
Abstract
Revolutionary progress in combinational antiretroviral therapy has transformed human immunodeficiency virus (HIV) infection into a chronic manageable disease; yet, there exists an uneasy truce between the virus and the immune cells, where inflammation is limited but infection continues to fester from latent reservoirs of the virus. Clinical studies have identified the major immune cell types that constitute the latent HIV-1 reservoirs as monocytes/macrophages and CD4+ T cells. Latency probing approaches have thrown some light on the interaction between the virus and the reservoir cells from the time of onset of infection. However, research combining latency reversal strategies and immunotherapies face daunting obstacles in clinical trials because of the lack of in-depth knowledge on viral pathogenesis and mechanisms of viral evasion, leaving us behind in the battle for HIV cure. This article reviews existing knowledge on the cells and mechanisms that contribute to the establishment and survival of HIV reservoirs in infected individuals.
Introduction
Evolution, dissemination, and frequent alterations in the virus–host interaction are central to human immunodeficiency virus (HIV) pathogenesis. Loss of CD4+ T lymphocytes by direct viral infection and indirect impairment of bystander cells leads to the subsequent loss of immune competence. Earlier studies have documented that nonsyncytium-inducing or chemokine receptor-5 (CCR5)-utilizing (R5-tropic) transmitted viruses appear first, followed by syncytium inducing or C-X-C chemokine receptor type 4 (CXCR4)-utilizing (X4-tropic) viruses, which appear in 50% of people later during infection, and finally the M-tropic strain, which uses R5, X4, or both, in a T cell depleted environment such as in advanced HIV disease. 1 –3 While antiretroviral therapy (ART) blocks active viral replication and partially restores the immune function, sudden cessation of treatment for a week results in rapid viral rebound.
These clinical observations indicate the existence of large viral populations that are insensitive to antiretroviral drugs. Unlike the herpes viruses, which generate viral proteins that are particularly necessary for the establishment and maintenance of viral latency, HIV latency is controlled by the infected cells' molecular mechanisms. Mainly, the location of the integration site where the provirus integrates to establish latency is epigenetically controlled by the virus. 4 Findings from studies on the SIV/macaque model indicate that active as well as resting CD4+ T cells serve as initial replication foci, which eventually transfer the virus to the proximal tissues via the proximal lymphoid organs, from where they disseminate systemically.
During the early stages of HIV infection, increased amounts of proinflammatory cytokines and chemokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1β, C-C motif chemokine ligand (CCL)3/macrophage inflammatory protein (MIP)-1α, MIP-1β (CCL4), and RANTES (CCL5), are produced and released by activated T cells in both plasma and lymph nodes, leading to rapid viral dissemination. 5 –7
HIV Reservoirs
The life cycle of HIV occurs in two phases: the early stage in which the virus enters the host cell and integrates its genome into the host DNA, and the late phase in which the integrated provirus initiates and completes viral replication. Most viruses participate in active infection while a few enter the quiescent state and become latent. The latent reservoirs can subsequently get reactivated through the interaction between the host and virus at the transcriptional level. A clear understanding of the sites of HIV persistence will pave way for the development of effective curative treatment strategies.
The primary reservoirs of HIV are reported to be the resting memory CD4+ T cells, 8 –16 microglial cells, 17,18 macrophages, 19 –23 and other cells-like astrocytes, 24 –27 and dendritic cells. 28,29 In this review, we will confine our discussion to the major HIV-1 reservoirs, that is, the CD4+ T cells and macrophages. The major challenges to finding a cure for HIV are the lack of unique surface markers to identify latently infected cells, immune escape mutations, and unidentified host immune mechanisms involved in viral clearance. 30,31
Role of T Cell Subsets in HIV-1 Infection and Latency Establishment
Latent HIV-1 reservoirs are established in resting CD4+ T cells early during acute infection. 8 HIV-1 infection is known to induce the production of alloantigens, which trigger inappropriate antigen presentation and cause exhaustion of innate immune cell function due to continued immune activation. 32 –34 Antigen presenting cells (APCs) present the viral antigen to T cells and help in the formation of an immunological synapse (IS) that triggers a cascade of positive/negative signaling pathways. HIV-1 viral DNA has been found in resting CD4+ T cell subsets that include naive cells, stem cell memory T cells, central memory cells (TCM), transitional memory cells (TTM), and terminally differentiated effector memory T cells.
However, TCM and TTM have the highest levels of total HIV-1 DNA and are viewed as the principal repositories of latent viruses. 10,35 Quantitative viral outgrowth assay, however, revealed that replication-competent HIV-1 was not consistently found in the TTM cell compartment, despite the presence of significant levels of viral DNA in TTM cells. 15
Saez-Cirion et al. in 2013 reported that naive T cells can also produce numerous virions upon treatment interruption in the later stages of the disease. 36 Constant replacement of dying CD4+ T cells as a result of high levels of viral infection in the periphery, particularly during early disease, is provided by the thymus. But as disease progresses, there is a drop in the number of CD4+ T cells in the periphery, due to viral replication in the thymus, resulting in the infection of memory cells in the thymus. 37 Homeostatic proliferation of these memory populations requires low-level signaling through the TCR and/or cytokines, including IL-7 and IL-15. 38
Some studies have reported that CD4/CD8 double-negative T cells can also actively harbor HIV-1. 14,39 It has also been found that expression of CCR6, CCR7, and CXCR3 chemokines significantly increases the size of the latent reservoirs in the early stage of HIV-1 infection. 9 CCL19 and CCL21, the chemokine ligand for CCR7, play a critical role in maintaining the stability of HIV-1 integrase which plays a major role in proviral integration and establishment of latency. 40 As infection progresses, HIV-infected lymph nodes attract CXCR3 expressing CCR5+CD4+ T cells via CXCL10 and CXCL11 chemokines. This approach of retaining HIV-infected T cells instead of destroying them impairs the peripheral T cell response and elevates the level of T cell susceptibility to HIV upon long-term exposure to high levels of the virus.
Macrophages in HIV-1 Infection and Latency Establishment
Monocytes and macrophages are an integral part of the immune system. The proportion of tissue-resident macrophages and monocyte-derived macrophages (MDMs) vary during homeostasis and inflammation. 23 Monocytes are classified into three subtypes in the peripheral blood, based on the surface expression of CD14 and CD16 molecules. Classical monocytes (CD14++/CD16−) constitute 90%, proinflammatory intermediate monocytes (CD14++/CD16++) constitute 5%, and nonclassical/patrolling monocytes (CD14+/CD16++) constitute 5% of the total monocyte pool respectively. 41 Activation and differentiation of peripheral blood monocytes influences their susceptibility to HIV-1 infection as well as their ability to move into tissues, thereby contributing to the progression of HIV-1 pathogenesis. 42
Monocytes use host restriction factors such as SAMHD1 and APOBEC3, as well as cellular microRNAs, to prevent viral infection from spreading. 43 –45 As a result, less than 0.1 percent of the monocytes harbor HIV-1 provirus that is capable of replication. 46 Classical monocytes express very low levels of CCR5 receptors, explaining their relative resistance to HIV-1 infection. 47 Intermediate monocytes, on the contrary, exhibit more CCR5 on their surface and have been shown to store proviral DNA in both treated and untreated HIV-1 infection. 46 The extent to which monocytes serve as HIV reservoirs and their significance as such continues to remain a mystery. However, their role as anatomical sanctuaries cannot be overlooked due to their presence in lymphoid as well as nonlymphoid organs.
Although studies have documented the presence of infected macrophages in the lamina propria of the intestinal, penile, urethral, and vaginal mucosa, 13,48,49 their role in the spread of infection is still under debate. Cribbs and colleagues were able to detect HIV-1 DNA and RNA in alveolar macrophages obtained from bronchoalveolar lavage of virally suppressed individuals, but were unable to determine whether the macrophages were actually infected with the virus or had ingested infected T cells, due to a limitation in the detection methodology, 21 Since detection of infected macrophages would not only be limited to in situ hybridization followed by gating on cd206 positive cells via flowcytometry but also be based on impaired phagocytic activity. 47 Kupffer cells, a type of tissue-resident macrophages present in the liver, are reported to be vulnerable to HIV-1 infection. The results of a phylogenetic study of the viral quasispecies revealed unique clustering/compartmentalization of HIV-1 variations in the liver. 50,51
HIV can also infiltrate the central nervous system (CNS) as early as 8 days postinfection. It crosses the blood-brain barrier formed by endothelial cells and astrocytes and end up within infected monocytes, 52 leading to increased glial cell activation and neuronal injury, particularly in combinational ART (cART)-naive individuals. 53 Although microglial cells and perivascular macrophages are the two important targets that migrate to the brain as part of the inflammatory response, it is still unclear which cell type supports the evolution and replication of macrophage-tropic viruses in the CNS. Veazey et al. discovered that tissue macrophages were infected by SIV following substantial depletion of CD4+ T cells in macaque lymphoid tissue. 54 Further, apoptotic cells are thought to be a part of the suppressive microenvironment that could help in the establishment of reservoirs in macrophage populations.
Immune Cell Cross talk Between Macrophages and CD4 T Cells in HIV-1 Latency
Cell-to-cell contact is thought to be essential for efficient transmission of the virus from MDMs to CD4+ T cells during antigen presentation, a process known as trans infection. 55 The synapse formed during the cell-to-cell interaction are of two types: the synapses at the dendritic cell and T cell junction called the IS and the synapse at the T cell-T cell junction referred to as the virological synapse (VS). 56,57 Trans infection can happen in three ways: (i) virus accumulation in tetraspanin-enriched compartments (TEMs/microvesicles); (ii) transfer of microvesicles to the cell surface; and (iii) transfer of microvesicles from the internal compartment through the VS upon contact with the target cell. 58 Viral proteins (Gag and Env), adhesion molecules, and actin are recruited to the VS in T cells. The interaction of intercellular adhesion molecule-1 and CD80/86 on the APCs with lymphocyte function-associated antigen 1 and CD28 on the T cells mediates IS formation.
Studies have shown that Cholesterol/Ganglioside GM1-enriched lipid rafts are required for VS formation and recruitment of Gag and Env. Actin remodeling via Rac-Pak-LIMK-cofilin pathway in T cells is known to increase in response to the presence of Env proteins in the cellular environment. Different cytoskeletal networks are involved in different target cells for efficient infection, assembly, and spread of HIV-1. 59 In macrophages, virus transfer/entry occurs through an endocytic mechanism in a clathrin-dynamin-dependent manner through Rac, Pak1, and Na+/H+ exchanger pathways.
Here, the viruses are taken into small clathrin-coated vesicles and are later released into the target cells with the help of dynamin, a cytosolic small GTPase responsible for breaking the internalized endocytic vesicle. 60 Transfer can occur concurrently from one infected cell to multiple uninfected cells at structures known as polysynapses. 61 To some degree, these findings call into question the view that VS is a privileged milieu that is sequestered from the host humoral immune response. 62
During HIV infection, inflammatory chemokines like CCL20 have been shown to increase ∼3.3-fold in serum. 63 In an SIV-macaque model, it was demonstrated that CCL20 was constitutively produced by the primary epithelial cells after vaginal exposure to SIV. This results in the recruitment of CCR6+ plasmacytoid dendritic cells to the site of infection and production of INF-α, CCL3 and CCL4 to inhibit viral infection. These molecules also recruit CCR5+CD4+ T cells, which are highly susceptible to SIV infection to the local area. 64 Thus, failure to inhibit viral entry eventually leads to rapid viral dissemination, reservoir establishment and disease progression. In vitro studies focusing on the microenvironment of infected CD4+ T cells and MDMs are needed to clearly understand cellular communications involved in reservoir establishment. Figure 1 shows the cross talk between macrophages and CD4+ T cells in the pathogenesis and establishment of latency in HIV-1 infection.
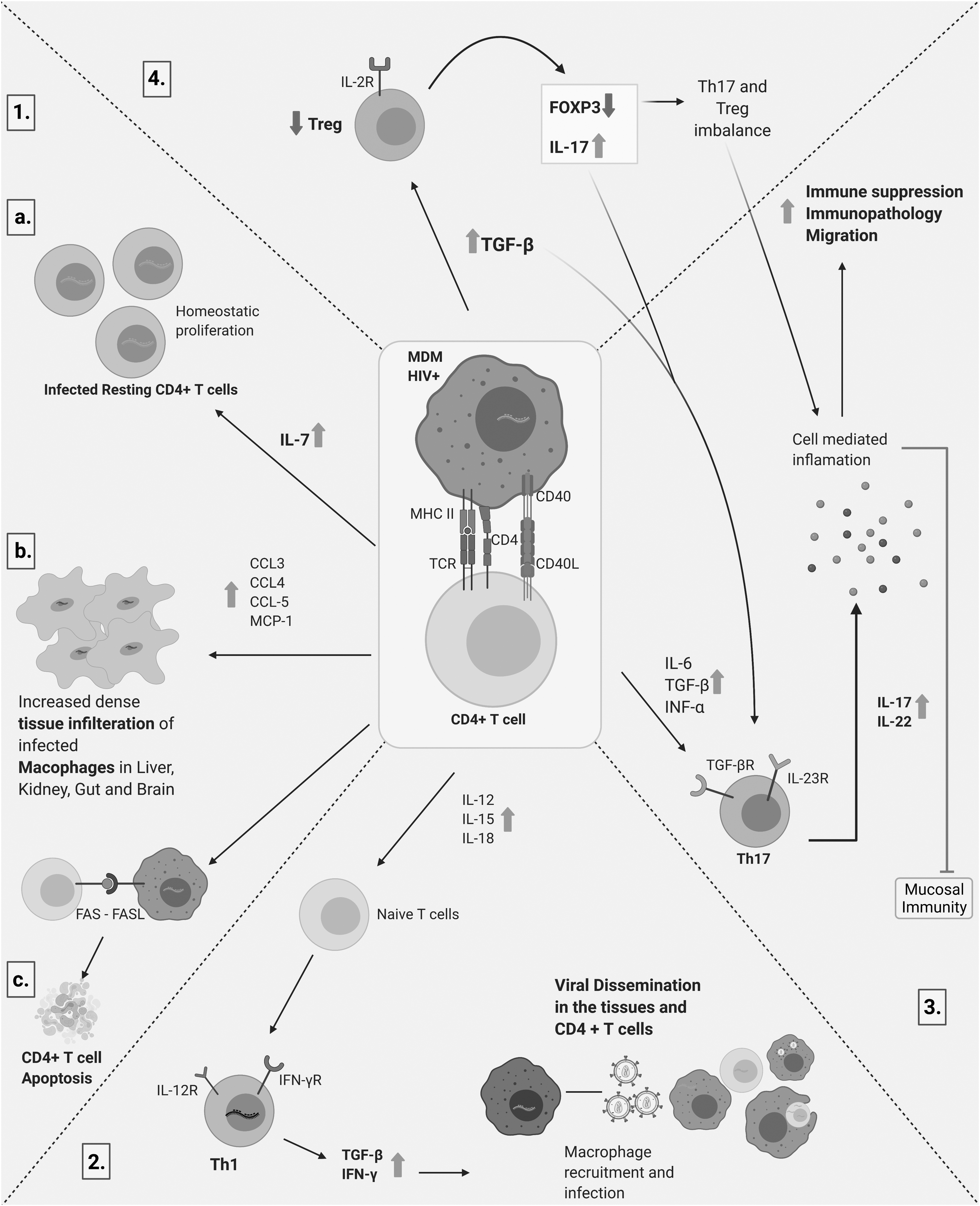
Cross talk between macrophages and CD4+ T cells in the pathogenesis and establishment of latency in HIV-1 infection. Infected macrophages are important viral reservoirs that upon entering tissues become densely packed and transformed into long-lived viral pools. (1.a.) Establishment and self-renewal of infected resting CD4+ T cells. (1.b.) The infected macrophages release chemokines and cytokines (CCL3, CCL4, CCL5, and MCP1) which attract CD4+ T cells and contribute to further viral dissemination. (1.c.) Macrophages infected with HIV-1 induce apoptosis in CD4+ T cells, thereby increasing the viral pool and minimizing viral neutralization. (2.) Tissue-specific macrophages further fuel viral dissemination via CD4+ T cells leading to the establishment of viral sanctuaries throughout the body. (3. and 4.) Cytokine/chemokine storm leading to an imbalance in mucosal immunity. Created with
Influence of HIV-1 Proteins on Establishment of CD4+ T Cell and Macrophage Reservoirs
Soluble HIV-1 proteins such as Nef, transactivator of transcription (Tat) and viral protein U (Vpu) modulate the cellular machinery and viral transcription. 65 –70 The role of HIV-encoded proteins in the establishment of latent reservoirs in CD4+ T cells and macrophages is schematically represented in Figure 2.
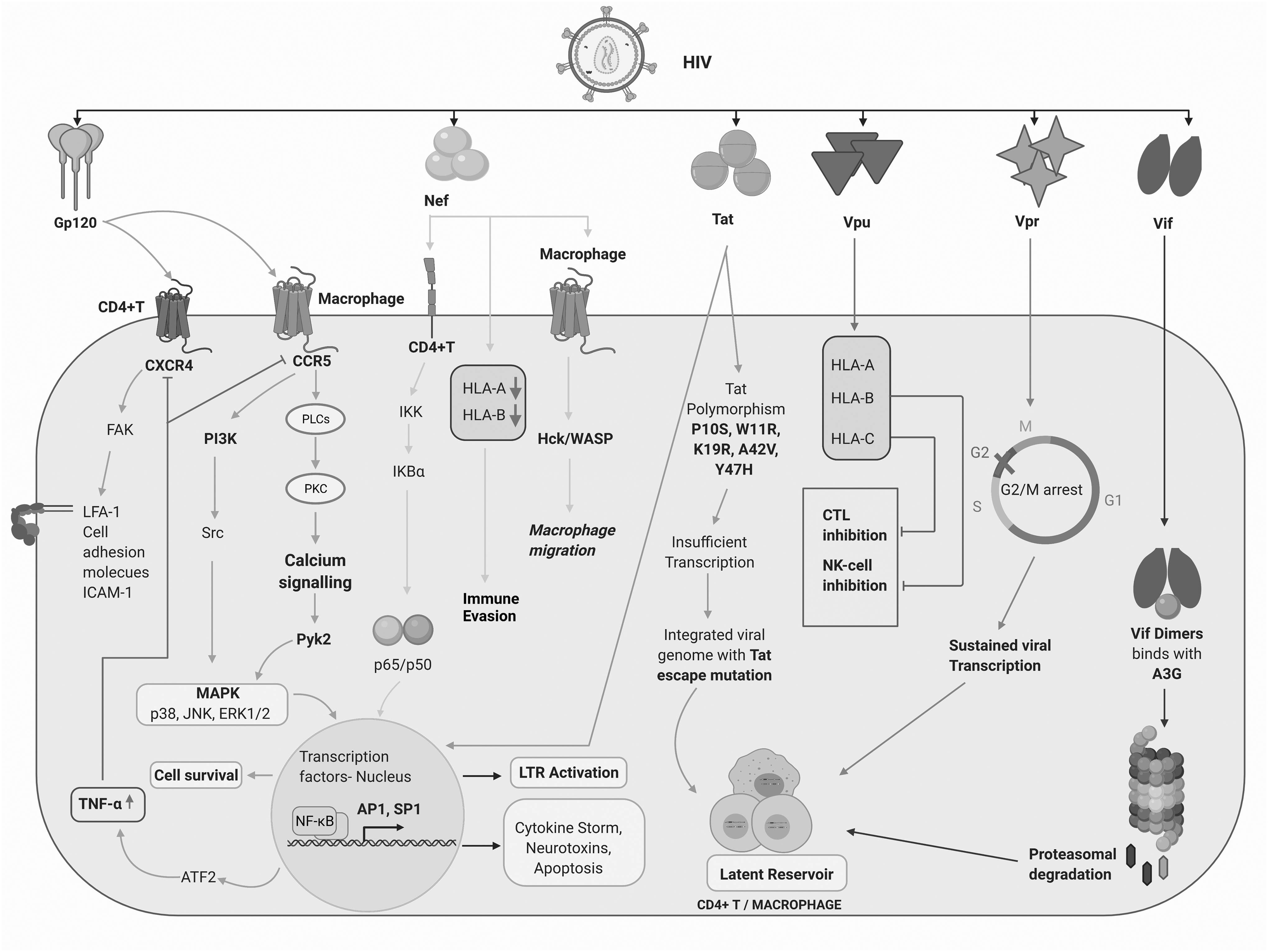
Role of HIV-1 Tat, Nef, and Vpu in the establishment of latent reservoirs in CD4+ T cells and macrophages. Indirect contribution of the structural protein Gp120 and regulatory/accessory proteins, Vpr and Vif, in active viral replication and latent reservoir establishment. Exogenously present HIV-1 proteins can penetrate cells and upregulate the production of proinflammatory cytokines and chemokines thereby fueling viral persistence. Furthermore, these proteins can bind to the long terminal repeats of HIV-1 and activate multiple transcription factors in CD4+ T cells and macrophages, including NF-κB, Sp-1, and AP-1, and result in the production of cytokine storm, neurotoxins, and cellular apoptosis. Vpr-induced cell cycle arrest may facilitate sustained viral transcription. Created with
Nef
Nef, a 27 kDa myristylated protein, is expressed early in the life cycle of HIV. It has been detected in the sera of AIDS patients in the range of 1 to 10 ng/mL 71 as well as in cultures of HIV-1-infected cells. It is usually internalized by MDMs and dendritic cells but not by T cells when added to cell culture. 72 Nef induces the prompt release of proinflammatory cytokines/chemokines, which in turn activate signal transducer and activator of transcription (STAT)-1 and STAT-3 signal transducers and transcription activators. 22 It also induces phosphorylation of three mitogen-activated protein kinase (MAPKs) that is, ERK1/2, c-Jun N-terminal kinase (JNK), and p38. 73 Nef in macrophages causes dysregulation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation and induces the production of proinflammatory cytokines and p50 homodimers. In HIV-infected macrophages, the translocation of p50 homodimer and p65/p50 heterodimer can lead to sustained long terminal repeat activation, which explains the silent transmission of the virus. 74
Recent studies on the Nef protein have shed light on the macrophage-CD4 T cell relationship in HIV-1 viral dissemination. Nef impairs T cell migration and modulates macrophage migration by modulating the podosome and hematopoietic cell kinase/Wiskott-Aldrichh syndrome protein (Hck/WASP) signaling pathways. It inhibits the amoeboid and 2D migration of macrophages but enhances mesenchymal migration. This leads to increased tissue infiltration of macrophages in the kidney, liver, gut, and brain tissues of transgenic mice. 67 Nef-mediated HLA downregulation is seen as a positive factor that influences the size of HIV latent reservoirs during cART. This downregulation and immune evasion influences reservoir dynamics during suppressive cART. 75 Nef promotes the expression of tumor growth factor-β1 (TGF-β1) that acts on the hippocampus and stimulates small mothers against decapentaplegic homology-2 phosphorylation resulting in the upregulation of CCL2, CD163, and glial fibrillary acidic protein. 76 CCL2 upregulation attracts perivascular macrophages into the brain causing an increase in inflammation, thereby leading to neurotoxicity and learning impairment.
Thus, Nef is considered to be a powerful migration modulator that favors trans infection by inhibiting T cell migration in lymph nodes and promoting virus dissemination through body tissues by enhancing macrophage 3D mesenchymal migration. 67
Tat
Tat is a viral encoded protein that binds to the transactivating element (TAR) and modulates viral gene expression and replication. It is reported to be detected in the sera of infected individuals, as well as in infected cell culture media. 77 Functional consequences of Tat include TNF-α release, which further results in the expression of various cytokines such as IL-6, TNF-β, TGF-β, as well as the expression of cytokine receptors such as that of IL-4 by macrophages and monocytes. 78 Tat replicates TNF-α's effects on viral replication by activating NF-κB, activator protein 1 and MAPK, including JNK/stress-activated protein kinase but not MEK. 79
Tat has also been shown to serve as a chemoattractant for monocytes and cause an enhancement in their invasive properties; it also induces the expression of CCR3, CCR5, and CXCR4 in monocytes and macrophages, thereby promoting HIV-1 infection. 80 It participates in a positive feedback mechanism to reactivate HIV from latency, but a decrease in Tat levels reduces HIV transcription and protein production to subthreshold levels, rendering the host cell inactive in viral production. 16,81 Several studies have shown that naturally occurring polymorphisms in Tat-like P10S, W11R, K19R, A42V, and Y47H (Fig. 2) result in impaired transactivation activity leading to a significantly enriched number of latently infected CD4+ T cells. 16,81 Besides, Tat in circulation can be captured by bystander cells, where it can induce apoptosis. 82
In monocytes, Tat suppresses STAT-1 activation triggered by gamma interferon (IFN-γ) and IFN-γ-induced autophagy in macrophages suggesting impaired antigen processing and elimination of pathogens. 83 In a recent study, selective degradation of Tat by autophagy showed blockage of HIV replication in CD4+ T cells. 84 It would be interesting, therefore, to examine the effects of specific degradation of Tat by autophagy upon establishment of HIV-1 latency.
Vpu
Volcic et al. very recently identified Vpu as a viral protein involved in the maintenance of HIV-1 latency through modulation of the NF-κB pathway, thus providing a strong rationale to screen for novel Vpu inhibitors as potential HIV-1 latency reversal agents (LRAs). 85 –87 However, whether their tethering activity favors cell-to-cell viral spread or inhibits overall viral dissemination is still a mystery.
Functional Role of Latent Reservoirs as Defined by Transcriptomics and Proteomics Studies
Exploratory transcriptomics and proteomics studies have helped to decode the alterations in immune cell functions during the establishment of latent reservoirs. Understanding how HIV modulates the normal physiology of the immune cells and enables immune escape has been a subject of great interest. A key question is how specific proteins are modulated during HIV infection and how such modulations affect viral gene expression and lead to the establishment of latent reservoirs. Insights into the mechanisms of latency and viral reactivation are essential for the design of rational strategies to reduce the size of the latent reservoir pool. A list of studies that have profiled proteins and transcriptional factors under conditions that promote latency establishment are tabulated in Table 1. 11,12,40,88 –108 These differentially expressed genes and markers are therefore candidate factors that might regulate latency. These data provide a beneficial resource for future mechanistic studies on HIV-1 latency.
Summary of the Functional Role of Latent Reservoirs Defined by Transcriptomics and Proteomics Analysis
PI and II - Phase I and Phase II Clinical trials.
APCs, antigen presenting cells; CNS, central nervous system; HIV, human immunodeficiency virus; INSTIs, integrase strand transfer inhibitors; LEDGINs, small-molecule integrase that target the binding pocket of LEDGF/p75; LTR, long terminal repeat; NFAT, nuclear factor of activated T-cells; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; NPC; PIC, preintegration complex; TAR, transactivating element; Tat, transactivator of transcription; TCR, T-cell receptor; TGF-β, tumor growth factor-β; TCM, central memory cells; TTM, transitional memory cells.
Challenges of Therapeutic Interventions
Viral clearance is impossible without targeting the latent reservoirs, especially of the monocyte/macrophage lineage. Several interventions and tools are being tried to minimize/eliminate the pool of latent viral reservoirs. Integrase strand transfer inhibitors (INSTIs) such as raltegravir, dolutegravir, elvitegravir, and a rare class of integration inhibitors called small-molecule integrase that target the binding pocket of LEDGF/p75 (LEDGINs) are reported to target latent reservoirs among CD4+ T cells and macrophages during the preintegration stage (Table 1). It has been noticed that a single point mutation in the INSTI target site in macrophages hinders the efficacy of the drug. 109 INSTIs or LEDGINs are ineffective once latency is established in the tissue sanctuaries. Thus, recent therapeutic interventions for latency focus on viral reactivation followed by eradication through ART known as the “Shock and Kill” strategy.
Latency reversing agents that target the histone deacetylases (HDACs) are crucial for suppressing HIV-1 proviral DNA expression. HDAC-inhibitors such as vorinostat, romidepsin, disulfiram, and chidamide are in phase I clinical trials and are showing promising levels of viral reactivation in HIV-1 patients on cART. 110 In all these clinical trials involving LRAs, viral load has been determined specifically in peripheral blood lymphocytes and not in tissue reservoirs such as microglial cells or rectal tissues, which are rather difficult to obtain. Moreover, studies have shown that low frequency of memory cytotoxic T cells (CTLs) reactivation in chronically infected persons due to persistent viral replication and generation of new immune escape variants. 111 –113
The CCR5 delta32 homozygous genotype (delta32/delta32) is known to confer protection against HIV-1 infection, as shown in a few clinical trials. Besides its limited success, the transplantation procedure is risky and the CCR5Δ32 mutation reduces protection against certain viral infections such as influenza and the West Nile fever. 114 –116 Gene-editing techniques such as antisense RNAs, ribozymes, dominant-negative mutants, TAR RNA decoys, and RNAi modalities are now being evaluated in preclinical and clinical contexts for the eradication of HIV. 117 However, the strategies that use recombinant proteins or RNAs to target latently infected cells require a unique way to target and deliver expression constructs or recombinant macromolecules to latently infected cells. Even so, their success may rely on the identification of relevant biomarkers to target these cells.
Immunotherapeutic interventions are aimed at improving infected cell death or clearance through LRA-induced viral replication or increasing innate immunity against HIV-infected cells. In this regard, LRAs in combination with IL-15 have been shown to induce latent cell reactivation and viral clearance by natural killer activity. 118 Chimeric antigen receptor (CAR) T cells show broad-spectrum viral-specific CTL activity. 119 However, the major drawback with CAR-T cells is the inability to deal with the rapidly evolving viral epitopes in HIV patients. 120,121 To address this issue, Herzig et al. developed a “MicAbody cCAR-T cell,” a bispecific CAR-T cell antibody with two ligands specific to NKG2D receptors viz. MIC—major histocompatibility complex class I chain-related ligand and ULBP ligand. The MicAbodies can be multiplexed to recognize diverse epitopes. When MicAbody cCAR-T cells pair with broadly neutralizing antibodies, they can destroy infected targets with great potency and prevent immune escape. 122
Long-acting slow-effective release ART (dubbed “LASER ART”) that uses nanoparticles to release highly active antiretroviral therapy drugs, thereby enhancing regimen adherence (e.g., NM23TC) 123,124 is the current strategy being studied for viral clearance and reservoir elimination. Lipid-drug nanoparticles not only increase the bioavailability of ART drugs in lymph nodes but also reduce toxicity for more than 14 days 125 and serve as nanocarriers (poly (lactic-co-glycolic acid) nanocarriers) to traffic and deliver LRAs to latent HIV reservoirs, thereby maximizing their therapeutic benefits. 126,127 An interesting proof of concept study by Jayant et al. showed that when vorinostat (a LRA) is packed with tenofovir assembled layer by layer in magnetic nanocarriers, it can reactivate and kill HIV in a sustained manner for 5 days across the blood-brain barrier. 126
Conclusion: The Missing Link in Latency Research
Despite significant advances and success in ART, eradication of HIV has continued to be an unreachable goal. Two serendipitous cases of HIV cure have been the silver lining of hope to this dark cloud. Decades of investment in HIV latency research have only focused on either understanding the molecular mechanisms of the disease or developing research tools required for clearing HIV from the body. While viral proteins such as gp120, viral infectivity factor, and Vpr have been extensively studied for their role in active infection, very few studies have documented their role in latency. Even so, some of these studies have quite a few limitations. Hence, an extensive study on the role of individual HIV proteins in the establishment of latency is needed.
It would be a worthwhile effort to comprehensively analyze the large amount of data generated through multiple studies on HIV latency to evocatively discover the underlying mechanisms of viral pathogenesis and cure. The promise of a single intervention therapy that has been brought closer to reality is the generation of HIV-resistant cells using viral vectors that can be used to deliver therapeutic gene payloads to hematopoietic stem and progenitor cells. Recent efforts on the transcriptomic and proteomic profiling of HIV-1-infected monocytes/macrophages and latent CD4+ T cell reservoirs have resulted in the identification of key genes/proteins that may be targeted for intervention strategies. More work in this line is definitely needed to understand the nature of the microenvironment and the processes that pave way for the trafficking of HIV during the establishment of latent reservoirs.
Footnotes
Acknowledgments
The authors thank Dr. J. Nancy Hilda, Dr. N. Sudhakar, and Mr. P.R. Asish for essential and constructive discussions. We apologize to those whose work we were not able to cite because of space constraints but acknowledge the contributions they have made to the field.
Authors' Contributions
L.E.H., G.K.C.P., and G.R. drafted, wrote, and revised the article. H.V. helped with the image construction and literature review.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
The first author (G.R.) acknowledges the Indian Council of Medical Research (ICMR), New Delhi, for Senior Research Fellowship (SRF No.HIV/Fellowship/1/10/2020-ECD-II).