig. 1). The analysis of the mosaic structure of both viruses, using SimPlot v. 3.5.1, complemented with analysis of short segments via maximum likelihood (Fig. 2), indicated that CH12 and CH80 shared identical mosaic structures, consisting of a predominantly subtype F1 genome with three fragments of subtype B: two in pol, between HXB2 positions 2470 and 2725 and between 2935 and 3696, respectively, and one in env, between HXB2 positions 6342 and 6447. Both viruses also clustered together in partial trees of all B and F subtype fragments along the genome (not shown). Two breakpoints (at positions 2470 and 3697) are coincident with CRF12_BF, CRF17_BF, and CRF38_BF (Fig. 3). Therefore, the results allow us to define a new CRF, which has been designated CRF44_BF. Coincident breakpoints and clustering in phylogenetic trees with other CRF_BFs of South American origin support the previously proposed model of a common origin of different BF recombinants in South America (excluding Brazil) that diversify by successive rounds of recombination along diverse lineages. 14
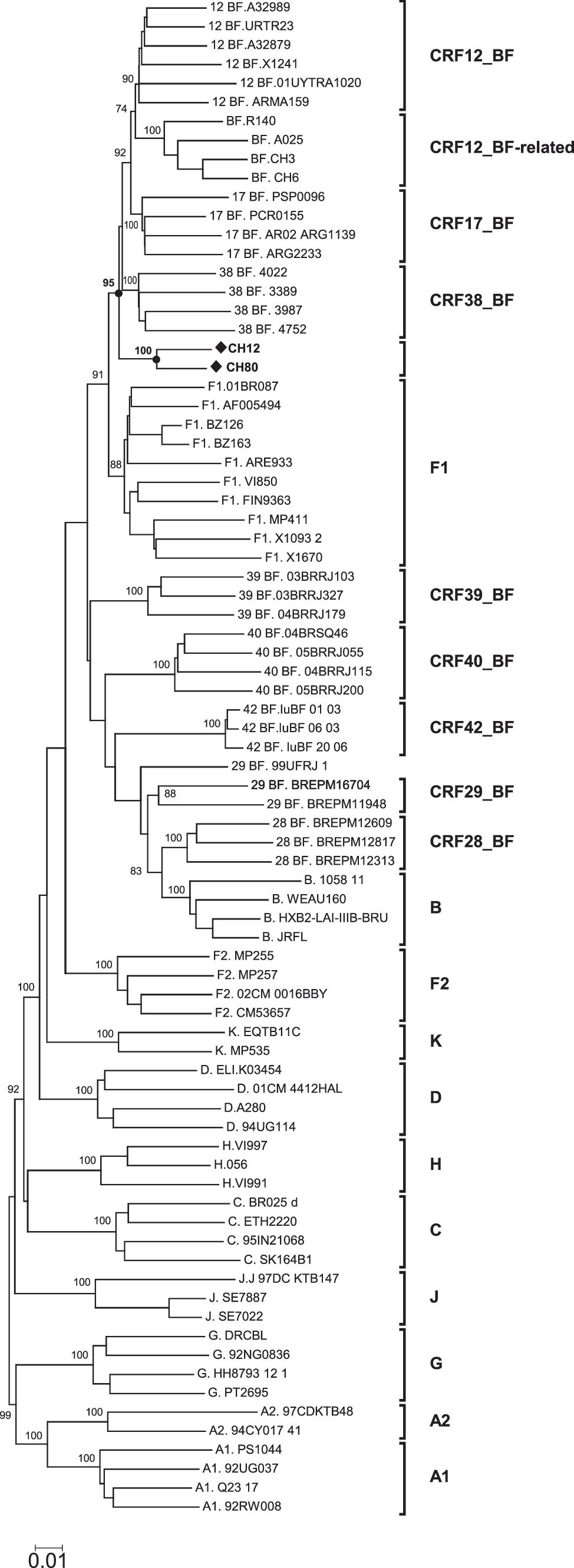
Phylogenetic tree of HIV-1 complete genomes including all CRF BFs reported to date. BF recombinants of the Chilean cluster are in bold type and marked with diamonds.
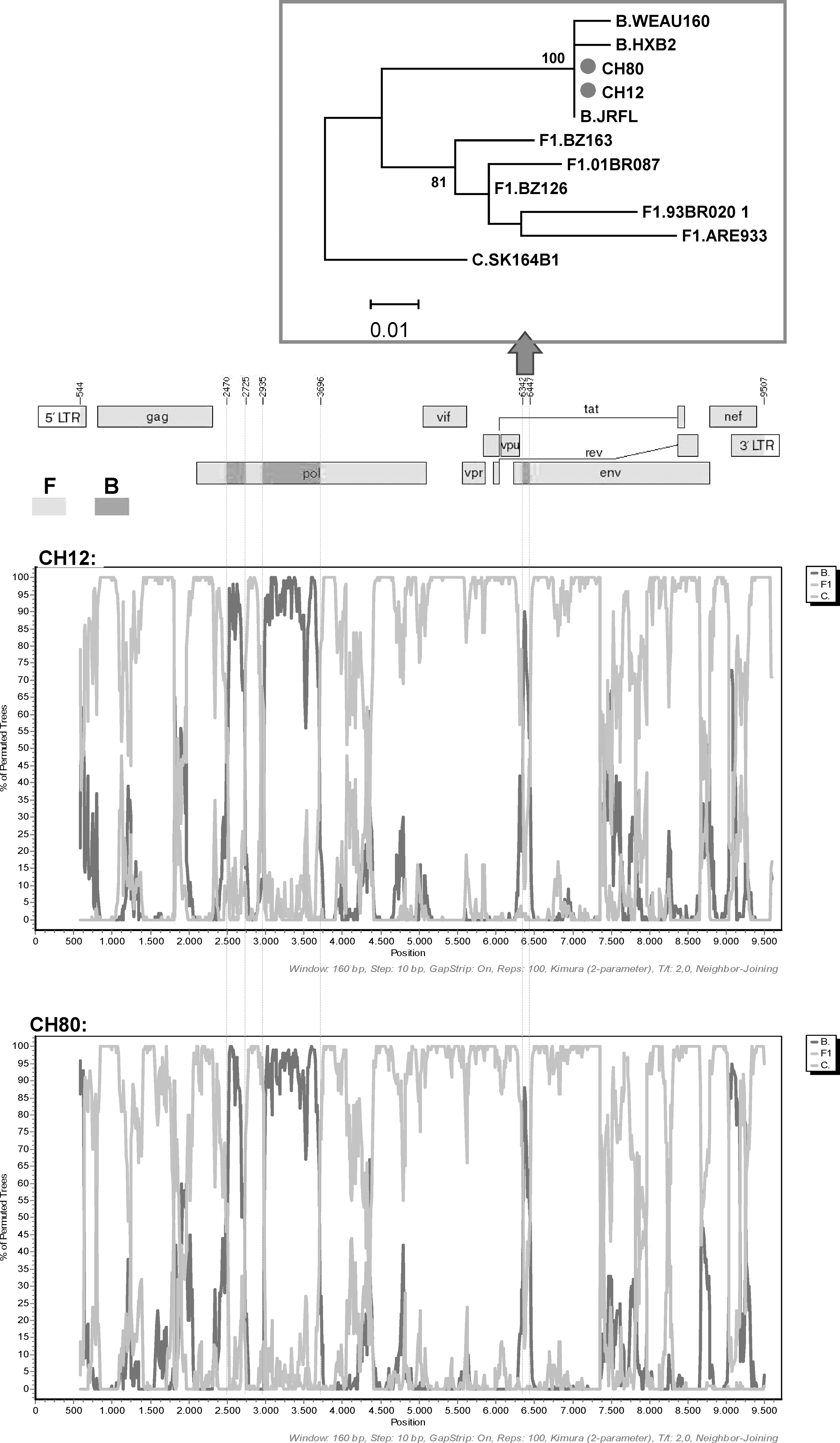
Bootscan analyses of the two near full-length genomes of samples CH12 and CH80. Bootscanning was performed using SimPlot v. 3.5.1, moving a window of 160 nt at steps of 10 nt to detect short recombinant fragments. A maximum likelihood (ML) phylogenetic tree of fragment 6342–6447, which supports the subtype B assignation performed by bootscan analysis, is shown. In ML trees of other short segments whose subtype assignation was unresolved by bootscan analysis, CH12 and CH80 failed to cluster with subtype references (results not shown).
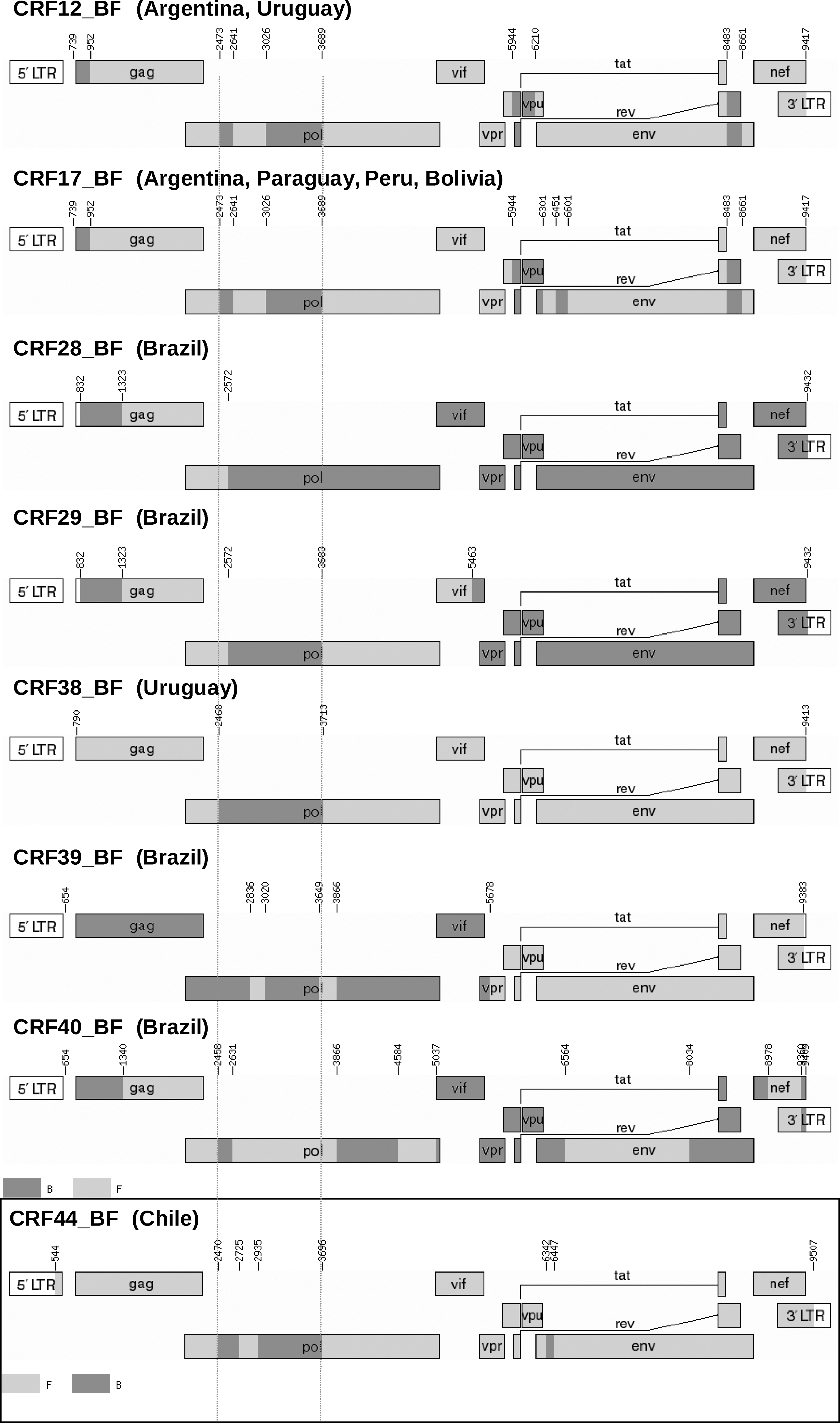
Mosaic patterns of all the CRF BFs from South America reported to date and comparison with CRF44_BF, characterized in this study. The two dotted red lines indicate two common breakpoints at HXB2 positions 2470 and 3696, shared by CRF12_BF, CRF17_BF, CRF38_BF, and CRF44_BF, which circulate in Argentina, Uruguay, Paraguay, Peru, Bolivia, and Chile. Only the CRFs circulating in Brazil (CRF28_BF, CRF29_BF, CRF39_BF, and CRF40_BF) do not share both breakpoints.
In summary, in this study we have identified and characterized a new HIV-1 genetic form circulating in Chile, which has been designated CRF44_BF. It is the eighth BF intersubtype CRF identified in South America, and it probably shares a common origin with other CRF_BFs (CRF12_BF, CRF17_BF, and CRF38_BF) circulating in Argentina and Uruguay. CRF44_BF has been identified in 10 individuals from the north and central regions of Chile with low frequency (7%). These viruses have been transmitted through mother-to-child, heterosexual, and homosexual routes.
The suggested higher fitness and replication capacity of some BF recombinants, 15 as well as their high prevalence and explosive expansion in some South American countries, highlight the importance of routine molecular epidemiological surveillance of BF recombinant viruses in these countries. Most BF recombinants described to date contain intersubtype breakpoints in the PR-RT region. Therefore, the genotyping of this fragment, performed for antiretroviral resistance testing, should be accompanied by phylogenetic analysis, which will allow identification of these recombinant forms.
A proper follow-up of the HIV-1 epidemic in a geographic region is fundamental to establish effective preventive campaigns, and knowledge of the evolutionary dynamics of the different subtypes and recombinants circulating is necessary for the implementation of antiretroviral drug therapies as well as for the design of an effective vaccine in that geographic area. This may be relevant with regard to the newly identified CRF, considering the lower susceptibility of subtype F viruses to some protease inhibitors 23 and the reported correlation of immune response to subtypes. 24
Sequence Data
The sequence has been deposited in GenBank under accession number FJ358521.
Footnotes
Acknowledgments
We are grateful to the Genomic Unit of Centro Nacional de Microbiología, Instituto de Salud Carlos III (Majadahonda, Madrid). This study was supported in part by Centro Nacional de Referencia de VIH/SIDA, Instituto de Salud Pública de Chile, Santiago, Chile. This work was presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention, Cape Town, 19–22 July, 2009. Poster number TUPEA070.
Author Disclosure Statement
No competing financial interests exist.