
Abstract
To determine viral subtypes and resistance mutations to antiretroviral treatment (ART) in untreated HIV-1 acutely infected subjects from Southwest Switzerland. Clinical samples were obtained from the HIV primary infection cohort from Lausanne. Briefly, pol gene was amplified by nested PCR and sequenced to generate a 1 kb sequence spanning protease and reverse transcriptase key protein regions. Nucleotide sequences were used to assess viral genotype and ART resistance mutations. Blood specimens and medical information were obtained from 30 patients. Main viral subtypes corresponded to clade B, CRF02_AG, and F1. Resistant mutations to PIs consisted of L10V and accessory mutations 16E and 60E present in all F1 clades. The NNRTI major resistant mutation 103N was detected in all F1 viruses and in other 2 clades. Additionally, we identified F1 sequences from other 6 HIV infected and untreated individuals from Southwest Switzerland, harboring nucleotide motifs and resistance mutations to ART as observed in the F1 strains from the cohort. These data reveal a high transmission rate (16.6%) for NNRTI resistant mutation 103N in a cohort of HIV acute infection. Three of the 5 resistant strains were F1 clades closely related to other F1 isolates from HIV-1 infection untreated patients also coming from Southwest Switzerland. Overall, we provide strong evidence towards an HIV-1 resistant transmission network in Southwest Switzerland. These findings have relevant implications for the local molecular mapping of HIV-1 and future ART surveillance studies in the region.
I
Thereby, the objective of this investigation was to determine viral subtypes and resistance mutations to antiretroviral treatment in untreated HIV-1 acutely infected subjects from Southwest Switzerland.
Clinical samples were obtained from an ongoing HIV primary infection clinical trial taking place at the Vaccine and Immunotherapy Center from Lausanne since 1998. This research protocol was approved by the Research Ethics Committee from the Faculty of Biology and Medicine of the University of Lausanne. Individuals enrolled in the study signed the informed consent and met the following criteria: 2–3 weeks from onset of HIV primary infection symptoms, incomplete seroconversion status verified by Western blot and a positive p24 antigen (Elecsys HIV Antigen Immunoassay, Roche Diagnostics, Basel, Switzerland). Whole blood EDTA samples collected at baseline were centrifuged to obtain plasma and extract viral RNA with the Qiagen (Alameda, CA) QIAamp Viral RNA Mini Kit. Then pol gene was amplified through a nested polymerase chain reaction (PCR) and sequenced to generate a 1–1.5 kb fragment. The genotyping algorithm from Virco (VirtualPhenotypeTM; Beerse, Belgium) was used in most cases. Alternatively, genotyping assessment was completed in a subset of samples by an in-house nested PCR protocol using amplification and sequencing primers previously described by Eyer–Silva et al.
3
Briefly, DNA samples (±1 μg) were amplified in a GeneAmp 9700 PCR system (Applied Biosystems, Foster City, CA) to obtain a 1 kb fragment spanning protease and reverse transcriptase regions (location from start of HXB2 genome 2250 → 3250). GeneAmp XL PCR kit (Applied Biosystems) was used to improve PCR performance and optimize the yield of long amplified products. A hot start consisting of 80°C (5 min), followed by 25°C (3 min), was initially performed on the lower first round mix sealed with AmpliWax (Applied Biosystems). The first round PCR was completed with an upper mix containing rTth DNA polymerase XL and DNA templates under the following thermocycling conditions: 94°C (1 min), followed by 28 cycles with 94°C (15 s) and annealing at 60°C (10 min), continued by final extension at 72°C (10 min). Next, a nested PCR run using AmpliTaq Gold DNA Polymerase Kit (Roche) took place as follows: 1 cycle with 95°C (3 min), 55°C (1 min), and 72°C (1 min), continued by 35 cycles with 95°C (1 min), 55°C (45 s), and 72°C (1 min) plus a final extension step of 10 min at 72°C. Sequencing reaction mixtures were assembled with BigDye Terminator v.3.0 Cycle kit (Applied Biosystems) and loaded to an ABI310 Genetic Analyzer. Nucleotide sequences were edited with BioEdit 7.0 and aligned by CLUSTAL X 2.0 alignment program with known reference strains of group M and circulating recombinant forms (CRFs) sequences pooled from the HIV-1 gene database (
Viral subtype was determined by Phylip DNADIST trees and bootstrap analysis. Drug resistance mutations were examined using the 2009 Stanford HIV drug resistance list (
Viral load was initially assessed by COBAS Amplicor HIV-1 Monitor version 1.5 and replaced later by real-time PCR (COBAS AmpliPrep/COBAS TaqMan version 1.0; Roche). Also CD4+ T cell counts were determined by multiparameter flow cytometry.
Isolates were collected from 30 individuals (29 men and 1 woman) assigned as primary HIV infection one through thirty (PHI-1 to 30), respectively. All subjects were at incomplete Western blot seroconversion window with a mean viral load of 7.3 Log HIV-1 RNA copies/ml (range, 4–8) and mean CD4+ T cell count of 428 cells/mm3 (range, 122–965). Risk transmission groups consisted of men having sex with men (66.6%), heterosexual intercourses (20%), IV drug users (10%), and one case (3.3%) of undefined transmission route.
Viral subtype assignments corresponded to clade B (n = 15), CRF02_AG (n = 4), F1 (n = 3), CRF11-like (n = 2), CRF01_AE (n = 2), G (n = 2), subtype D (n = 1), and one untyped recombinant isolate PHI-16 (Fig. 1). To better assess CRF11-like sequences (PHI-11 and 25) and PHI-16, we plotted this 3 pol sequences to a deduced alignment consisting of clades A1, J, CRF11_cpx, and D to determine similarity boundaries of PHI-16, after testing the consensus alignment provided in NCBI HIV-1 genotyping tool (
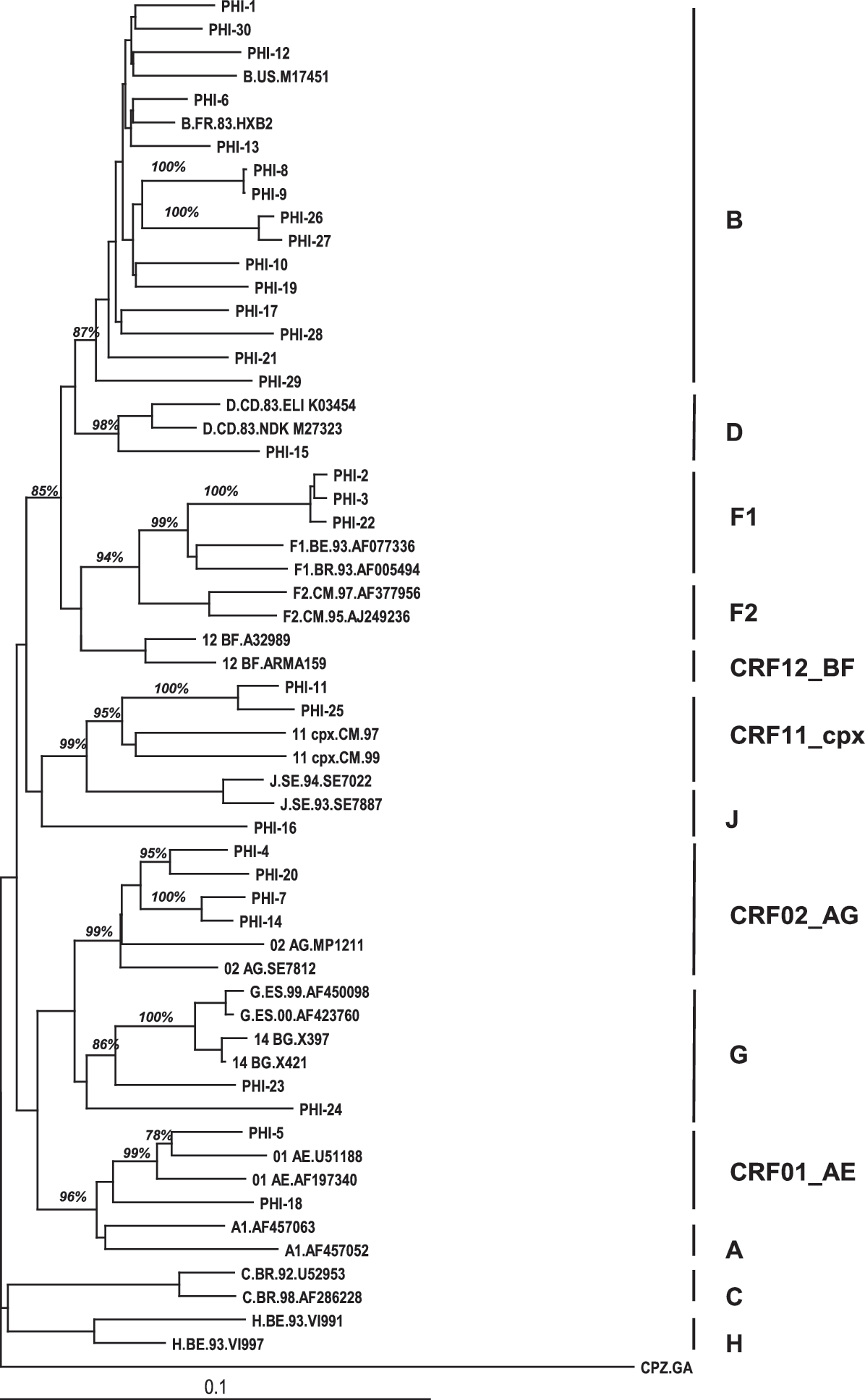
Neighbor-joining tree of HIV-1 sequences from the primary infection cohort from Lausanne with group M and CRF reference sequences pulled from the Los Alamos HIV database.
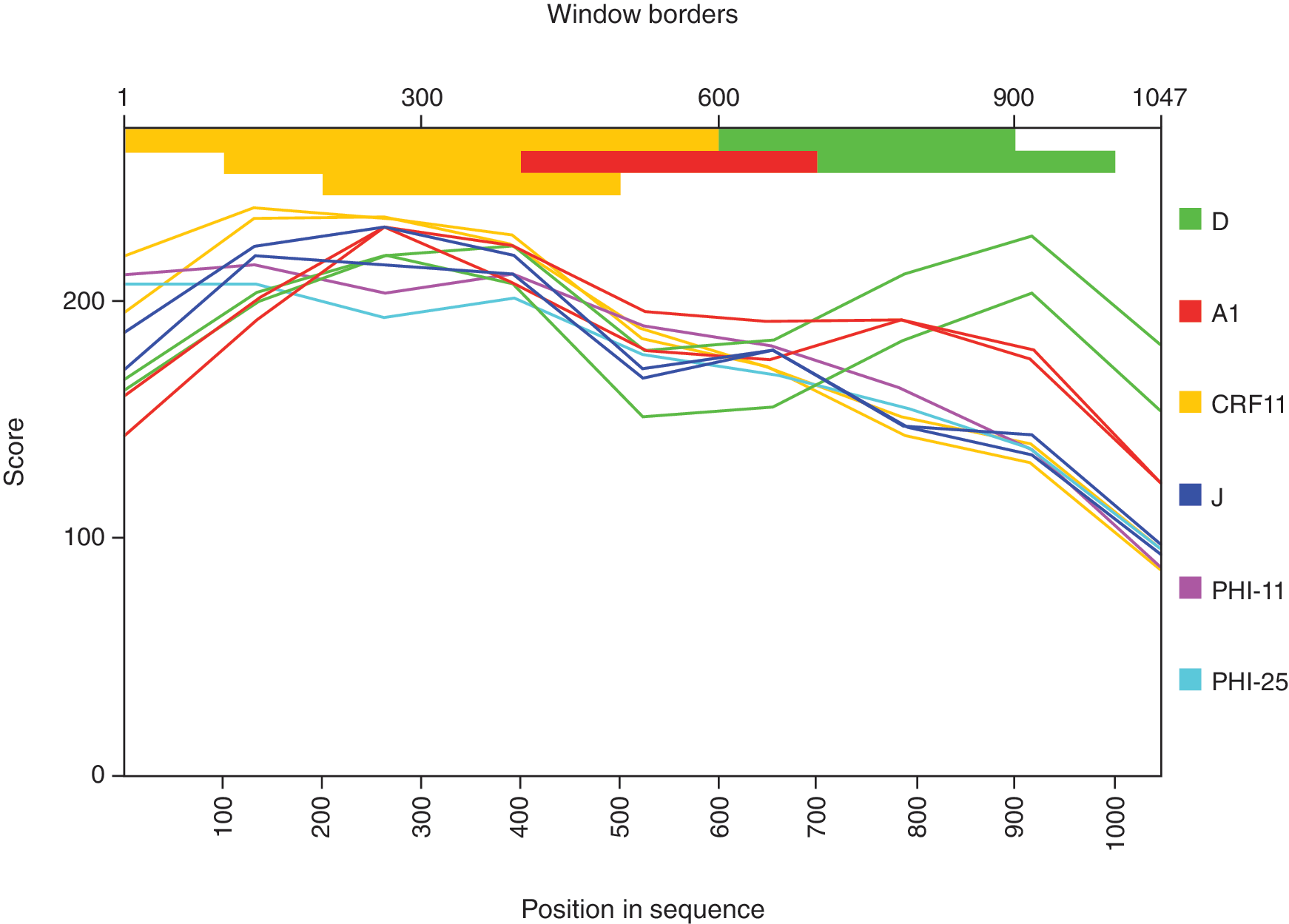
Similarity plot of HIV-1 pol genotype isolated from participant PHI-16 compared to other CRF11-like cohort sequences and reference sequences. (Color image can be found at
No major resistant mutations to PIs were detected. However, minor mutation 10V and accessory mutations 16E and 60E were found in all F1 clades (PHI-2; 3 and 22).
The RT amino acid sequences analysis revealed NNRTI major resistant mutation 103N in all clade F1 viruses. Also 103N was detected in one B clade (PHI-29) and in the D strain from the cohort (PHI-15). Moreover, PHI-15 also harbored resistance mutations to NRTIs as TAMs 41L and 215Y (Table 1).
Prot, protease; RT, reverse transcriptase.
To determine whether these PHI-F1 sequences were indeed a founder effect or a hint towards a transmission network, we extended the phylogenetic analysis to other HIV-1 F1 infections genotyped at the routine laboratories from Lausanne (n = 11) and Geneva (n = 13) during 1999 to 2007. A group of 24 partial pol sequences were analyzed. Sequences obtained from Geneva consisted of two separated fragments spanning reverse transcriptase and protease, respectively.
The resulting subtype F1 nucleotide alignment was used to determine ratios of synonymous to nonsynonymous base substitutions (ds/dn) with Los Alamos HIV database website tool SNAP
5
and plotted with Highlighter (
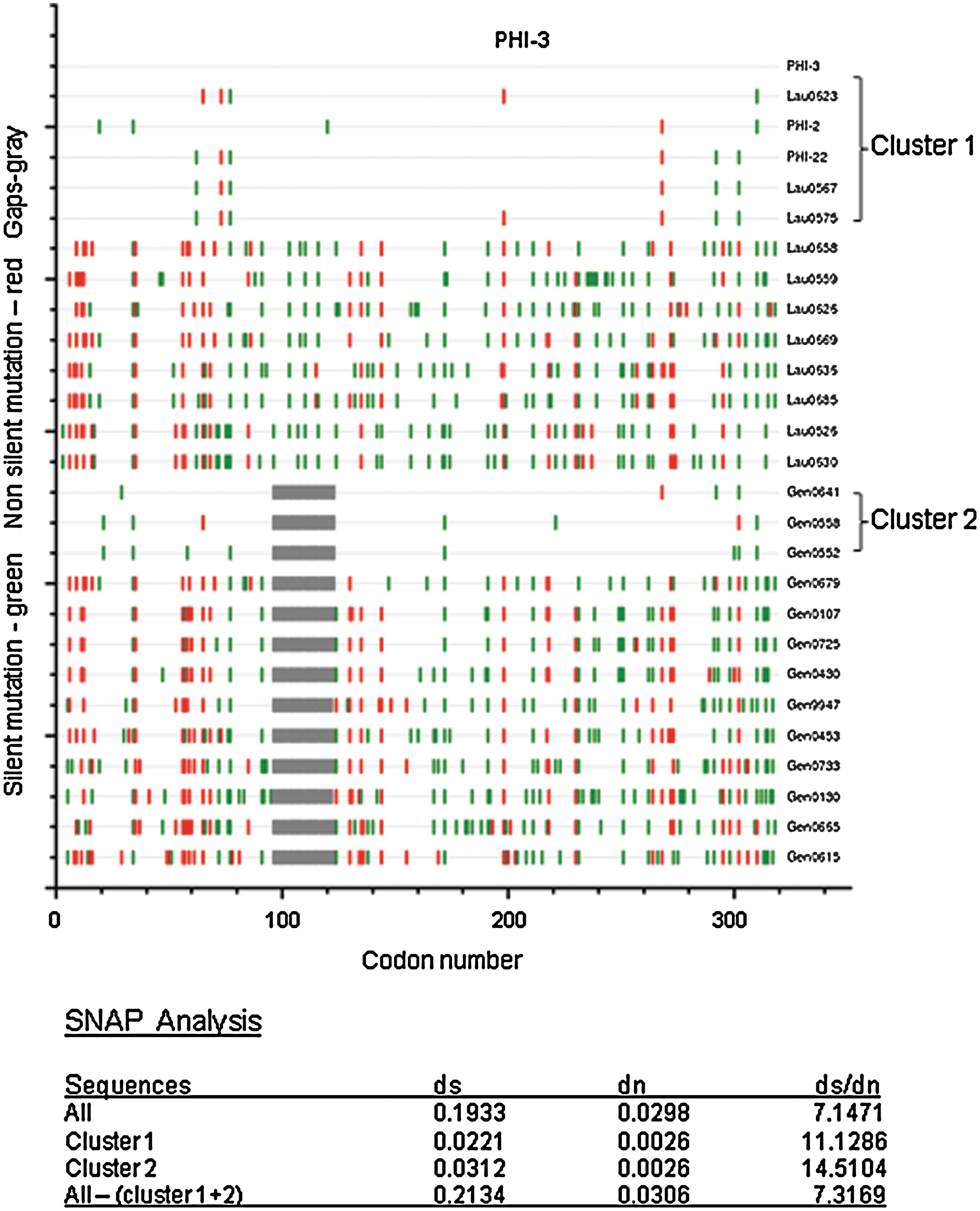
Codon-aligned sequences plot and ds/dn ratios of HIV-1 pol F1 strains from Southwest Switzerland compared to PHI-3 Lausanne cohort sequence. (Color image can be found at
Then we checked for the presence of drug resistance related mutations and polymorphisms previously found in the PHI-F1 sequences cluster 1 and 2 members. Interestingly, 103N was found in 5/6 sequences and 10V in the entire set. Additionally, all other accessory mutations were confirmed too.
Resistance mutations to NNRTI 103N, 181C, and 184V/I were previously reported as minority quasi-species in patients from Southwest Switzerland with early therapy failure using allele-specific real-time PCR in pretreatment plasma samples; but the study was limited to 4 clinical cases.
6
However, the Swiss HIV Cohort Study (SHCS) reported recently an increased rate of NNRTI resistance (3.5%) in newly diagnosed HIV infected individuals, underscoring 103N substitution as the second most common mutation from this drug class with a rate of 37% among all individuals harboring NNRTI resistance.
7
All together, these reports provide evidence to the continuous and increasing transmission of 103N mutation in the local epidemic. The 9-mer K103N mutant peptide 101–109 KK
Moreover, the extended analysis of the other HIV-1 F1 sequences obtained from untreated and nonrelated infected individuals during the same period in Geneva and Lausanne revealed the presence of two clusters with tightly related sequences as defined by nucleotide motifs and drug resistance patterns.
This study has determined a high transmission rate (16.6%) for NNRTI resistant mutation 103N in a cohort of acutely HIV infected individuals from Lausanne. Half of these new HIV infections consisted of non-B subtypes or recombinant forms. Four of the 5 men harboring F1 or D clade 103N resistant viruses were diagnosed during 2005 and one later case corresponds to a B clade infection from 2007. They all contracted HIV infection through unprotected sexual intercourse with occasional sexual partners. Three of these 5 men (two MSM and one heterosexual man) carried a highly homogeneous F1 clade distinct from previously reported F1 local sequences. Despite the small number of acutely infected patients considered in this study and the multiple clades detected, we provide strong epidemiological and molecular evidence towards an HIV-1 subtype F1 resistant strain transmission in Southwest Switzerland.
Moreover, the presence of 103N mutation in newly HIV-1 B and non-B clade infections reveals cross-clade antiretroviral resistance transmission (ART) at the local epidemic level.
These findings have relevant implications for the local molecular mapping of HIV-1 and future ART surveillance studies in the region.
Footnotes
Acknowledgments
We are grateful to all the patients who participated in this study. We thank Patricia Pochon and Myriam Giudoux from the Service of Immunology and Allergy, Centre Hospitalier Universitaire Vaudois, for technical support and graphic layout assistance, respectively. We also thank the Swiss HIV Cohort Study for some of the HIV pol sequences records used in the phylogenetic analysis and SmartGene GmBH for provisional access to the HIV analysis module.
Finally, we like to acknowledge Dr. Brian Foley from the Los Alamos HIV databases for fruitful discussion of the genotyping data.
Sequence Data
Nucleotide sequences of pol gene were obtained in all cases and deposited at the Genbank database under accession numbers GQ131596-GQ131625, respectively.
Author Disclosure Statement
No competing financial interests exist.