
Abstract
In this preliminary study we show that in 2008, 3 years after antiretroviral therapy was introduced into the Karonga District, Malawi, a greater than expected number of drug-naive individuals have been infected with HIV-1 subtype C virus harboring major and minor drug resistance mutations (DRMs). From a sample size of 40 reverse transcriptase (RT) consensus sequences from drug-naive individuals we found five showing NRTI and four showing NNRTI mutations with one individual showing both. From 29 protease consensus sequences, again from drug-naive individuals, we found evidence of minor DRMs in three. Additional major and minor DRMs were found in clonal sequences from a number of individuals that were not present in the original consensus sequences. This clearly illustrates the importance of sequencing multiple HIV-1 variants from individuals to fully assess drug resistance.
M
Karonga District is the northernmost district of Malawi and HIV-1 subtype C has been reported to be the most predominant subtype in this District. 7 ART drugs were introduced in Malawi first in the context of prevention of maternal-to-child transmission (PMTCT) through antenatal clinics (using a mother and child nevirapine regime, which is ongoing). Full ART first became available at public clinics in the two major cities in Malawi in 2003, in the regional capital in 2004, and at Karonga district hospital in June 2005, at which time Karonga residents who had been seeking care outside the district were transferred to local services. ART delivery has been largely based on clinical assessment with CD4 counting becoming available in January 2008 at one clinic. Viral load assessment and routine drug resistance testing are not available and the baseline genotype of the genes, which are the targets for ART, are unknown. The objective of this study was to investigate the presence of polymorphisms associated with drug resistance found in the reverse transcriptase (RT) and protease (PR) regions and to explore the intrapatient diversity of HIV-1 in these gene regions at a time when most individuals seen are drug naive and transmission of drug resistance from ART-treated individuals should be uncommon.
DNA was extracted from blood samples from 71 individuals living in Karonga District, Malawi using the QIAamp DNA Blood Mini Kit (QIAGEN Ltd). Extracted DNA was subjected to nested polymerase chain reaction (PCR) amplification of the HIV-1 PR and RT genes employing the primers previously described. 6 The PCR mixture contained 1.5 mM MgCl2, 1.5 units of Taq (Roche Expand High Fidelity PCR system), 0.8 mM each dNTP, and 5 pM primers (Eurofins MWG Operon). The cycling conditions were as follows: “hotstart” at 98°C for 1 min, primary denaturation at 94°C for 2 min followed by 35 cycles with 1 min denaturation at 94°C, 1 min annealing at 42°C, 4 min extension at 72°C, and final extension for 7 min at 72°C. For each gene region from 10 samples two PCR amplifications were pooled and the mixture cloned using the TOPO Cloning Kit (Invitrogen). Colony PCR and sequencing were carried out on 30 colonies per sample. Sequences obtained were edited in SeqMan (Lasergene, DNASTAR, Inc) and aligned using MacClade 4.0 (Sinauer Associates).
Preliminary phylogenetic analyses were first carried out to identify the subtype status of all sequences generated. Subsequently all consensus and clonal sequences that were subtype C were assembled into multiple alignments for each gene region using MacClade 4.0. Phylogenetic trees were reconstructed using the LANL (
Of the 71 DNA samples, amplification and sequencing were successful from 62 for RT and from 53 for PR. Three RT and six PR sequences were excluded from analysis because of poor quality and four individuals were infected with non-subtype C virus (no DRMs were present in these non-C sequences). For both gene fragments from subtype C strains phylogenetic analyses with and without DRMS present showed identical tree topologies indicating that sharing the same DRM was not, by itself, responsible for individual sequences clustering together. Figure 1 shows the relationships between sequences with DRMS included.
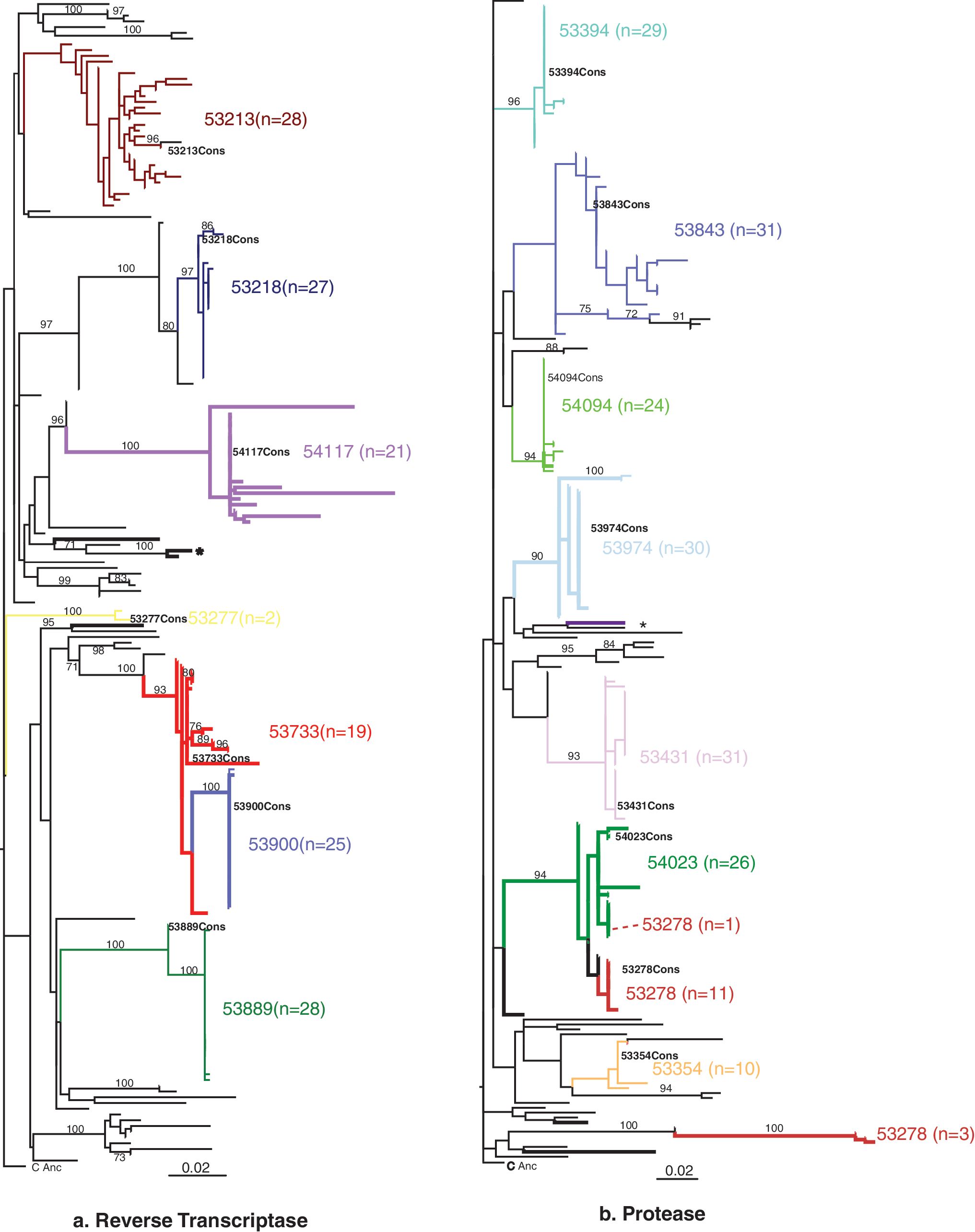
Maximum likelihood trees generated from reverse transcriptase
RTsequences included sequences from 40 individuals who were drug naive and from 17 who had been on ART. None of the drug-exposed individuals showed any DRMs, however, five (5/40) drug-naïve individuals showed nucleoside reverse transcriptase inhibitor (NRTI) resistance mutations and four (4/40) showed non-NRTI (NNRTI) resistance mutations with both types of drug resistance mutations found in one individual (Table 1). The NRTI mutations found were V75LV (one individual) and V118I (four individuals) with five NNRTI mutations identified (A98G, K101Q, V106LV, E138A, and G190R) (Table 1). V118I occurs in ∼2% of untreated persons infected with subtype C and with increased frequency in persons receiving multiple NRTIs.
9
It causes low-level resistance to 3TC and possibly to other NRTIs when present with other mutations.
10
This was the only mutation found in both previous studies of drug resistance mutations in Malawi, being present in 4 of 21 individuals studied by Petch et al.
3
and 15 of 96 individuals with confirmed virologic failure by Hosseinipour et al.
11
Of the other drug resistance mutations found K101Q minimally reduces susceptibility to each of the NNRTIs, whereas mutation at position 75 (V75T/M/A/I) is associated with reduced NRTI susceptibility (
Mutations involved in resistance to NRTI and NNRTI drugs and minor mutations involved in resistance to protease inhibitors are shown. The four shown in bold were previously drug exposed prior to enrolling in the study.
According to the Stanford database G190R is a highly unusual mutation and is flagged as an NNRTI resistance mutation. Little seems to be known about the effects of this mutation as yet in subtype C but G190A/S/E/Q/T/V/C are NNRTI resistance mutations. Similarly, although V106A causes high-level resistance to NVP and DLV and low to intermediate resistance to EFV, V106L is a rare polymorphism and its association with NNRTI resistance is less clear; E138A is responsible for decreased ETR response and A98G reduces NVP susceptibility by two- to threefold (
Cloning was successful for seven samples (Table 2). From individuals whose consensus sequence showed neither NRTI nor NNRTI drug resistance mutations, none of the clonal sequences generated showed drug resistance mutations (Table 2). All cloned sequences obtained from individual 53733 contained the NRTI V118I mutation that was found in the consensus sequence and one clonal sequence showed an additional L74K NRTI-resistant mutation. The consensus sequence retrieved from individual 54117 showed a G190R (NNRTI) mutation and also had four stop codons at positions 71, 88, 212, and 239 (according to HXB2 RT gene). All clones obtained from this individual showed the same four stop codons and the same G190R mutation. This study utilized proviral DNA to generate RT and PR sequences due to the difficulty of working with RNA in this context (i.e., getting samples from a large population study in rural Africa). HIV-1 with a G–A hypermutation at positions 88 and 212 has previously been found in resting CD4 cells and the resulting DNA is reported to be degraded or defective. 12,13 It is possible that the plasma of this individual would yield functional HIV-1. It is also possible that the individual may be infected with a defective virus and that this might affect disease progression.
Drug-exposed individuals are in bold.
Consensus sequence was different from each of the clones.
All clones and consensus contained four stop codons.
The phylogenetic relationships of the RT sequences from most individuals were straightforward with both the clonal and consensus sequences grouping together in monophyletic groups, with high bootstrap support (Fig. 1a). The clones and the consensus sequence from 53733 appear to be ancestral to the virus infecting individual 53900 with both of these individuals infected with viral strains containing the same NRTI DRM (V118I). There is no known epidemiological link between these individuals. The laboratory work on these samples was carried out by different people and therefore contamination is not likely in this case. Of the individuals for whom the PCR products were not cloned there is only one instance of clustering of sequences containing DRMs with high bootstrap support (see the asterisk in Fig. 1a). One of these individuals is a male and the other is a female. These individuals are in the same reporting group and thus may live geographically close together.
Of 45 subtype C protease sequences, 16 were from drug-exposed individuals and 29 were from drug-naive individuals. Seven consensus sequences showed minor PI mutations including three from drug-naive individuals (Table 1). Cloning was successful for eight samples, three for which the consensus had shown minor resistance sites and five for which the consensus showed no drug resistance mutations. Three drug-exposed individuals (53278, 54302, and 53974) showed minor PR resistance mutations in clonal sequences that were not seen in the consensus sequences, whereas 53278 also showed evidence of major PR resistance mutations in the sequenced clones (Table 3). Furthermore, this pattern was repeated among the drug-naive individuals with major and minor DRMs present in clones that were not present in the consensus sequence (Table 3). Clones of 53278 showed major mutations D30N, M46I, and I84T. D30N causes high-level resistance to NFV and potential low-level resistance to ATV. M46I decreases susceptibility to IDV, NFV, FPV, LPV, and ATV when present with other mutations. I84V causes intermediate to high-level resistance to various PIs. Minor mutations at position 10 (L10I/V/F/R/Y) are associated with resistance to most PIs when present with other mutations and mutation at this site occurs in 5–10% of untreated persons including subtype C-infected individuals in Africa. 4,5,11,14 Mutation at position 74 (T74S) is associated with reduced NFV susceptibility whereas mutation at position 76 (L76V) reduces susceptibility to FPV, IDV, LPV, and DRV and increases susceptibility to SQV, ATV, and TPV. 14,15
Drug-exposed indivudals are in bold.
For most individuals phylogenetic relationships between the sequences were uncomplicated; however, while 10 clones and the consensus sequence from individual 53278 clustered closely and all showed the T74S minor PI mutation, sequences of three variants clustered together in the tree with sequences retrieved from a different individual with whom she has no known epidemiological links (Fig. 1b) rather than with other 53278 sequences. These three sequence variants were those that showed major PR inhibitor resistance mutations. Furthermore, none of these three variants showed the original T74S mutation but did show another minor PI mutation (G48R). One additional clone sequence was found clustering with clonal sequences from another female, 54023, again with whom she had no known link.
This work clearly shows the presence of both NRTI and NNRTI drug resistance mutations and the presence of minor and major PR resistance mutations in a number of drug-naive individuals in rural Malawi in 2008 3 years after ART became widely available in the district. We have also shown the importance of sequencing multiple HIV-1 variants from individuals to fully assess drug resistance, as a number of individuals have shown major and/or minor drug resistance mutations in sequenced clones that were not present in the consensus sequences. Sequencing multiple clones of RT and PR from 16 individuals showed that one of them (female, aged 52, 53278 in Fig. 1b) was dual infected and showed evidence of being infected with viruses containing DRMs. The dual infection was confirmed by sequencing of multiple clones of the gag gene as part of another project, which showed a similar pattern of multiple lineages for this individual (Seager, unpublished). In terms of identifying possible transmission events involving HIV-1 with DRMs, apart from the dually infected individual mentioned above, there are very few cases of clustering of sequences from individuals with DRMS.
Our analyses showed that the presence of DRMs alone was not responsible for the pattern of relationships shown on the trees indicating common ancestry rather than convergence. However, given the short length of gene fragments used to explore drug resistance and in many cases having sequence evidence from only one gene, such results are not reliable reports of transmission and sequencing of additional loci is underway to confirm transmission in each case.
The presence of drug-resistant mutations in drug-naive individuals may prevent successful treatment of certain individuals. This knowledge may also inform policymakers in planning future therapy strategies as this indicates that additional therapy combinations may be required. Although antiretroviral drugs were made more widely available in Karonga District in June 2005, prior to this NVP was available for the PMTCT in antenatal clinics and full ART would have been available to a number of individuals elsewhere in Malawi in 2002. Thus, it is also possible that some individuals assumed to be drug naïve did not disclose (or in the case of PMTCT were not aware of ) ART exposure, and did not appear in the ART study cohorts. Furthermore, the presence of DRMs in drug-naive persons may represent natural polymorphisms and may not be suggestive of transmission of DRMs. The impact of such natural polymorphisms on the development of drug resistance in those people at commencement of ART is unknown and such individuals need to be monitored. Indeed the impact of such polymorphisms on the HIV-1 subtype C-infected population and the speed of more widespread drug resistance are also unknown. Therefore it may be important to further examine some of the individuals and mutations found during this study in view of a long-term treatment strategy in the district.
Sequence Data
All sequences have been deposited into GenBank, accession numbers HQ159410–HQ159841, and alignments employed in this work are available from the authors on request.
Footnotes
Acknowledgments
This material is based upon works supported by Science Foundation Ireland under Grant No. 07/RFP/EEEOBF424 and 08/UR/B1350. The Karonga Prevention Study is funded primarily by the Wellcome Trust, with contributions from LEPRA. Permission for the study was received from the National Health Sciences Research Committee, Malawi, and the Ethics Committee of the London School of Hygiene and Tropical Medicine, UK.
Author Disclosure Statement
No competing financial interests exist.