
Abstract
The prevalence of HIV type 1 among men who have sex with men (MSM) is increasing in China. We report here a novel HIV-1 recombinant form (CRF01_AE/CRF07_BC) detected from a male patient infected with HIV-1 by homosexual behavior in Zhejiang Province of eastern China. The near full-length genome analyses showed that the unique HIV-1 recombinant isolate (16ZJ305) has two recombinant breakpoints observed in the env and tat/rev gene regions. To date, several novel CRF01_AE/CRF07_BC recombinant forms have been identified, which may imply an active transmission network of HIV-1 infection among MSM. Further studies of the molecular epidemiology of the HIV-1 epidemic among MSM are necessary to gain a better understanding of the transmission network and track the genetic evolution.
H
Here, we detected a novel HIV-1 second-generation recombinant form (CRF01_AE/CRF07_BC), designated as 16ZJ305, by near full-length genome (NFLG) analyses, isolated from an HIV-positive consenting male patient among the MSM population in Zhejiang Province, which is different from the CRF01_AE/CRF07_BC recombinant viruses previously reported in China.
In this study, an HIV-1-positive plasma sample was collected from a 27-year-old male merchant (Patient 16ZJ305) in the provincial capital city of Zhejiang Province, Hangzhou. 16ZJ305 is a Chinese citizen of Han ethnicity, residing in Xiaoshan District, which is the biggest district of Hangzhou. He is an individual businessman after graduation from college. He was diagnosed as HIV positive on April 30, 2016 after he had suffered a long-term oral ulcer and was recruited from the Zhejiang Provincial Center for Disease Control and Prevention (ZJCDC). A self-report by 16ZJ305 shows that he was infected through homosexual behavior. 16ZJ305 was highly active antiretroviral therapy treatment naive before his blood sample was collected. His CD4+ T cell count was 6 cells/μL and viral load was 70,800 copies/mL on May 17, 2016 when the plasma was collected. Written informed consent was obtained from the subject before sample collection, and was approved by the institutional review board of the First Affiliated Hospital, School of Medicine, Zhejiang University.
The amplification and sequencing of the new generated NFLG were performed as previously described.
5
Recombination breakpoints were determined using RIP and jpHMM (
The NFLG sequence amplified from Patient 16ZJ305 was 9,001 nucleotides (nt) in size (relative to the HXB2 nucleotide numbering system: positions 638 to 9642) and the blast search data show that no evidence of sample contamination was detected. As shown in Figure 1, 16ZJ305 clustered with CRF07_BC, CRF08_BC, and C subtype reference sequences (bootstrap value 100%), but formed a distinct monophyletic branch different from CRF07_BC, CRF08_BC, and C subtype reference sequences.
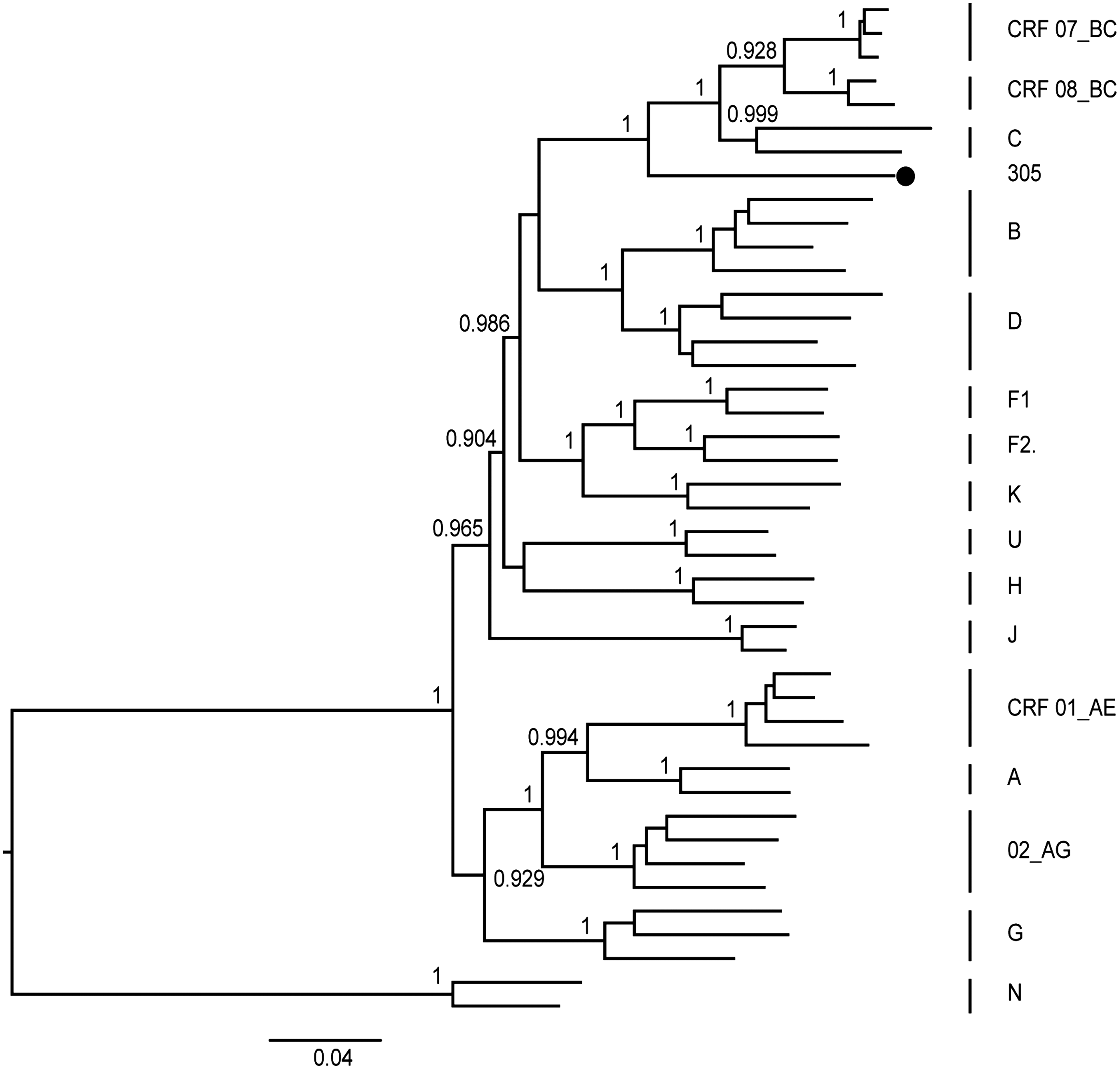
Phylogenetic tree analysis of the near full-length genome nucleotide sequence of the subject, 16ZJ305. The tree was constructed through the maximum likelihood method with 1,000 bootstrap replicates. The solid black circle denotes 16ZJ305. All reference sequences of the HIV-1 group M subtypes and CRFs were downloaded from the HIV database (
SimPlot analysis shows that the NFLG sequence of 16ZJ305 was composed of CRF01_AE and CRF07_BC, with one region of CRF01_AE inserted into a CRF07_BC backbone (Fig. 2A). And bootscanning analysis of the NFLG sequence reveals that the two unique recombination breakpoints corresponded to HXB2 nucleotide positions 6398 and 8742, located in tat/rev and env gene regions, respectively, and divided the NFLG into three regions: region I (HXB2, 638-6397), CRF07_BC; region II (HXB2, 6398-8741); region II I (HXB2, 8742-9642) (Fig. 2B). Similar results were obtained using online software RIP (
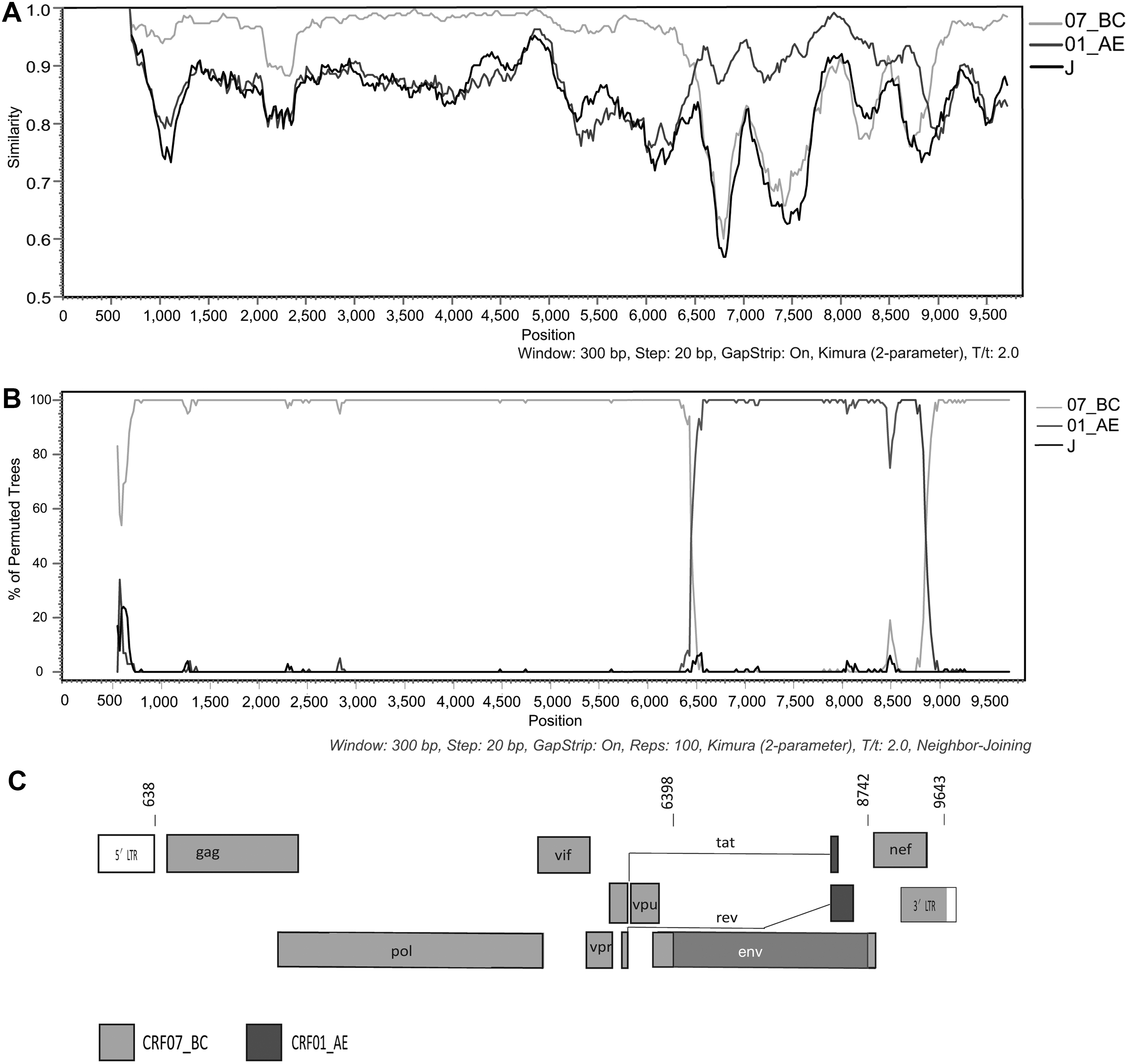
Recombinant analyses of the near full-length genome nucleotide sequence of 16ZJ305.
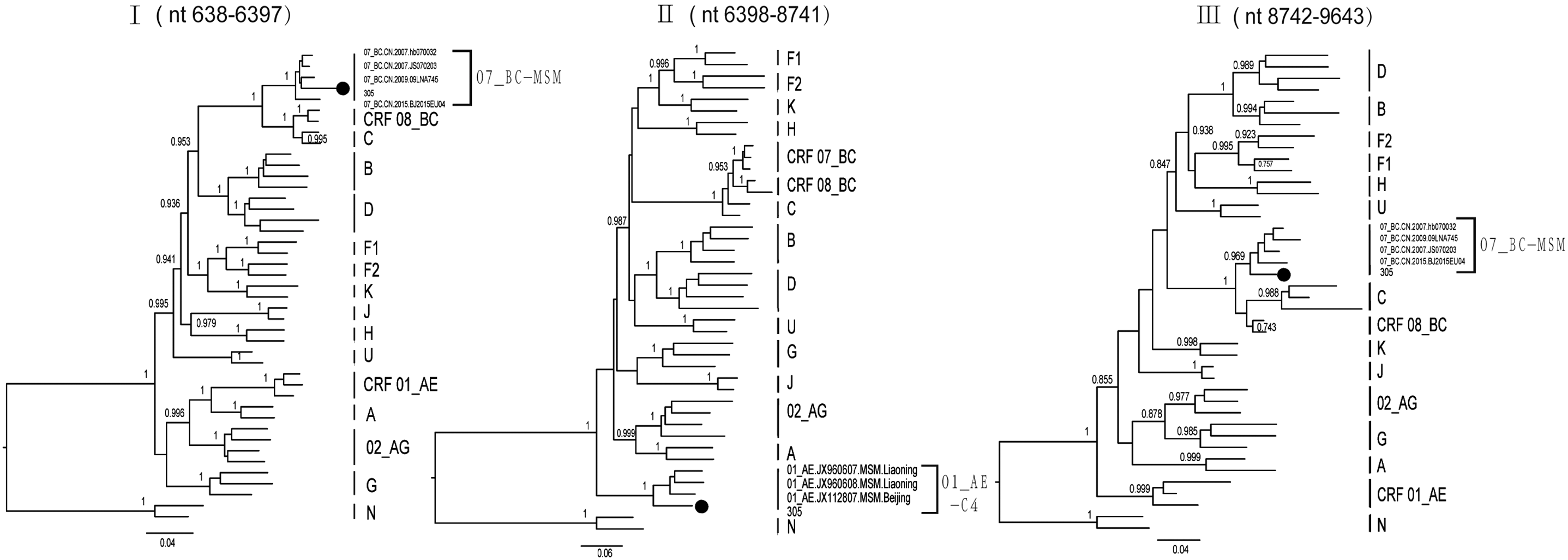
Subregion tree analysis of 16ZJ305 futher confirmed the two recombinant breakpoints identified by bootscanning analysis (Fig. 3). It also demonstrated that the CRF01_AE region (II) in 16ZJ305 belonged to CRF01_AE subcluster 4 lineage, but formed a distinct monophyletic branch from CRF01_AE - Cluster4. A potential reason for this could be that there was a very short region of CRF 07_BC inserting into CRF01_AE region (HXB2 8339-8401). The CRF07_BC regions (I and III) in 16ZJ305 clustered with CRF07_BC reference sequences (bootstrap value 97.7% and 100%), and belonged to CRF07_BC Cluster 1. In summary, the parental origins of the CRF01_AE and CRF07_BC regions of novel second-generation recombinant (16ZJ305) could be from CRF01-4 and CRF07-1.
Recombination between different HIV-1 subtypes is a primary mechanism that contributes to the genome complexity of HIV-1, and new recombinant strains are frequently found in regions where various subtypes are prevalent. CRF01_AE and CRF07_BC are two predominant HIV-1 circulating strains among MSM in Zhejiang. 7 To date, two NFLG sequences of HIV-1 CRF01_AE/CRF07_BC second-generation recombinants have been identified in Zhejiang Province. 15zj032 comprised three CRF01_AE and four CRF07_BC fragments, 8 whereas 15zj016 with three fragments of CRF07_BC was inserted into the CRF01_AE backbone. 9 In the study, a novel circulating recombinant forms (CRF) recombinant strain 16ZJ305 was characterized with a CRF01_AE fragment inserted into a CRF07_ BC parental backbone. Although the three novel second-generation recombinants are unrelated variants, they may indicate that diverse recombination forms exist between CRF01_AE and CRF07_BC prevailing in the Zhejiang Province. Since CRF_01AE and CRF_07BC have become the two major strains of HIV-1 in China especially among young MSM populations, 10 increasing recombinant strains of two strains have been reported in Beijing, Guangxi, Jilin, Sichuan, and Shanxi. 5,9,11 –13 The emergence of new second-generation recombinants indicated the presence of complex transmission networks of different HIV subtypes/CRF infections among sexual transmissions in this region. Therefore, an effective and continuous HIV-1 molecular epidemiologic monitoring among high-risk populations is indispensable, which is of great importance in understanding the dynamics of the HIV-1 epidemic in China.
Footnotes
Sequence Data
The nearly full-length genomic sequence of 16ZJ305 is available under GenBank accession no. MH684584.
Acknowledgments
This work was supported by the International Cooperative Grant (2016YFE0107600) and the National Natural Science Foundation of China (grant no. 81500491). We would like to thank Brian Foley from Los Alamos National Laboratory for advice on URFs nomenclature.
Author Disclosure Statement
No competing financial interests exist.