
Abstract
Transcriptional regulation is critical for the human immunodeficiency virus 1 (HIV-1) life cycle and is the only step at which the virus amplifies the content of its genetic information. Numerous known and still unknown transcriptional factors, both host and viral, regulate HIV-1 gene expression and latency. This article is a comprehensive review of transcription factors involved in HIV-1 gene expression and presents the significant implications of nuclear factor kappa B (NF-κB) and the HIV-1 transactivator of transcription (Tat) protein. We include recent findings on chromatin remodeling toward HIV transcription and its therapeutic implication is also discussed. The current status of small-molecular-weight compounds that affect HIV transcription is also described.
Introduction
For three decades, HIV-1 has continued to be a global health threat and its impact has been increasing. HIV, the causative agent of acquired immunodeficiency syndrome (AIDS), was first isolated in 1983. 1 Cumulative efforts from experts of the field have generated effective treatment regimens that considerably slow disease progression to AIDS. 2 –5 To date, a number of chemotherapeutic regimens against HIV-1 have been developed and used in clinical applications against the disease. However, many treatment options have failed due to a drug's low oral bioavailability, frequent side effects that are often fatal, and the rapid emergence of resistant viral strains. 6 –8 Moreover, the persistence of latent HIV-infected reservoirs has remained a formidable obstacle in the eradication of the virus. 9 –11
In a model of HIV viral replication dynamics (Fig. 1), there is a rapid turnover of both plasma virions and CD4+ lymphocytes during HIV-1 infection. Although daily virus production exists, a decay of infected cells and CD8+ T cell-mediated immune response that clears infectious viruses simultaneously occur. 12,13 Taking these into account, it indicates that HIV clearance can be ultimately achieved. This also predicts that the viral quasispecies would be generated during this amplification process, and also predicts that salient inhibition of viral replication would not only limit explosive virus replication but also eventually lead to substantial repression of virion production, thus preventing the emergence of drug-resistant viral clones.
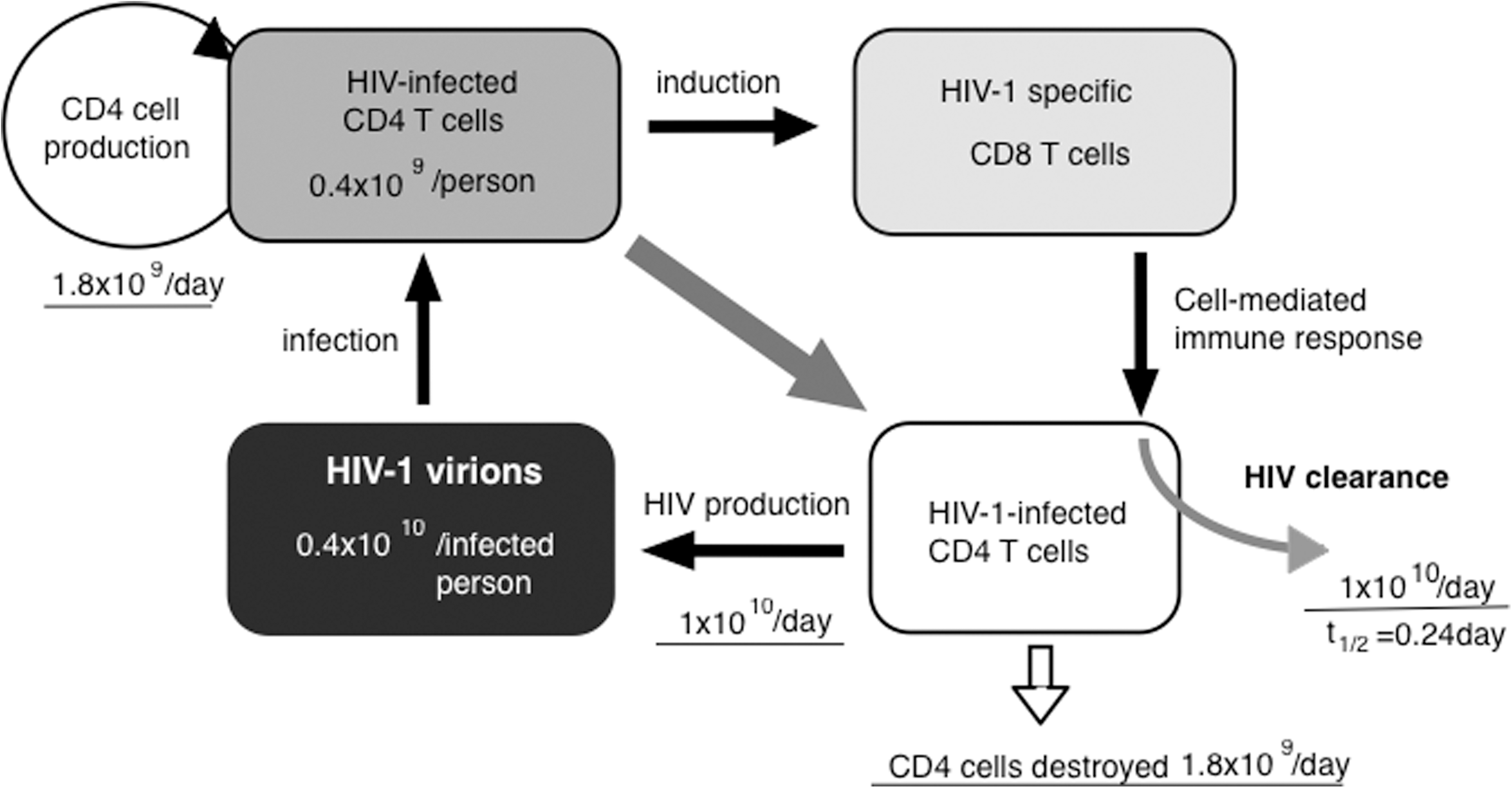
A schematic model of HIV-1 viral replication dynamics. An infected individual is estimated to harbor 0.4×10 10 virions that can infect 0.4×109 of his or her CD4 T cells. Despite this event, it is believed that the body is continually replenished with uninfected CD4 cells (1.8×109 cells/day). HIV infection triggers a cell-mediated immune response via CD8 T cell induction that can eventually facilitate viral clearance of approximately 1×1010 virions per day. Amid an approximately 1×1010 daily virus production, a corresponding 1.8×109 HIV-infected cells also undergo destruction. This diagram has been based on the findings by Perelson et al. 184
Transcription of the HIV-1 provirus is crucial for viral expression. Briefly, after the virus enters and integrates its proviral DNA into the host genome, HIV utilizes the host cell's transcriptional machinery to replicate itself. The HIV-1 long terminal repeat (LTR) contains numerous binding sites for many cellular transcription factors that lead to either upregulating or downregulating of viral gene expression and its replication. 14 However, HIV can establish latency wherein a nonproductive transcription is attributed to a repressive chromatin environment, 15 –18 insufficient levels of Tat protein for viral transactivation, 19,20 occurrences of transcriptional interference, 21 and absence of host transcription/elongation factors. 22 Accumulated evidence reveals that HIV latency can be epigenetically regulated through histone protein modifications and other posttranslational events. 23 HIV-1 transcription is therefore regulated by the intricate interplay of negative and positive transcription factors of the host and the virus. Among the numerous factors involved, this review focuses on the host transcription factor NF-κB and the viral transcription factor Tat, as well as the chromatin environment, which can be therapeutically exploited to treat HIV infection. Recently, Sigal et al. 24 have reported that multiple infection of HIV to a single cell involves cell-to-cell transmission irrespective of drug resistance, thus further highlighting the significance of viral transcription. A steadily growing number of agents regulating these two transcription factors and chromatin-related factors are presented. A potential novel approach against HIV latency is also described.
Transcriptional Landscape of HIV
The HIV-1 LTR (Fig. 2), consisting of regulatory elements for transcriptional initiation and polyadenylation flanks the integrated proviral DNA within the host genome. 25 Each LTR copy, located in both the 5′- and 3′-end of proviral DNA, is divided into three regions: U3, R, and U5. The U3 region is further functionally segmented into a modulatory region (nt −454 to −104), an enhancer (nt −105 to −79), and a basal promoter (nt −78 to −1). 14 A negative regulatory element (NRE) has been proposed to exist between nt −340 and −184 of the modulatory region because deletions of this region augmented HIV-1 LTR-directed transcription and viral replication. 26,27 There are at least three distinct transcription factors involved in HIV-1 replication at NRE: activating protein 1 (AP-1), nuclear factor of activated T cells (NFAT1), and Myb. However, since these transcription factors normally act as positive regulators, the reason behind the negative regulatory role of NRE on HIV transcription has not yet been fully elucidated. One possible reason is due to the presence of chromatin-modifying factor(s) such as histone deacetylases (HDAC) proteins or other corepressors proteins. 28 –30 Two NF-κB binding sites are found at the enhancer region of most HIV-1 isolates and are pivotal in the activation pathway of viral replication. 14 The stem-bulge-loop structure transactivation response (TAR) element is found within R (nt +1 to +60) at the 5′ end of the HIV viral mRNA, 31 which is crucial for Tat-mediated transactivation. 31,32 Multiple binding sites for several transcription factors occur in the nucleosome-free region of the HIV-1 promoter including three SP1-binding sites, TATA box, ligand-binding protein 1 (LBP-1)/Yin Yang 1 (YY1), and AP-4 sites. 33 –35 Although these factors were earlier reported to be positive regulators of transcription, they have lately been recognized to exert antagonistic action on transcription as a result of their capacity to recruit HDACs. For example (as shown in Fig. 2), Coull et al. found that the LSF-YY1 complex cooperatively recruits HDAC1 to repress HIV gene expression. 33

Schematic representation of the HIV-1 long terminal repeat (LTR). Some of the transcription factor landmarks where activators or repressors bind to regulate HIV-1 gene expression. Putative nucleosomal formations are described. The proposed action of AP-4 based on Imai and Okamoto 35 is depicted in the inset.
Several studies on the chromatin environment of the HIV-1 LTR in latently infected cells revealed that two nucleosomes, namely nuc-0 and nuc-1, are constitutively positioned in the 5′ LTR 15,16 and act as barriers to transcription. Nuc-1 position separates two nucleosome-free regions corresponding to the enhancer/promoter region (nt 200 to 465) and the regulatory region located downstream of the transcription start site. 36,37 Since the positive modular region of LTR is located upstream of the nucleosomal positions, arithmetic summation of transcription factors cannot simply explain the promoter activity. Consideration of additional factors such as chromatin status must be explored.
The LTR-associated nucleosomal architecture is disrupted by various stimuli such as tumor necrosis factor alpha (TNF-α) to undergo modifications such as acetylation and phosphorylation that foster an environment conducive to active transcription. Upon stimulation, a dynamic change of transcriptional competence of the proviral DNA occurs. Since this induction process cannot solely be ascribable to the signal-activated positive transcription factors as discussed above, the same signaling cascade might also be involved in the induction of global changes in chromatin structure. The relevance of the chromatin structures and roles of their posttranslational modifications on HIV-1 transcription and the development of a new anti-HIV therapeutic approach will be further discussed below.
Inhibitor Compounds for Transcriptional Activators
Among the cis elements present at the LTR, positive transcription factors NF-κB and Tat are significantly implicated in HIV-1 transcription and its replication. Being highly efficient inducers of HIV-1 expression (Fig. 3), both are potential targets for therapeutic intervention against the virus.
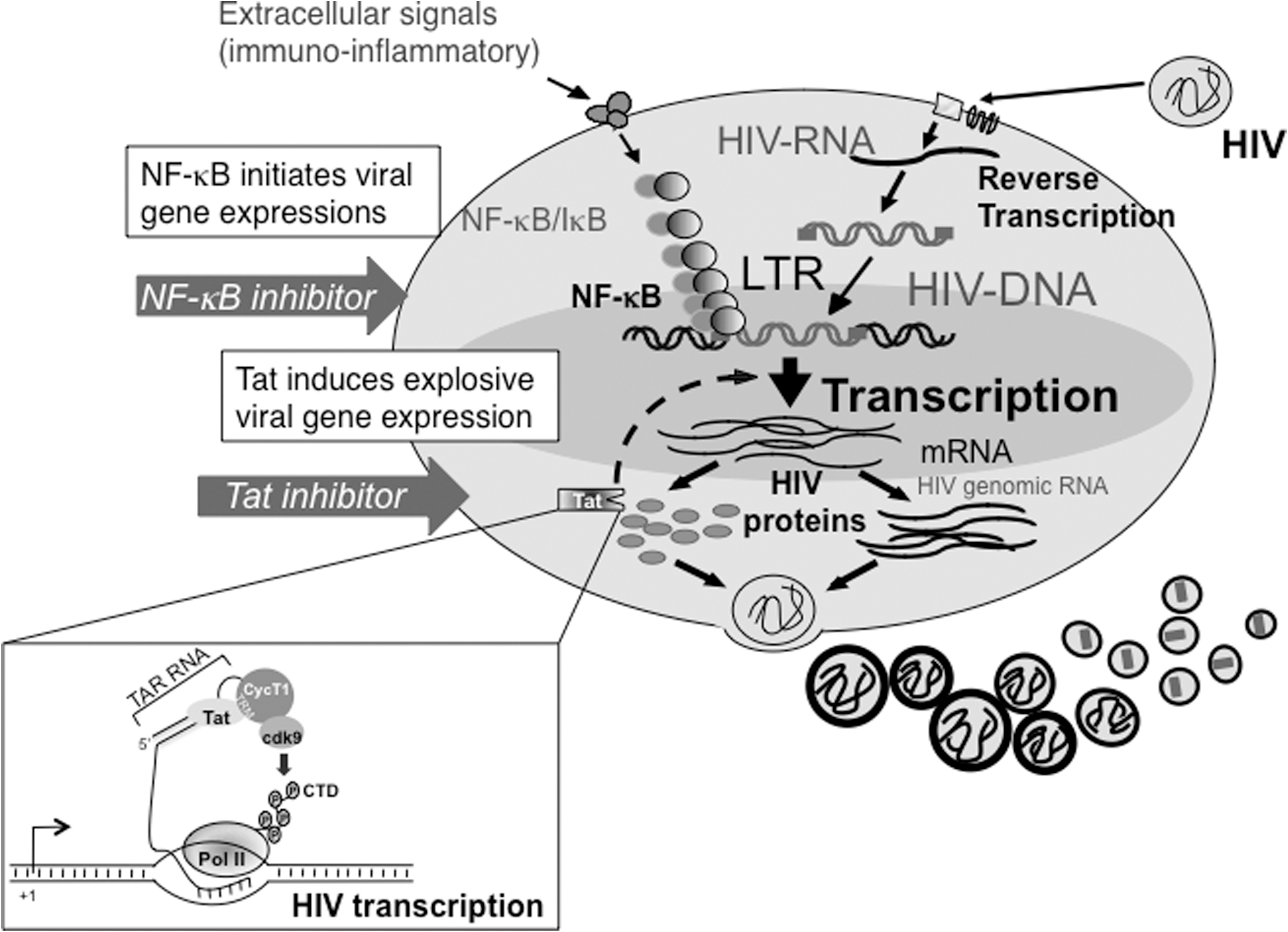
Schematic diagram of transcriptional activation of latent HIV-1. HIV-1 latency is maintained as a form of proviral DNA integrated into the host genome with little or no viral replication. Following extracellular signals, a cascade of events occurs involving NF-κB-mediated transcription and HIV-1-specific activator Tat. Tat increases transcription of the HIV-1 LTR through binding to TAR, formed at the 5′-end of all the viral mRNA species and Cyclin T1, a subunit of host transcription elongation factor P-TEFb (inset). 84,85,185 Actions of these transcription factors dramatically lead to robust HIV-1 gene expression and its replication.
NF-κB
NF-κB is a ubiquitously expressed inducible transcription factor that regulates various cellular processes and viral gene expression including that of HIV. It belongs to the Rel/NF-κB protein family, consisting of two subunits including p65/RelA, RelB, c-Rel, p50/p105, and p52/p100. NF-κB can thus form homodimers or heterodimers, the majority of which are p50/p56 heterodimers. In resting cells, NF-κB is cytoplasmically retained by IκB in its inactive state. 38,39 Activation of NF-κB by a wide array of signals dissects this signaling pathway into two (schematically depicted in Fig. 4): the classical/canonical pathway and the alternative/noncanonical pathway. 40,41
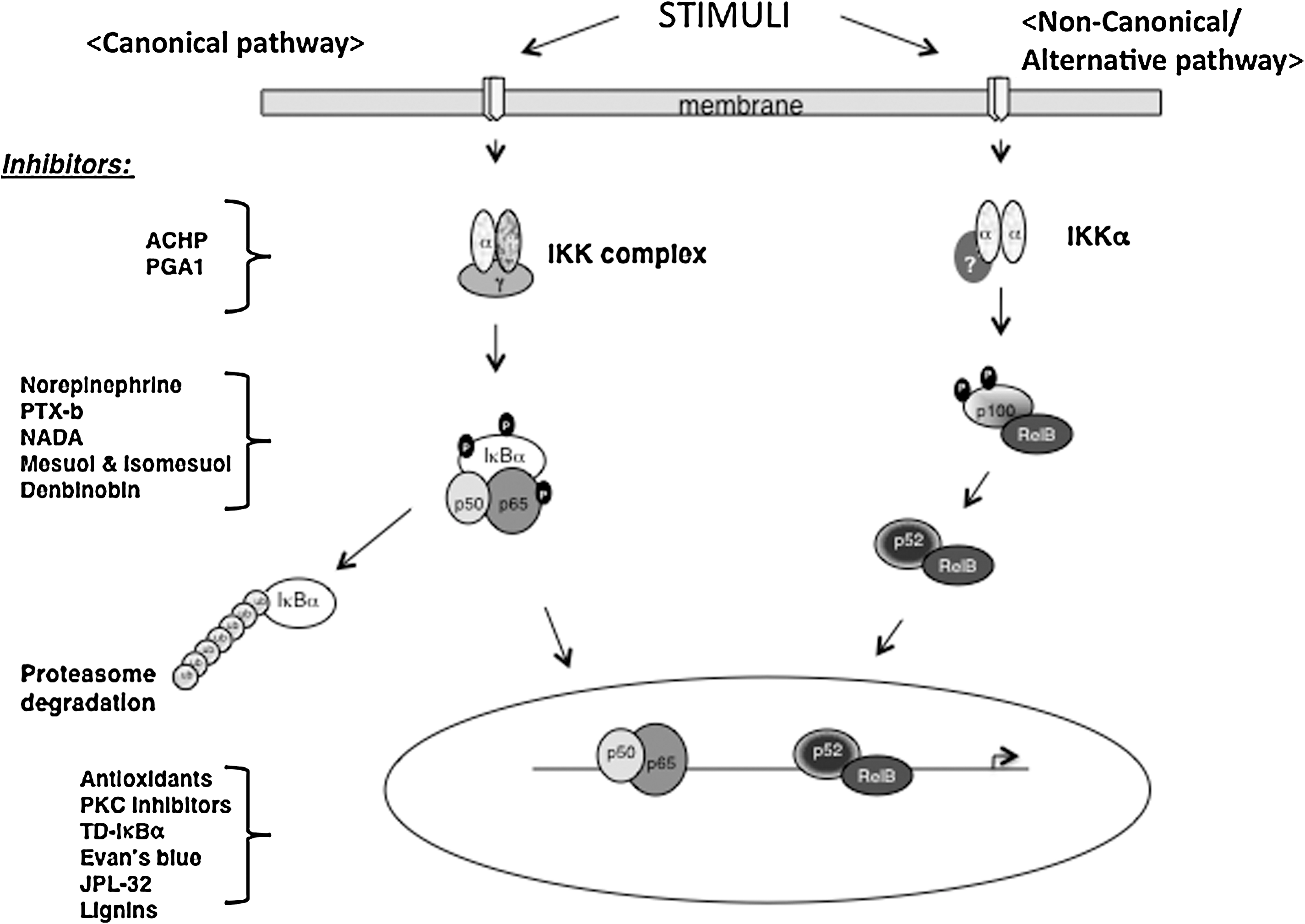
The two NF-κB activation pathways. This diagram illustrates the canonical and alternative signaling pathways leading to the activation of NF-κB. Identified target steps and inhibitors of pathways are depicted. Although most components of the IKK complex are known, little is known regarding the IKKα complex in the noncanonical pathway.
In the canonical pathway, IκB phosphorylation by IKKβ primes the protein for signal-induced degradation resulting to the release of free NF-κB. Meanwhile, in the noncanonical pathway, the IKKα-mediated phosphorylation and processing of p100 generates p52/RelB homodimers. We found that IKKα phosphorylates p65 at Ser-536, thus making NF-κB more competent for transcriptional activation. 39 In either pathway, the unmasked NF-κB proteins translocate to the nucleus to transcribe target genes.
NF-κB is a crucial regulator of HIV-1 transcription. It has the ability to orchestrate gene expression in concert with other transcription factors. In fact, minimal amounts of NF-κB could trigger efficient transcription of the viral regulatory factor such as Tat, generating massive replication of the virus. A number of compounds have been reported to inhibit HIV-1 replication in both acutely and chronically infected cells through interference with the NF-κB signaling cascade. 42
Upstream pathways that lead to NF-κB activation can be deregulated using IKK inhibitors. There are a number of specific inhibitors against IKKα and IKKβ. For instance, ACHP [2-amino-6[2-(cyclopropylmethoxy)-6-hydroxy-phenyl]-4-(piperidin- 4-yl)nicotinonitrile] 43 and cyclopentenone prostaglandin A1 (PGA1) 44 are specific for IKKβ whereas noraristeromycin (NAM) 45 is highly specific for IKKα. ACHP specifically inhibits IKKβ, and to a lesser extent IKKα but not IKKγ, and was shown to remarkably suppress the TNF-α-mediated HIV-1 production in latently infected OM10.1 cells. 43 PGA1 could inhibit herpes simplex virus (HSV)-induced IKK and NF-κB activities, block HIV-1 LTR-driven gene expression, and prevent HSV-induced HIV-1 replication in coinfected cells. 44 NAM was similarly found to inhibit the TNF-mediated HIV replication from latently infected cells. 45 Interestingly, whereas NAM effectively suppressed viral replication from chronically HIV-1-infected Molt4 cells, ACHP did not show any effect. 43,45 These compounds may have potential as adjuncts to conventional anti-HIV-1 therapy.
In addition, a trans-dominant mutant of IκBα (TD-IκBα) dramatically reduced both NF-κB binding activity and HIV-1 LTR gene expression and were most likely associated with the observed impairment of de novo HIV-1 replication, further validating the concept of NF-κB-mediated HIV transcription. 46 Similarly, norepinephrine, a catecholamine neurotransmitter, inhibits HIV infection partially through NF-κB inactivation by increasing IκBα expression. However, as contradictory reports regarding the effect of norepinephrine and/or other catecholamines on NF-κB exist, further investigations are required to clarify this point. 47
Antagonists/modulators of NF-κB phosphorylation, p65 nuclear translocation, and DNA binding activities of NF-κB proteins have also been evaluated as possible HIV inhibitors. N-Arachidonoyldopamine (NADA) blocked the phosphorylation of the p65 subunit causing downregulation of TNF-α-mediated NF-κB activation without affecting either the degradation of IκB or p65 DNA binding activity. It showed inhibitory activity against vesicular stomatitis virus-pseudotyped HIV-1 infection. 48 Although less effective as compared with others, Evans Blue (EB) could abolish the DNA binding ability of NF-κB at concentrations of 100 μM. 49 EB was shown to interact with the DNA binding region of p50, thus preventing its binding to the enhancer region of the HIV-1 LTR. Another report indicates that the B oligomer of pertussis toxin (PTX-B) could impair HIV-1 LTR-driven luciferase gene expression in U937 promonocytic cells. This effect was shown to be independent of the actions of either IκBα or Tat, and was not restricted to the HIV-1 LTR promoter. PTX-B activity was mediated through suppression of the phosphorylation and nuclear translocation of p65. 50
Several other chemicals extracted from natural compounds have been shown to inhibit NF-κB and HIV, although their precise mechanisms of action are yet to be identified. For instance, plant-derived substances such as mesuol and isomesuol, two 4-phenyl coumarins isolated from Marila pluricostata, inhibited HIV replication in Jurkat cells by interfering with the phosphorylation and transcriptional activity of the NF-κB p65. 51 Denbinobin, a naturally occurring 1,4-phenanthrenequinone from a variety of Cannabis sativa, exhibited inhibitory effects in Jurkat cells by affecting IκB phosphorylation and its degradation. 52 Cepharanthine, a biscoclaurine alkaloid isolated from Stephania cepharantha Hayata, strongly inhibited HIV-1 replication in a chronically infected monocytic cell line by suppressing NF-κB activation. 53 Lignins, especially the low-molecular-weight molecular form, strongly inhibited NF-κB and HIV-1 replication. Small molecules having lignin-like structural moiety may have benefit for the anti-NF-κB and anti-HIV approach due to its higher cell permeability. 54
As highly oxidative environments induce NF-κB activity and HIV replication, the anti-HIV effects of antioxidants were explored. N-Acetyl-l-cysteine, 55,56 trimidox (TD), 56 cepharantine, 53 α-lipoic acid, 57 α-tocopherol (vitamin E), 58,59 and butylated hydroxyanisole (BHA) 58 were found to block cytokine-stimulated HIV-1 LTR gene expression and viral replication. The prototype pyridine N-oxide derivative, JPL-32, suppressed the TNF-α-induced reactivation of HIV-1 in latently infected cell lines. JPL-32, whose 50% effective concentration was about 0.24 μg/ml, inhibited neither the release and degradation of IκBα nor the nuclear translocation of NF-κB. It, however, oxidized the thiol groups on the “redox-sensitive” cysteine residue within the p50 subunit of NF-κB, resulting in the inhibition of DNA binding of nuclear NF-κB. 60,61 Interestingly, a latency-reversing agent 5-hydroxynaphthalene-1,4-dione (5HN) has been found to activate latent HIV-1 through radical oxygen intermediates and NF-κB without affecting NFAT or protein kinase C (PKC), demonstrating a strategy that does not induce global T cell activation. 62
Other events leading to activation of NF-κB-dependent gene transcription may also represent valuable therapeutic targets. Fasudil hydrocholoride, a serine/threonine inhibitor that is clinically used in the treatment of vasospasm after subarachnoid hemorrhage, could efficiently block TNF-mediated HIV replication in latently infected monocytic cell lines. 63 The PKC inhibitors known include rottlerin, 64 staurosporine, Go 6976, and pentoxifylline (PTX) [1-(5′-oxohexyl)-3,7-dimethylxanthine]. 65 Rottlerin could inhibit HIV-1 replication in chronically infected MT-2 (IC50=5.2 μM), Jurkat (IC50=2.2 μM), and peripheral blood lymphocytes (IC50=4.4 μM). It could dephosphorylate PKC at Thr-538 in the kinase catalytic domain, thus inhibiting PKC translocation to the lipid rafts and eventually repressing NF-κB and HIV. 64 The indolocarbazole inhibitor Go 6976 and its derivatives were reported to abrogate PKC-mediated HIV-1 reactivation in latently infected cells. 65
Since NF-κB regulates a wide array of host cellular genes, any therapeutic strategy attempting to restrict or allow HIV-1 transcription and gene expression by exploiting the NF-κB signaling pathway must be undertaken with precaution to avoid potential adverse effects. Aberrant NF-κB activity has been associated with many chronic diseases and different types of cancer. 66 –69 The canonical NF-κB pathway has been generally associated with innate immunity and cell survival, and its inactivation can lead to inflammation. 69,70 On the other hand, the noncanonical/alternative NF-κB pathway is essential for lymphoid organogenesis and B cell function. 71 –73 Deregulation of this pathway can contribute to the development of several lymphoid malignancies. 74,75 Thus, correction of aberrant NF-κB activation by appropriate inhibitors would not only block HIV-1 reactivation but also prevent the occurrence of such malignancies.
Tat
The HIV transactivator of transcription (Tat) protein is a virus-encoded 86–101 amino acid regulatory protein that is essential for the replication and pathogenicity of the virus. 76,77 It has six different regions including the N-terminal putative α-helical region, a cysteine-rich region (amino acids 22–37); a core sequence (37–48); a basic RNA-binding region (49–59); and a glutamine-rich region located at the carboxyl-terminus end. Another mutational analysis has divided Tat into two functionally domains: residues 1–47 as an activation or cofactor-binding domain containing a highly conserved sequence and residues 49–59 as a basic domain that is required for both RNA binding and nuclear transport of Tat. 78
Tat commences HIV-1 transcription by recruiting the positive transcription elongation factor (P-TEFb) complex, a heterodimer of human cyclinT1 (hCycT1) and cyclin-dependent kinase 9 (CDK9), to the transactivation response element (TAR) located in the 5′ region of HIV viral mRNAs (Fig. 3, inset). The Tat/TAR/P-TEFb complex binding to the HIV-1 promoter allows CDK9, apart from inducing autophosphorylation of P-TEFb, to hyperphosphorylate the C-terminal domain of RNA polymerase II resulting to efficient transcriptional elongation. 79 –85 Interestingly, Tat action requires the presence of strong cellular transcriptional activators such as Sp1 and NF-κB. 86,87 In the absence of Tat or cellular activation signals, transcription fails to further proceed before RNA pol II reaches the poly(A) signal within the 3′ LTR (known as “premature termination”) 83 because the negative transcription elongation factor (N-TEF), consisted of two subunits of negative elongation factor (NELF) and 5,6-dichloro-1-β-d-ribofuranosyl-benzimidazole (DRB)-sensitive inducing factors (DSIF), associates with RNA pol II resulting in abortive short viral transcripts. 88 –90
The extreme importance of the Tat-mediated transactivation cascade in HIV replication, as well as in the emergence of HIV-1 drug-resistant strains through the generation of abundant viral pseudospecies, makes it one of the most wanted targets for transcriptional therapy against the disease. Several approaches have been attempted and designed targeting specific viral and cellular factors that modulate the process. However, although initial studies were successful in identifying Tat inhibitors such as DRB 91 and 7-chloro-5-(2-pyrryl)-3H-1,4-benzodiazepine-2(H)-one (Ro-5-3335), 92 none of these compounds was considered for clinical use due to the narrow therapeutic window and toxicity issues. Thus, the search of Tat-specific inhibitor compounds is still desperately needed.
Agents against Tat protein directly or indirectly bind to Tat and could inhibit its extracellular uptake and, consequently, its HIV-LTR transactivating activity. 93 The quinoline derivatives are one of the prospective lead compounds for the development of Tat inhibitors. We previously reported that fluoroquinoline derivatives 7-(3,4-dehydro-4-phenyl-1-piperidinyl)-1,4-dihydro-6-fluoro-1-methyl-8- trifluoromethyl-4-oxoquinoline-3-carboxylic acid (K-37) and 8-difluoromethoxy-1,4-dihydro-6-fluoro-7-(3,4-dehydro-4- phenyl-1-piperidinyl)1-[4,(1,2,4-triazol-1-yl)methylphenyl]- 4-oxoquinoline-3-carboxylic acid (K-38) were found to be potent inhibitors of HIV-1 replication with an EC50 of 27 and 3.8 nM for HIV-1IIIB in peripheral blood mononuclear cells (PBMCs), respectively. In chronically infected cells, such as OM10.1, K-38 proved to be more potent (EC50=17 nM) compared to the previously identified K-12 (EC50=50 nM). K-37 and K-38 could also inhibit cytokine production and ICAM-1 expression. However, both compounds showed more cytotoxicity than K-12 in various cell lines as well as PBMCs. 94,95 Novel quinoline derivatives have been reported to inhibit Tat-dependent HIV-1-LTR chloramphenicol acetyltransferase (CAT) gene expression and had potent inhibition activity against SIV replication with low cytotoxicities. 96 Molecular modeling in silico suggested that these compounds could change the Tat conformation by binding to it, eventually disrupting Tat–TAR complex formation. Indirect inhibitory effect on Tat by PTX-B has also been reported. 97
Conceptually, disruption of the Tat–TAR interaction would inevitably inhibit the Tat transactivation function. Most compounds, reported so far, could block Tat-mediated transcription by acting as a competitor to Tat binding to the bulge region of TAR RNA. Based on their chemical properties, these agents were divided into peptide-based, oligonucleotide-based, and small molecules compound. 98 These compounds were designed analogous to the Tat RNA-binding domain, peptides containing modified sequences of Tat or amino-glycoside-arginine conjugates. 37,95,98
Other compounds could suppress Tat function by blocking or modifying one component of Tat's interacting partner P-TEFb. Flavopiridol, a CDK inhibitor, hindered Tat transactivation by restraining P-TEFb kinase activity. 99,100 The clinical application of this drug, however, was limited by its toxicity. A series of flavopiridol analogues has recently been described as having antiviral potency comparable to that of flavopiridol, but with significantly reduced cytotoxicity. 101 DRB successfully blocked Tat-activated transcriptional elongation in vitro and Tat transactivation in cell culture. 102 The use of small-molecular-weight compounds or mutant proteins to impair cyclin-dependent kinases activities 103 –106 or use of anti-hCycT1 human single chain antibodies 107 could also abrogate HIV-1 replication. CDK inhibitors highly selective against CDK9 appear to be desirable but still require further investigations.
Tat also undergoes multiple posttranslational modifications because of its ability to bind deacetylases and acetyltransferases. 6,108,109 Apparently, the acetylation-deacetylation state of Tat can regulate its transcriptional activity, making its modification state a good resort to inactivate Tat. Additionally, pretreatment of resveratrol, an SIRT1 inhibitor, attenuated Tat-mediated HIV-1 transactivation through modulating the redox status of the cell. 110
Although its precise mechanism of action is yet to be identified, the 1-8-naphthyridone derivative (HM13N) has been shown to inhibit HIV-1 Tat-mediated transcription. It displayed very potent and selective anti-HIV activity in acutely (EC50 0.0.2-0.3 μg/ml), chronically (EC50 0.032 μg/ml), and latently infected cells (0.003-0.004 μg/ml). Because this compound did not show any tendency to select for resistance mutations, one of host factors is considered as its target. 111 In addition, derivatives 5 and 6 of a first series of 2-phenylquinolones (2-PQs) designed to obstruct the Tat–TAR interaction exhibited a residual dose-dependent anti-HIV activity (IC50=75 and 80 μM, respectively) coupled with lower or null cytotoxicity (CC50=90 and >100 μM, respectively). 112
Recently, GSK3-beta inhibitors were identified as inhibiting HIV-mediated transcription and protecting against Tat-induced neurotoxicity. 6BIO and its derivative, 6BIOder, exhibited an IC50 of 40 nM and 0.5 nM in TZM-bl cells and astrocytes, respectively. 113 Target molecules of these compound are yet to be identified.
Although several of the Tat inhibitors offer great potential to be a drug candidate, none has emerged relevant for clinical use due to narrow therapeutic windows. An anti-Tat approach may be further restrained by the presence of NF-κB, which can support replication of even a Tat-defective HIV. 114 Nevertheless, because of its pivotal role in viral replication, Tat is still highly regarded as an attractive target and may hold significant promise in a combination strategy against HIV. Recently, the x-ray crystallography of P-TEFb in complex with Tat has been reported 115 and this development should foster the structure-based design of transcriptional inhibitors.
Transcriptional Inhibitors Acting Through the Remodeling of Chromatin Structure
Epigenetic control of HIV latency
Several host transcription factors have been identified to downregulate HIV-1 transcription. NF-κB p50 recruits histone deacetylases (HDACs) to the LTR and represses HIV-1 transcription. 17 Upon appropriate stimulation signal, the free, active NF-κB heterodimer (p50:p65) translocates into the nucleus and displaces the p50 homodimers constitutively bound to LTR. This event results in the recruitment of histone acetyltransferases, like CBP/p300 and P-TEFb, which ensure efficient transcriptional initiation and elongation. 116,117
AP-4, a ubiquitously expressed transcription factor of the basic helix-loop-helix leucine-zipper (bHLH-Zip) subgroup of bHLH proteins, is constitutively present on the silent HIV-1 promoter in latently infected cells. 35 It negatively regulates HIV-1 transcription through recruitment of HDAC1 to the promoter as well as by masking the TATA-binding protein to the TATA box. Other host transcription factors, such as the LSF-YY1 complex, 118 C-promoter binding factor 1, 119 and Sp1-c-Myc complex, 120 also recruit HDAC proteins to the HIV-1 LTR and exert repressive effects on viral transcription.
Apart from HDAC recruitment, some regulatory proteins act on other transcription factors to downregulate HIV transcription. The cellular factor LBP1 prevents the binding of TFIID, thus restricting the processivity of RNA polymerase II on the HIV-1 promoter. This effect of LBP-1 on HIV-1 transcription is relieved by Tat, which enhances transcription. 121
HDAC as a potential therapeutic target
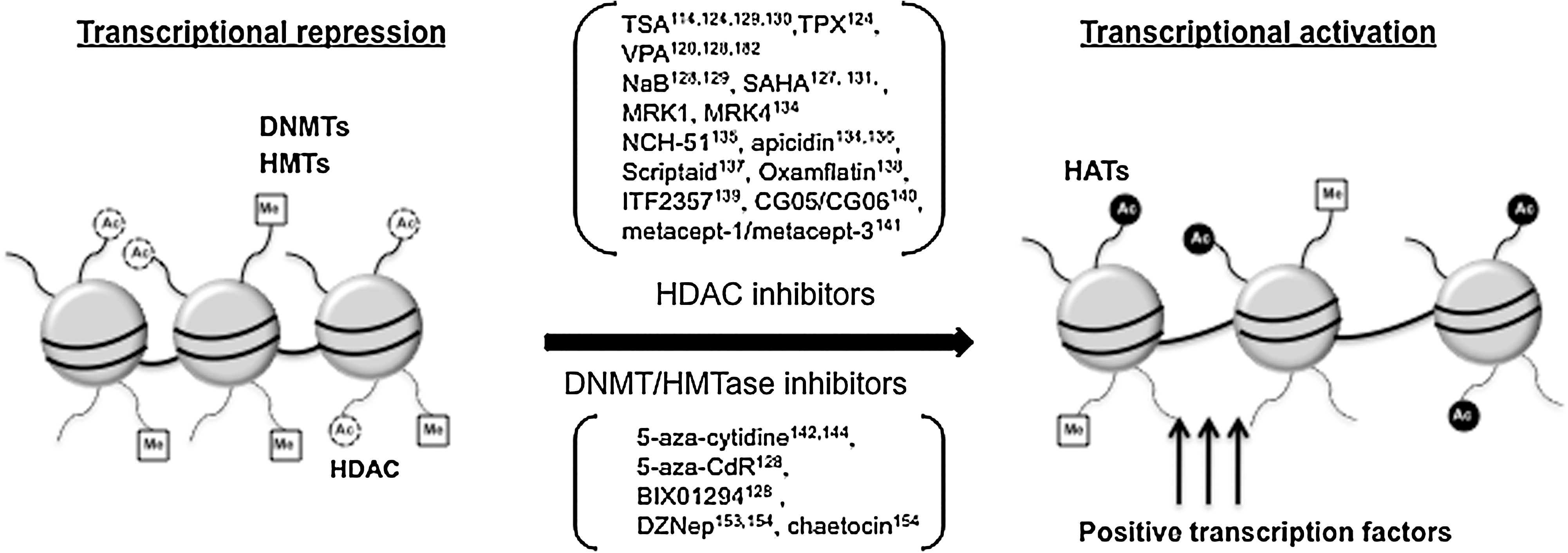
The advent of epigenetics has paved the way to elucidate the mechanisms underlying HIV latency. Dynamic modifications of chromatin structure and nucleosome remodeling have been implicated in HIV-1 transcription. Hyperacetylation of histones is correlated with transcriptional activation, whereas hypoacetylation of histones by HDAC proteins is associated with transcriptional repression. 122 –124 HDACs are lysine deacetylases that repress transcription by modification of the lysine tails of histones within chromatin nucleosomes through deacetylation. HDACs are grouped into four classes: class I HDACs (HDAC1, 2, 3, and 8) are proteins orthologous to the yeast Rpd3 enzyme; class II is subdivided into IIa (HDAC4, 5, 7, and 9) and IIb (HDAC6 and 10); HDAC11 is the sole member of class IV and has properties of both class I and II; and class III deacetylases represent the nicotinamide adenine dinucleotide (NAD)-dependent sirtuins. 125,126
Nucleosome 1 (nuc-1), located at the −20 to +140 position of the viral LTR, impedes HIV-1 transcription in silent proviruses. The deacetylation of histones conferred by HDACs diminishes the accessibility of transcription factors to the viral DNA, thus generating a repressive state. In addition, HDACs are able to interact with a variety of nonhistone proteins, including transcription factors, in mediating transcriptional repression. However, this restrictive state can be reversed to a permissive state by inhibiting the action of HDACs that results in the concomitant accumulation of positive regulators such as TATA-box binding protein (TBP) to the LTR. Interestingly, we found the presence of an AP-4-HDAC1 corepressor complex at the LTR. Due to the close proximity of TBP to AP-4 binding sites, we hypothesized that AP-4 could have masked TBP binding to the HIV-1 TATA box (the inset of Fig. 2), thereby causing full inactivation. 34 Recently, a consensus matrix attachment region element has been found in the HIV-1 LTR where the scaffold/matrix attachment region-binding protein 1 (SMAR1) protein binds and reinforces transcriptional silencing by recruiting the HDAC1–mSin3 complex and by tethering the LTR to the nuclear matrix. 29
Several studies have established that treatment of HDAC inhibitors (HDACis), such as trichostatin A (TSA), trapoxin (TPX), valproic acid (VPA), sodium butyrate (NaB), or suberoylanilide hydroxamic acid (SAHA), in various latently infected cell lines and primary cells could induce HIV-1 gene expression. 124,127 –131 HDACis induce global hyperacetylation of histones and loss of HDAC occupancy in the viral LTR resulting in HIV promoter activation in the absence of NF-κB induction. 124,131 –133 Inhibitors selective for class I HDACs were found to be more efficient activators of the LTR than inhibitors that target class II HDACs. 134
In addition, a number of new chemicals have recently been reported to revert HIV-1 latency by inhibiting HDAC. These chemicals include NCH-51, 135 apicidin, 136 scriptaid, 137 oxamflatin, 138 ITF2357, 139 CG05/CG06, 140 and metacept-1 and -3. 141 For instance, we reported induction of HIV-1 replication from latently infected cell lines by HDACi NCH-51, where SAHA was modified by substituting hydroxamic acid with nonhydroxamate moiety and the acylated thiol group to obtain better pharmacokinetics and less toxicity. 135
Methyltransferases as potential therapeutic target
Recent evidence incriminates methylation, another posttranslational or epigenetic modification, as a regulator of HIV-1 transcription and latency. Methylation is associated with both activation and silencing. Methyltransferases can transfer methyl groups to arginine, lysine, histidine, and proline as well as carboxyl groups. 122 The bulk of the studies reported so far involved protein Lys and Arg methyltransferases.
Transcriptional inactivation by methylation was initially illustrated by Bednarik et al. 142 showing the reactivation of a stably integrated HIV-1 LTR-CAT plasmid by the methyl inhibitor 5-aza-cytidine. Later studies further implicated protein methylation with HIV transcription. 143 –146 The use of a general methyltransferase inhibitor with an adenosine analog, adenosine periodate, was demonstrated to have increased virion production in less infective cell lines. 147
Although histone methylation does not affect DNA/histone interactions, it serves as a recognition template for effector proteins modifying the chromatin environment. 148 Trimethylation of Lys-9 of histone H3 (H3K9) mediated by the histone-lysine N-methyltransferase suppressor of variegation 3-9 homologue 1 (SUV39H1) has been correlated with HIV-1 silencing by recruiting heterochromatin protein 1 homologue-γ (HP-1 γ). 149,150 Upon stimulation, trimethylated H3 levels as well as HP-1 γ and HDAC occupancy at the HIV-1 LTR were abolished. 151 Concomitant recruitment of the HDAC1, HDAC2, SUV39H1, and the HP1 proteins to the viral promoter by corepressor CTIP2 [chicken ovalbumin upstream promoter-transcription factor (COUPTF)-interacting protein 2] establishes a repressive chromatin structure, thus silencing HIV-1 proviruses. 152
We have previously reported the involvement of H3K9 methytransferase G9a in the transcriptional quiescence of latent HIV-1 provirus by promoting repressive dimethylation at H3K9. 153 siRNA against G9a and use of the G9a inhibitor, BIX01294, reactivated HIV-1 gene expression in pNL4-3-transfected cells and in latently infected ACH-2 and OM10.1 cells. Meanwhile, Friedman et al. 153 recently found that knocking down HKMT Enhancer of Zeste 2 (EZH2), the enzyme responsible for trimethyl histone Lys-27 (H3K27me3) synthesis, and treating latently infected cells with the EZH2-specific HKMT inhibitor 3-deazaneplanocin A (DZNep), would lead to the reactivation of silenced proviruses. 154
Apart from histone methylation, protein and DNA methylation also influence HIV-1 replication. Interestingly, the viral factor Tat has been demonstrated to be methylated on Arg-52 and -53, and Lys-50 and -51 by PRMT6 155,156 and SETDB1, 157 respectively. Methylation on these specific amino residues inhibited Tat-dependent transcription. DNA methylation impedes the recruitment of SP1 and NF-κB proteins to the LTR and promotes viral latency. 158 –160 H3K27 trimethylation and CpG island methylation also hinder proviral integration. 161,162
Flush Out Therapy
HIV latency poses a major obstacle in the eradication of the virus from infected individuals. Several therapeutic strategies have been attempted without promising results. The massive progress in understanding the mechanisms underlying the epigenetic regulation of gene expression reveals new concepts for treating diseases such as HIV.
A few experts in the field proposed the use of activators that will flush HIV out from its hiding places, thus making the reactivated virus susceptible to antiretroviral therapy (ART). The use of HDACis or methyltransferase inhibitors that modify chromatin conformation has shown promising results (Fig. 5). Margolis and colleagues demonstrated this proof-of-concept theory where the coadministration of VPA together with enfurvitide, an HIV fusion inhibitor, resulted in the depletion of latently infected CD4+ T cells from aviremic HIV-infected individuals. 163
Epigenetic regulation of HIV-1 latency. DNA methylation and methylation/deacetylation of precisely positioned nucleosomes on the HIV-1 LTR induce proviral silencing as a consequence of a repressive chromatin state. Inhibitors of methyltransferases and deacetylases are indicated. References of various inducers and inhibitors involved in this epigenetic modification are indicated.
Different activators of proviral latency, either in synergy with HDACis or alone, have been described. The non-tumor-promoting NF-κB inducer prostratin, in combination with either VPA or SAHA, led to a synergistic activation of HIV latent virus in promonocytic U1 cells and J-Lat T cells accompanied by the remodeling of nuc-1. 164,165 Although prostratin reactivates HIV through PKC activation and induction of NF-κB and Sp1, it could also downregulate HIV receptors, giving it an advantage in decreasing the risk of reinfection. 166 The combination of TSA with an NF-κB inducer, such as TNF-α, also transactivates the HIV-1 promoter in U1 cells. 129,167
Hexamethylbisacetamide (HMBA), a hybrid bipolar compound, remodels the HIV-1 promoter and induces HIV expression in latently infected cells by phosphorylating the C-terminal domain of RNAP II in a Tat-independent but CDK9-dependent manner. 168,169 However, HMBA neither inhibits HDACs nor increases histone acetylation. 170 Contreras et al. 171 demonstrated that HMBA could also activate the Akt/PI3k pathway leading to the concomitant disruption of the HEXIM1-7SKsnRNA-P-TEFb complex, which results in the recruitment of the active P-TEFb in the HIV-1 promoter to stimulate viral transcription elongation.
The use of cytokines with an ART cocktail in disrupting proviral quiescence has resulted in contrasting results. The initial finding of Chun et al. 172 could isolate the virus neither in peripheral blood CD4+ T cells nor in lymph node biopsies from HIV patients receiving interleukin (IL)-2 and ART. Meanwhile, the Stellbrink et al. 173 study concluded that combined treatment with IL-2 and ART could accelerate normalization of CD4 T cell counts but had no beneficial effect on viral production or latency. Recently, IL-7, a pleiotropic cytokine, has been found to positively induce HIV proviral reactivation from resting CD4+ T cells from ART-treated HIV-infected individuals through the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway. 174
An HIV-1-reactivating protein factor secreted by the nonpathogenic bacterium Massilia timonae was recently found to induce a high but nonsustained peak of NF-κB, whereby it generated minimum amounts of Tat protein that triggered HIV-1 transactivation and subsequent HIV-1 expression. Efficient reactivation of latent HIV-1 provirus was achieved without activating multiple NF-κB-regulated proinflammatory genes, thus a positive feedback amplification was unobserved. 19
Other strategies for the selective killing of infected cells employed either a combination of antibodies against CD3 and IL-2 or of an immunotoxin (such as DAB389CD4 or OKT3) and an activator (IL-2), but had shown limited success due to toxicity. 175 –178
While the feasibility of this approach has been demonstrated, others have failed to show a decay of infected resting CD4+ T cell latent reservoir following VPA treatment. 179 –182 Thus, it remains controversial and warrants further investigation.
Conclusions
ART therapy has been successful in controlling HIV infection. However, prolonged use of antiretroviral therapy would result in adverse effects such as toxicity problems. Viral rebounds from latent reservoirs arise upon cessation of ART. Hence, the cure for HIV infection apparently relies on the elimination of the persistent HIV proviral genome.
HIV infection is under the control of both viral and host factors. Transcriptional targeting of either factor has its benefits and ill effects. For example, direct targeting of viral elements may have the benefit of specificity. Meanwhile, strategies that target host cellular proteins are considered plausible ways to circumvent the problem of mutations and the generation of drug-resistant viral clones. However, such approaches may be compromised with unwanted effects that could be fatal to the host. The risk–benefit balance is always an issue of the transcriptional inhibitors targeting the host factors. However, the benefits outweigh the risks if such inhibitors were applied for short-term therapy, such as flush out therapy. Notably, Wolschendorf et al. 19 recently showed that HIV-1 reactivation can be attained without cytokine gene induction by applying only a short NF-κB pulse, indicating that events governing gene regulation might be separated. Also, we may be able to learn the adverse effects of transcriptional inhibitors, especially HDACis, from the oncology field because it is often the case that these inhibitors show antitumor activities, and the clinical trials against cancer precede those against HIV infection. In fact, a phase I study has been reported for valproate. 183 It has become evident that to fully eradicate the virus, a cocktail of interventions is needed. Thus, a better understanding of the intricate play of host and HIV-related transcription factors together with their effects on viral dynamics in vivo would open new avenues for the treatment of AIDS and for the development of novel therapeutic agents with high efficacy and safety.
Footnotes
Author Disclosure Statement
No competing financial interests exist.