
Abstract
We report here a novel HIV-1 second-generation recombinant form (CRF01_AE/CRF07_BC) composed of CRF01_AE and CRF07_BC, identified among men who have sex with men (MSM) in Jilin, with four breakpoints observed in the pol, vif, and vpr genes. The CRF01_AE regions of the recombinant were clustered with the CRF01_AE lineage, which is mainly circulating among MSM in northern China, with the support of 100% bootstrap value, indicating that the parental origin of the CRF01_AE regions was from MSM, in which recombination events may be more likely to occur. To the best of our knowledge, this is the first detection of a novel HIV-1 second-generation recombinant form (CRF01AE/CRF07_BC) in Jilin, which indicates active transmission networks of HIV-1 infection among MSM in the region. Therefore, it is necessary to continue monitoring the molecular epidemiology of HIV-1 among MSM in Jilin to obtain a better understanding of the transmission and potential public health impact of HIV-1 among MSM in the region.
C
Reports about China in recent years have showed that the proportion of HIV infection among men who have sex with men (MSM) has increased rapidly, and CRF01_AE, CRF07_BC, and subtype B (U.S.-European origin) were the three predominant genotypes among MSM in China. In this study, we detected a novel HIV-1 second-generation recombinant form (CRF01_AE/CRF07_BC) among MSM in Jilin by near full-length genome (NFLG) sequence analyses, which is different from the CRF01_AE/CRF07_BC recombinants previously reported.
The subject, from whom JL070032 was obtained in this study, was a Chinese citizen and was confirmed to be HIV-1 antibody positive in 2006. The subject, residing in Changchun city of Jilin province, was recruited during our nationwide cross-sectional HIV-1 molecular epidemiology survey in 2007, from the Changchun Center for Disease Control and Prevention (CDC). The subject was a 38-year-old male infected with HIV-1 with only one high-risk factor (homosexual behavior). His CD4+ T cell number and viral load information were not available when his blood sample was collected. The study was approved by the institutional review boards of the National Center for AIDS/STD Control and Prevention, China CDC. Written informed consent was obtained from the subject at the time of sample collection.
Viral RNA was extracted from plasma using the QIAamp Viral RNA Mini kit (Qiagen, Valencia, CA) and reverse transcribed by SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. The NFLG sequence was obtained, using the near-endpoint diluted cDNA template and single-genome amplification (SGA) methods with one data set of primers previously described.
7
Amplicons were purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA) and then sequenced directly by an ABI 3730XL automated sequencer using BigDye terminators (Applied Biosystems, Foster City, CA). All sequenced data were cleaned and assembled using Sequencher v.5.1 (Gene Codes Corporation, Ann Arbor, MI). To avoid potential laboratory cross-contamination, the NFLG was queried against all sequences obtained in the laboratory. The NFLG sequence was then aligned against all known HIV-1 group M reference sequences representing subtypes/subsubtypes (A1, A2, B, C, D, F1, F2, G, H, J, and K) and CRFs relevant to our study in China (CRF01_AE, CRF07_BC, and CRF08_BC), as well as all NFLG sequences of CRF01_AE, obtained from the Los Alamos HIV Sequence Database (
The NFLG sequence from JL070032 was 9,091 bp (HXB2 nucleotide sequence numbering: 575–9,628) in size, spanning the noncoding region (NCR), the gag, pol, env, tat, rev, vif, vpr, vpu, and nef genes, and a 3′ part of the 5′ long terminal repeat (LTR) and a 5′ part of the 3′ LTR. The nucleotide sequence alignment results indicated that no evidence of sample cross-contamination was detected. NFLG phylogenetic analyses showed that the NFLG sequence of JL070032 clustered with CRF01_AE reference sequences, but formed a distinct monophyletic branch distantly related to CRF01_AE (Fig. 1). Similarity plot analyses were performed using all known HIV-1 group M reference sequences representing subtypes/subsubtypes (A1, A2, B, C, D, F1, F2, G, H, J, and K) and CRF01_AE, CRF07_BC implemented in SimPlot v.3.5.1 to characterize the genomic structure; the results showed that the genomic structure was composed of CRF01_AE and CRF07_BC with a backbone of CRF01_AE (Fig. 2). Bootscanning and informative site analyses were performed using three CRF01_AE sequences (JL100007, JL100014, and LN070010), three CRF07_BC sequences (98CN009, XJDC6441, and XJDC6431), and three subtype J sequences (SE9280, 04CMU11421, and KTB147) as references implemented in SimPlot v.3.5.1; the results revealed that the breakpoints corresponded to HXB2 nucleotide sequence positions 4448, 4788, 5509, and 5835, respectively (Fig. 2). Similar results were obtained using online software jpHMM-HIV.
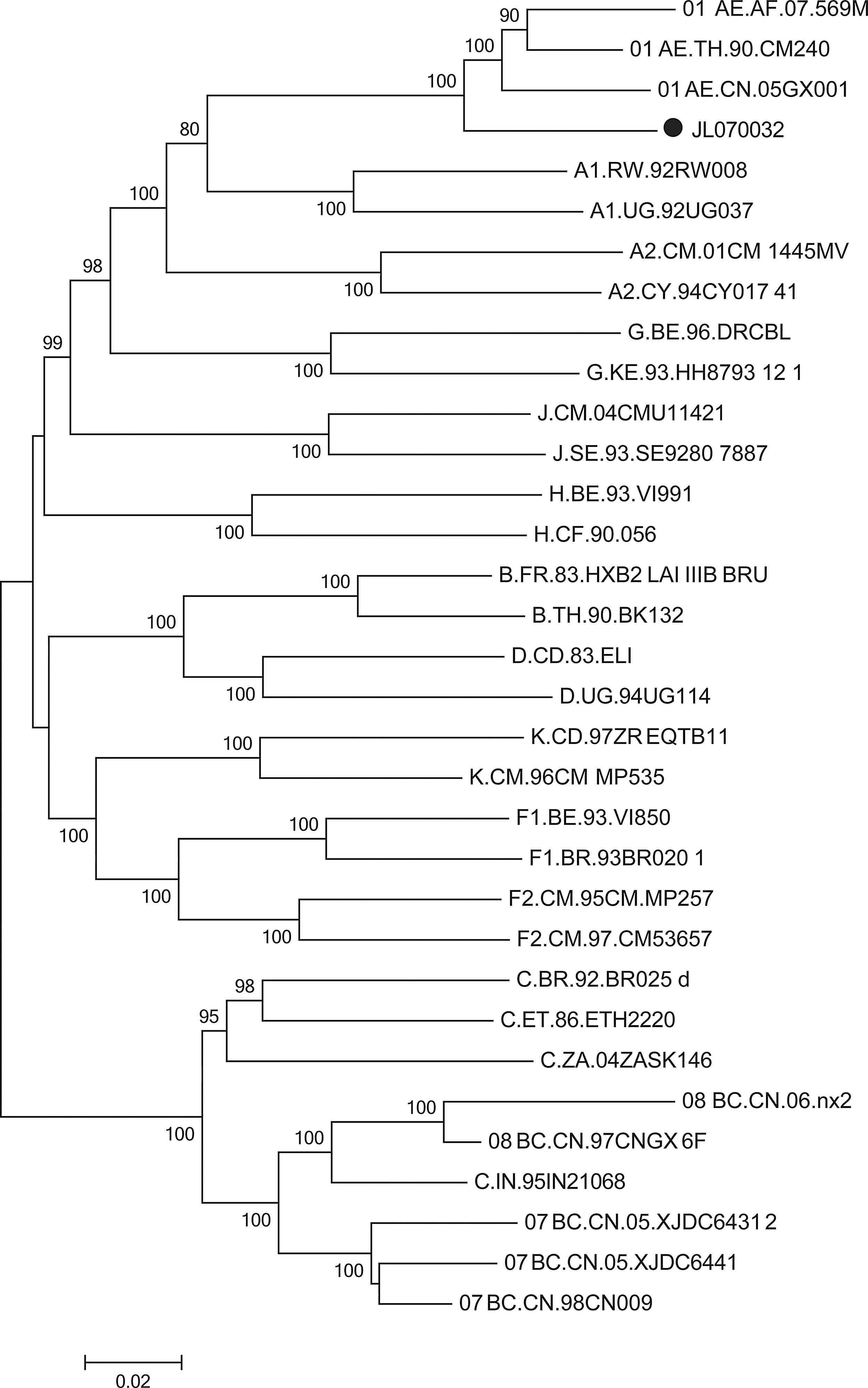
Phylogenetic analyses of the near full-length genome (NFLG) nucleotide sequence of JL070032. All known HIV-1 group M reference subtypes/subsubtypes and available circulating recombinant forms (CRFs) were initially used to construct the neighbor-joining phylogenetic tree; some references were later removed for clarity. A solid circle (●) marks JL070032 throughout the article. The stability of the phylogenetic nodes was assessed by bootstrap analysis with 1,000 replications, and only bootstrap values ≥70% are shown at the corresponding nodes. The scale bar represents 2% genetic distance.
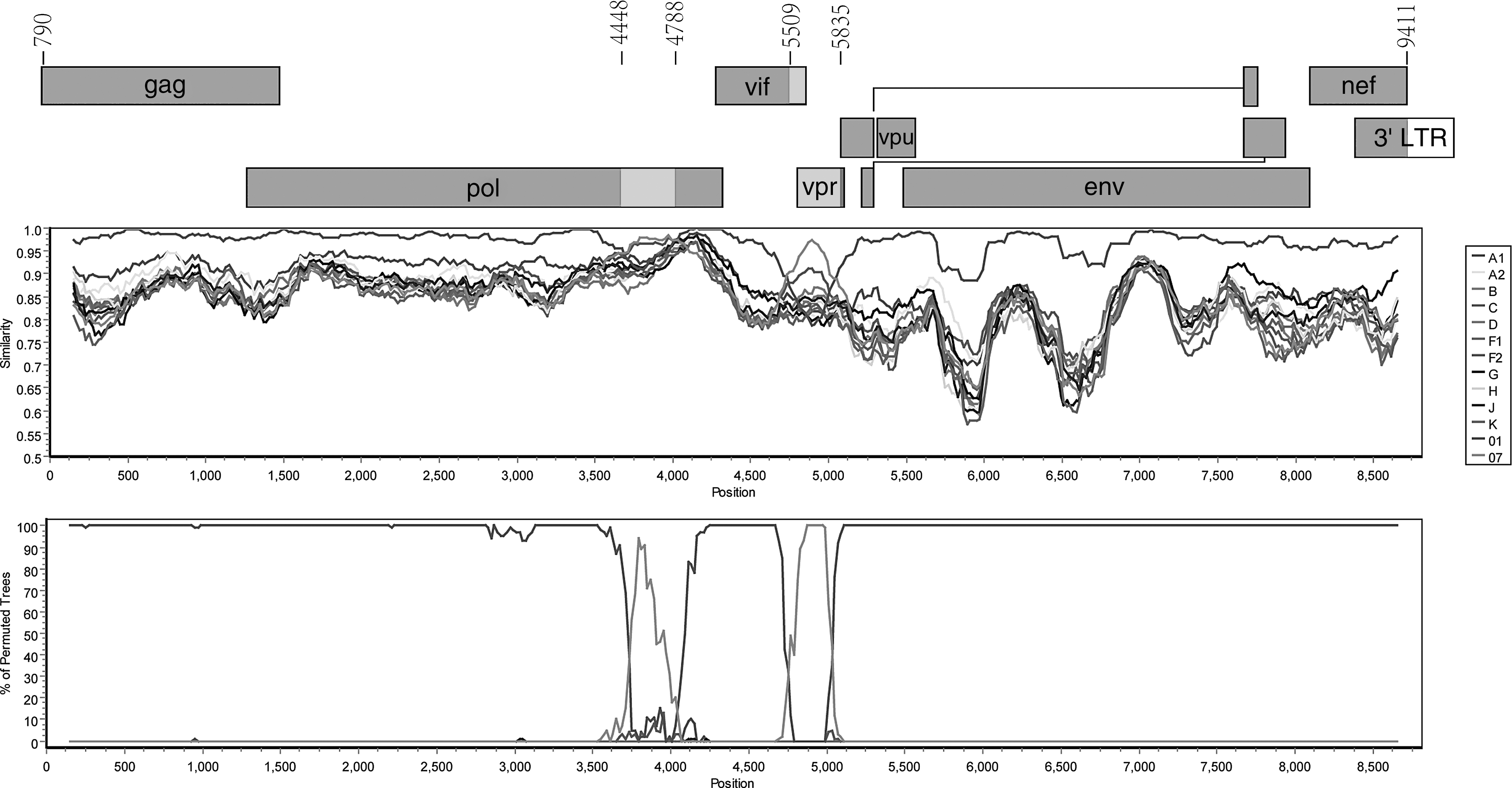
Recombinant analyses of the novel HIV-1 strain JL070032. The NFLG map of JL070032 is shown at the top of the figure with breakpoints based on HXB2 nucleotide sequence numbering. In the center, similarity plot analyses were performed with all known HIV-1 group M genotypes representing subtypes/subsubtypes (A1, A2, B, C, D, F1, F2, G, H, J, and K) and CRF01_AE and CRF07_BC. At the bottom is bootscanning and information site analyses of the newly identified strain JL070032. Crossover points of the two curves denote the recombinant breakpoints that divided the genome into five regions (I–V). The recombinant analyses were performed with a sliding window of 300 bp and a step size of 20 bp. 01=01_AE and 07=07_BC.
Our previous study identified at least seven distinct clusters of CRF01_AE lineage circulating among various high-risk populations and geographic regions in China, resulting from multiple founder events, introduced independently during the early-to-late 1990s. 12 Therefore, it is tempting to speculate that the three CRF01_AE regions of JL070032 originated from the CRF01_AE lineage (designated CRF01_AE cluster 5), which is mainly circulating among the MSM population in northern China (especially in Beijing, Liaoning, and Jilin).
We further performed subgenomic analyses to determine the parental origin of the three CRF01_AE regions. The three regions were initially aligned against all known HIV-1 group M reference sequences representing subtypes/subsubtypes (A1, A2, B, C, D, F1, F2, G, H, J, and K) and CRF01_AE and CRF07_BC obtained from the Los Alamos HIV sequence database, as well as 75 NFLG sequences of CRF01_AE from our laboratory previously described. 12 For clarity, we selected three NFLG sequences from each well-supported distinct phylogenetic CRF01_AE cluster or subcluster (cluster 4 was composed of two CRF01_AE subclusters). Finally, a total of 24 NFLG sequences of CRF01_AE lineage sampled mainly from three high-risk groups [intravenous drug users (IDUs), heterosexuals, and MSM] in 10 provinces across China during 2002–2010 was used in this study. Subregion tree analyses confirmed the parental origin of each region of JL070032: region I (HXB2, 790 to 4447), CRF01_AE; region II (HXB2, 4448 to 4787), CRF07_BC; region III (HXB2, 4788 to 5508), CRF01_AE; region IV (HXB2, 5509 to 5834), CRF07_BC; and region V (HXB2, 5835 to 9411), CRF01_AE. Subregion tree analyses also demonstrated that the CRF01_AE regions were indeed clustered with the CRF01_AE lineage, which is mainly circulating among the MSM population in northern China, with the support of a 100% bootstrap value, indicating that the parental origin of the CRF01_AE regions was from the MSM population, in which recombination events may be more likely to occur (Fig. 3).
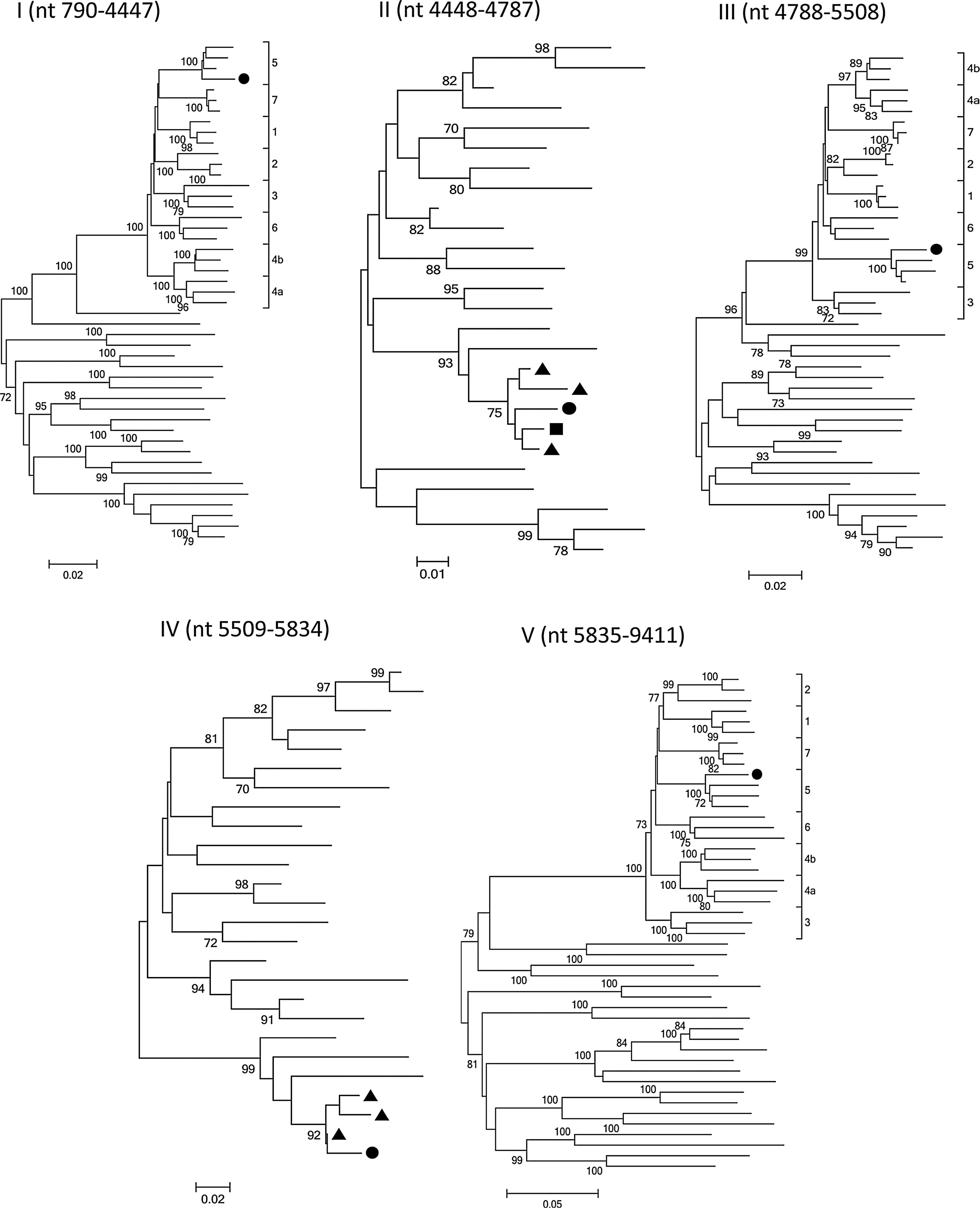
Subregion tree analyses of the five regions
In addition, preliminary epidemiologic data showed that the subject from whom JL070032 was obtained had reported only one high-risk factor (homosexual behavior), confirming that the CRF01_AE regions indeed originated from CRF01_AE of the MSM population circulating in northern China. Interestingly, region II clustered with reference sequences representing CRF07_BC and one subtype C sequence (95IN21068) with a 75% bootstrap value, suggesting that it may have been derived from a region of CRF07_BC that originated from subtype C. It is also noted that region IV clustered with reference CRF07_BC with a 92% bootstrap value, suggesting that it may contain one breakpoint of CRF07_BC.
In this study we first identified the NFLG sequence of a novel HIV-1 second-generation recombinant form (CRF01_AE/CRF07_BC) composed of CRF01_AE and CRF07_BC among the MSM population in Jilin, China. The new recombinant strain, JL070032, consists of four breakpoints combined with two short regions of CRF07_BC origin into a backbone of CRF01_AE. Region II was located in the 3′ end of the HIV-1 pol gene and region IV in the 3′ end of the HIV-1 vif gene and the majority of the vpr gene. Of interest, the routine HIV genotyping method using the gag (1,056 bp, HXB2 nucleotide sequence numbering: 781–1836) and env (540 bp, HXB2 nucleotide sequence numbering: 7002–7541) genes, both of which were identified in JL070032 as CRF01_AE, failed to define the genotype correctly, which may indicate that the HIV-1 epidemic in China is more complex than what we detected in high-risk populations and new challenges will occur in determining the HIV-1 genotype in molecular epidemiologic research.
The emergence of the novel second-generation recombinant form (CRF01_AE/CRF07_BC) identified among the MSM population in Jilin indicates active transmission networks of HIV-1 among the MSM population in the region. Therefore, it is necessary to further monitor the molecular epidemiology of the HIV-1 epidemic among the MSM population in Jilin.
Sequence Data
The nucleotide sequence of JL070032 used in this study is available in GenBank with accession number KC990127.
Footnotes
Acknowledgments
This study was supported by National Science and Technology Major Projects for Infectious Diseases Control and Prevention (2008ZX10001–004, 2012ZX10001–002, and 2012ZX10001–008), National Natural Science Foundation of China (81020108030 and 81261120379), International Cooperative Grant (2009DFB30420) and SKLID Development Grant (2012SKLID103).
Author Disclosure Statement
No competing financial interests exist.