
Abstract
Proper folding and assembly of major histocompatibility complex (MHC) class I complexes are essential for optimal peptide loading and subsequent antigen presentation. MHC class I folding involves the coordinated formation of multiple disulfide bonds within MHC class I molecules. However, the regulation of disulfide bond formation during the early process of MHC class I folding is uncharacterized. Here, we show that protein disulfide isomerase (PDI) catalyzes the disulfide bond formation of MHC class I molecules and thereby facilitates the assembly of MHC class I heavy chain with β2-microglobulin (β2m). Depletion of PDI but not ERp57 by RNAi interfered with the disulfide bond formation in the MHC class I molecules. In the absence of PDI, the association of free class I heavy chain with calnexin increased, whereas the assembly of MHC class I heavy chain–β2m heterodimers was delayed. These observations suggest that PDI-catalyzed disulfide bond formation of MHC class I molecules is an event downstream of the interaction of class I molecules with calnexin and upstream of their interaction with β2m. Thus, our data establish a critical function for PDI in the early assembly of MHC class I molecules. Antioxid. Redox Signal. 11, 2553–2561.
Introduction
MHC class I molecules undergo successive quality control with the assistance of molecular chaperones in the ER. Early events in MHC class I assembly involve interactions between MHC class I heavy chain (HC) and calnexin, the oxidoreductase ERp57, and β2-microglobulin (β2m) (1). Calnexin binds to newly synthesized free HC via a lectin interaction, facilitates the folding of MHC class I HC, and prevents aggregation (33 –35). MHC class I HC has two disulfide bonds in the α2 and α3 domains. ERp57, which is recruited by calnexin, plays a role in the disulfide bond formation of MHC class I HC (13, 41). Depletion of ERp57 impairs the α3-domain disulfide bond formation in mouse cells (41).
However, it is still controversial whether the catalytic activity of ERp57 is required for the quality control of MHC class I molecules. The redox state of MHC class I HCs is not influenced in ERp57 knockout mice (15). The redox activity of ERp57 is not necessary for MHC class I peptide loading (24). The majority of ERp57 in the peptide-loading complex (PLC) exists in the conjugated form with tapasin (25). MHC class I HC folding is linked to the assembly of MHC class I molecules because only fully disulfide-bonded class I HCs efficiently assemble with β2m (30, 39). The later stages of MHC class I assembly begin with the association of MHC class I HC with β2m. The MHC class I HC-(2m heterodimer is recruited into the PLC, where it can load optimal peptide with the help of chaperones such as transporter associated with antigen processing (TAP), tapasin, calreticulin, and ERp57 (12, 22). Unassembled MHC class I HC interacts poorly with the TAP complex (2, 31) and cannot present antigenic peptides at the cell surface (29).
The formation of a mixed disulfide between class I HC and components of the PLC such as PDI (23) and tapasin (3) occurs after MHC class I assembly into the PLC. These findings suggest that even after incorporation of oxidized MHC class I molecules into the PLC, the thiol-based redox regulation might play a role during late MHC class I assembly and peptide loading. Recently, we reported that protein disulfide isomerase (PDI) is a component of the PLC and affects optimal peptide loading by regulating the redox state of the α2 disulfide bond (23). PDI, a member of the PDI family, has two catalytic domains that contain an active-site CXXC motif (a and a') and two noncatalytic domains (b and b') (9). The catalytic domains are required for the PDI function as a thiol disulfide oxidase, reductase, and isomerase (5, 6, 18). The PDI-C36,39S point mutation, in which the catalytic site of the α domain is destroyed, failed to restore folding of MHC class I HCs in endogenous PDI-depleted cells (23). It is especially interesting that PDI forms a disulfide intermediate with MHC class I HC, not only within the PLC, but also outside the PLC (23). This suggests that PDI may be involved in the early stages of MHC class I assembly.
Here we demonstrate that depletion of PDI impairs the early oxidative folding and assembly of MHC class I molecules. PDI interacts with not only (2m-assembled MHC class I HC but also free MHC class I HC. Furthermore, we show that in the absence of PDI, the interaction of free MHC class I HC with calnexin is increased, whereas the interaction between MHC class I HC and β2m is reduced in PDI-depleted cells. Collectively, our data demonstrate the involvement of PDI in the early folding and assembly of MHC class I molecules.
Materials and Methods
Cell lines and retroviral infection
The HeLa cell line and retroviral packaging cells (Phoenix amphotropic cells) were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% FBS (HyClone Laboratories, Logan, UT), 2 mM
Constructs
A retrovirus-based vector (pSuper-Retro) for siRNA expression was purchased from OligoEngine Co. (Seattle, WA). Retrovirus/siRNA-expressing vectors siGFP, siPDI-ORF, siPDI-UTR, and siERp57 were constructed according to the manufacturer's instructions. Oligos containing the sequence of small interference RNA were synthesized as follows: siGFP (forward, 5′-GATCCCC GGTTATGTACAGGAACGCA TTCAAGAGA TGCGTTCCTGTACATAACCTT TTTTTA-3′; reverse, 5′-AGCTTAAAAA AAGGTTATGTACAGGAACGCA TCTCTTGAA TGCGTTCCTGTACATAACC GGG-3′); siPDI-ORF (forward, 5′-GATCCCC GGACCATGAGAACATCGTC TTCAAGAGA GACGATGTTCTCATGGTCCTT TTTTTA-3′; reverse, 5′-AGCTTAAAAA AAGGACCATGAGAACATCGTC TCTCTTGAA GACGATGTTCTCATGGTCC GGG-3′); siPDI-UTR (forward, 5′-GATCCCC GATGAACTGTAATACGCAA TTCAAGAGA TTGCGTATTACAGTTCATC TTTTTA-3′; reverse, 5′-AGCTTAAAAA GATGAACTGTAATACGCAA TCTCTTGAA TTGCGTATTACAGTTCATC GGG-3′); siERp57 (forward, 5′-GATCCCC GGAATTGTCAGCCACTTGA TTCAAGAGA TCAAGTGGCTGACAATTCC TTTTTA-3′; reverse, 5′-AGCTTAAAAA GGAATTGTCAGCCACTTGA TCTCTTGAA TCAAGTGGCTGACAATTCC GGG-3′). An myc-tagged PDI cDNA construct was generated by PCR and inserted into the pcDNA 3.1 vector (Invitrogen, Carlsbad, CA). The myc tag was placed in front of the C-terminal ER retention signal (KDEL). Mutant (C36,39,380,383S) and wild-type PDI constructs were cloned into the mammalian expression vector pcDNA3.1 vector (Invitrogen).
Antibodies
The mAb W6/32 recognizes only complexed MHC class I HC and (2m. The mAb HC10 that was raised against denatured MHC class I HC recognizes only free MHC class I HC (32) and was kindly provided by J. Neefjes (The Netherlands Cancer Institute, Amsterdam, The Netherlands). The ERp57 mAb, anti-(2m antibody BBM.1, and anti-MHC class I antibody H-300 were purchased from Santa Cruz Biotechnology. Rabbit polyclonal PDI antibody was raised against recombinant PDI purified from Escherichia coli.
Pulse chase and immunoprecipitation
Cells (5 × 106) were starved for 40 min in medium lacking methionine, labeled with 0.1 mCi/ml [35S]methionine (TranS-label; NEN, Boston, MA) for 5 min with 10 mM DTT, and chased in normal medium without dithiothreitol (DTT) for the indicated times. Cells were lysed by using 1% Nonidet P-40 (Sigma-Aldrich, St. Louis, MO) in PBS with 10 mM N-ethylmaleimide (NEM) and a protease inhibitor mixture for 30 min at 4°C. After preclearing cell lysates with protein G-Sepharose (Amersham Pharmacia Biotech, Piscataway, NJ), primary antibodies and protein G-Sepharose were added to the supernatant and incubated at 4°C with rotation for 1 h. The beads were washed 3 times with 0.1% Nonidet P-40 in PBS. Proteins were eluted from the beads by boiling in SDS sample buffer and separated with 10% SDS-PAGE. The gels were dried, exposed to BAS film for 14 h, and then analyzed with the BAS-2500 PhosphorImaging System (Fuji Film, Tokyo, Japan).
Coimmunoprecipitation and Western blot analysis
Cells were lysed in 1% digitonin in a protease inhibitor–supplemented buffer containing 25 mM HEPES, 100 mM NaCl, 10 mM CaCl2, and 5 mM MgCl2 (pH 7.6). Lysates were precleared with protein G-Sepharose (Amersham Pharmacia Biotech, Little Chalfont, UK) for 1 h at 4°C. For immunoprecipitation, samples were incubated with the appropriate antibodies for 4 h at 4°C before protein G-Sepharose beads were added. Beads were washed 4 times with 0.1% digitonin, and bound proteins were eluted by boiling in SDS sample buffer. Proteins were separated with 10% SDS-PAGE, transferred onto a nitrocellulose membrane, blocked with 5% skim milk in PBS containing 0.1% Tween 20 for 1 h, and probed with the appropriate antibodies for 4 h. Membranes were washed 3 times with PBS containing 0.1% Tween 20 and incubated with HRP-conjugated streptavidin (Pierce, Rockford, IL) for 1 h at 4°C. Immunoblots were visualized with enhanced chemiluminescence (ECL) detection reagent (Pierce, Rockford, IL).
Results
Depletion of PDI slows oxidation of the MHC class I heavy chain
In addition to being detected within the PLC, the disulfide intermediates between PDI and MHC class I HC also were detected outside the PLC (23). We therefore hypothesized that PDI could play a role in the early stages of MHC class I assembly before the incorporation of MHC class I molecules into the PLC. To test this hypothesis, we used RNA interference to knock down PDI. Because PDI has a long half-life (36) and is abundant in the ER, knockdown efficiency was low. To increase the efficiency of delivering of siRNA, we used a retroviral infection system. We initially confirmed that pSuper-Retro vector itself does not affect expression of the components of the PLC and MHC class I–mediated antigen presentation (Fig. 1A; compare lane 1 with 2). After enrichment of transfectants by selection in 1 μg/ml puromycin for 2 weeks, cells expressing GFP-, PDI-, or ERp57-specific siRNA were used for experiments. GFP siRNA was used as a control.
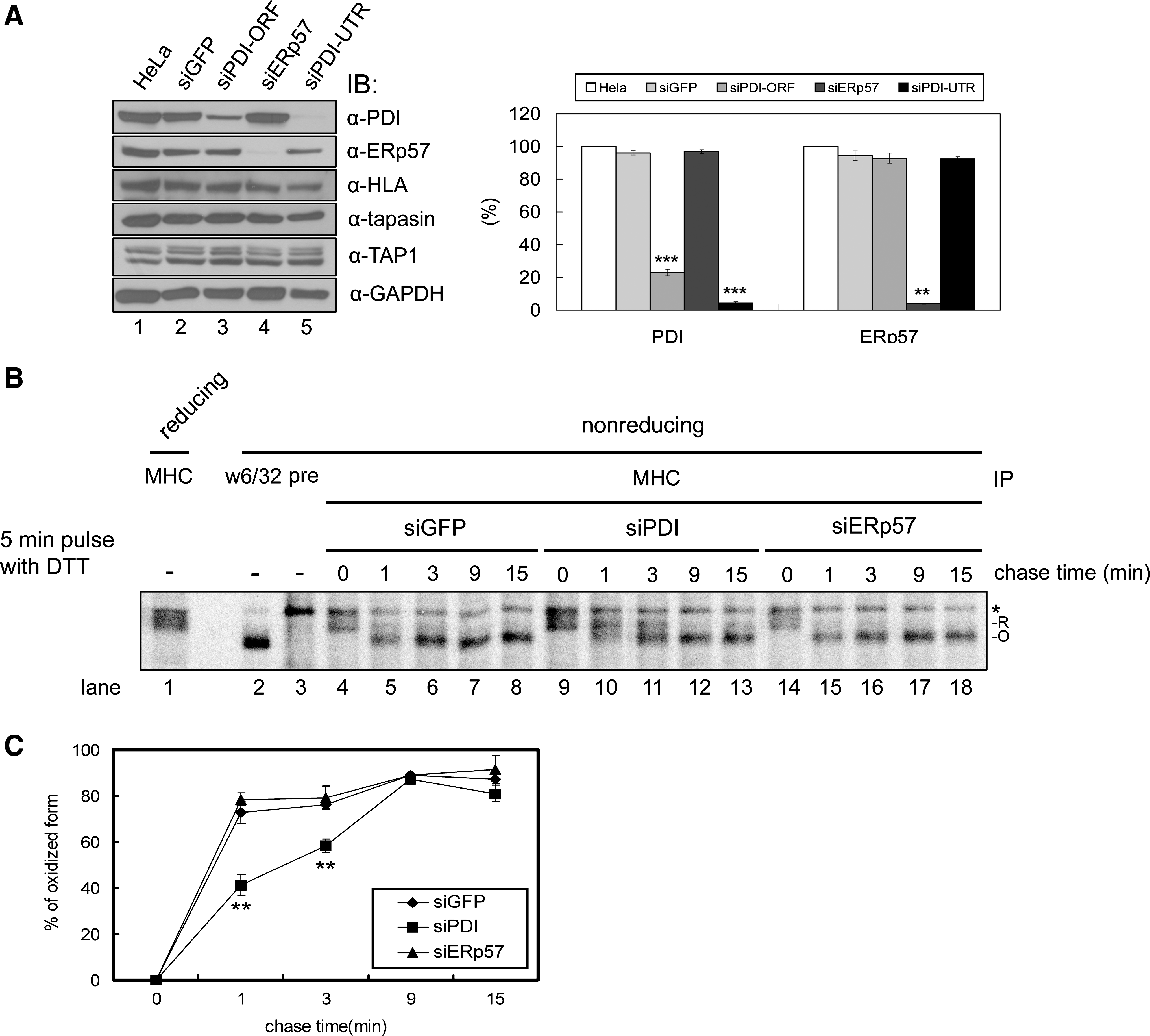
In previous report, we used PDI siRNA that was designed to target the ORF of PDI (siPDI-ORF) (23). To assess better the potential involvement of the catalytic activity of PDI in the early oxidative folding of MHC class I molecules, we constructed a new PDI siRNA targeting 3' UTR of PDI (siPDI-UTR). Because siPDI-UTR reduced the expression of PDI more efficiently than siPDI-ORF (Fig. 1A; compare lane 3 with 5), we used siPDI-UTR (hereinafter referred to as siPDI) throughout this study. The knockdown efficiency of PDI and ERp57 by siRNA was 80–90% (Fig. 1A).
To determine whether depleting PDI affects the disulfide bond formation of MHC class I HC in the early stages of assembly, we labeled cells with a short pulse of [35S]methionine and immunoprecipitated cell lysates with MHC class I–specific antibody. Disulfide bond formation of MHC class I was then monitored with SDS-PAGE under nonreducing conditions. However, the rapid oxidation of MHC class I HC in HeLa cells made it difficult to monitor the change in the redox state (data not shown). To overcome this difficulty, we used a DTT-labeling method (19). This method enables it to retain the reduced form of class I HC until the initiation of the chase. HeLa cells were pulsed for 5 min in the presence of DTT and then chased for 0, 1, 3, 9, and 15 min after washing (Fig. 1B).
We observed three major bands on nonreducing gel. The top band (*) represents a nonspecific band, because it has the same mobility with a band observed in immunoprecipitaiton with preimmune serum (Fig. 1B; compare lane 3 and others). MHC class I HCs were separated into two bands (R and O) according to their redox states under nonreducing conditions. Because the upper band shares mobility similar to that of reduced MHC class I HC under reducing conditions (Fig. 1B, lane 1), it represents the reduced form of class I HC. The lower band represents the oxidized form of class I HC because it shares mobility similar to that of the W6/32-recognized pool of MHC class I molecules, which is fully oxidized under nonreducing conditions (Fig. 1B, lane 2). As expected, most class I HCs were reduced at the 0-min chase (Fig. 1B, lanes 4, 9, and 14). MHC class I HCs were completely oxidized after the 3-min chase in HeLa cells expressing GFP or ERp57 siRNA, whereas the reduced form of MHC class I HC remained at the same time in the PDI knockdown cells (Fig. 1B; compare lane 11 with lanes 6 and 16). In contrast, in cells depleted of ERp57, a structural and functional homologue of PDI, the folding of MHC class I HCs was similar to the folding in control cells (Fig. 1B; compare lanes 14 to 18 with 4 to 8). Taking into account these results and the previous data that PDI knockdown does not affect the expression of other cell-surface glycoproteins (23), the involvement of PDI in the early oxidative folding of MHC class I molecules appears to be specific.
PDI interacts with free MHC class I heavy chains
Physical interaction of PDI with target proteins is essential for its function as an oxidase (11). Previously, we showed that PDI forms disulfide intermediates with MHC class I HC within as well as outside of TAP complexes (23). However, it remains unknown whether PDI interacts with free HC. To test this possibility, we lysed HeLa cells in 1% digitonin and immunoprecipitated the lysates with either the mAb HC10 that recognizes only free HCs or the mAb W6/32 that recognizes only complexed MHC class I HC and β2m. The immunoprecipitates were resolved with SDS-PAGE under reducing condition, followed by immunoblotting for PDI. Interestingly, we detected a substantial interaction between free HC and PDI (Fig. 2, lane 5). The isotype-matched control mouse immunoglobulin exhibited no reactivity with PDI (lane 3). Moreover, the mAb HC10 epitope represents the PxxWDR motif in α1 domain of MHC class I HC (26) that is absent in PDI.
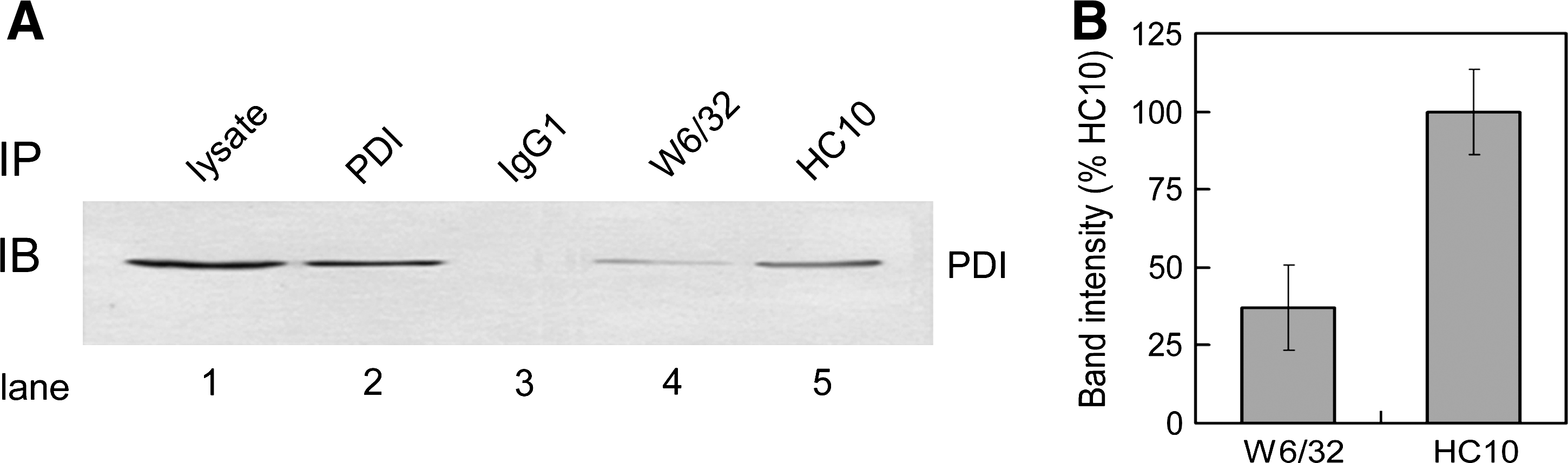
Thus, the observed interaction of free HC with PDI is unlikely due to the cross-reactivity of mAb HC10. A physical association between PDI and the MHC class I HC-β2m heterodimers, albeit slightly weak, was readily detected (lane 4). These data indicate that PDI binds free HC and that this binding may be required for the oxidation of MHC class I HC. Presumably, an interaction between PDI and the MHC class I HC-β2m heterodimer represents an event occurring within the PLC.
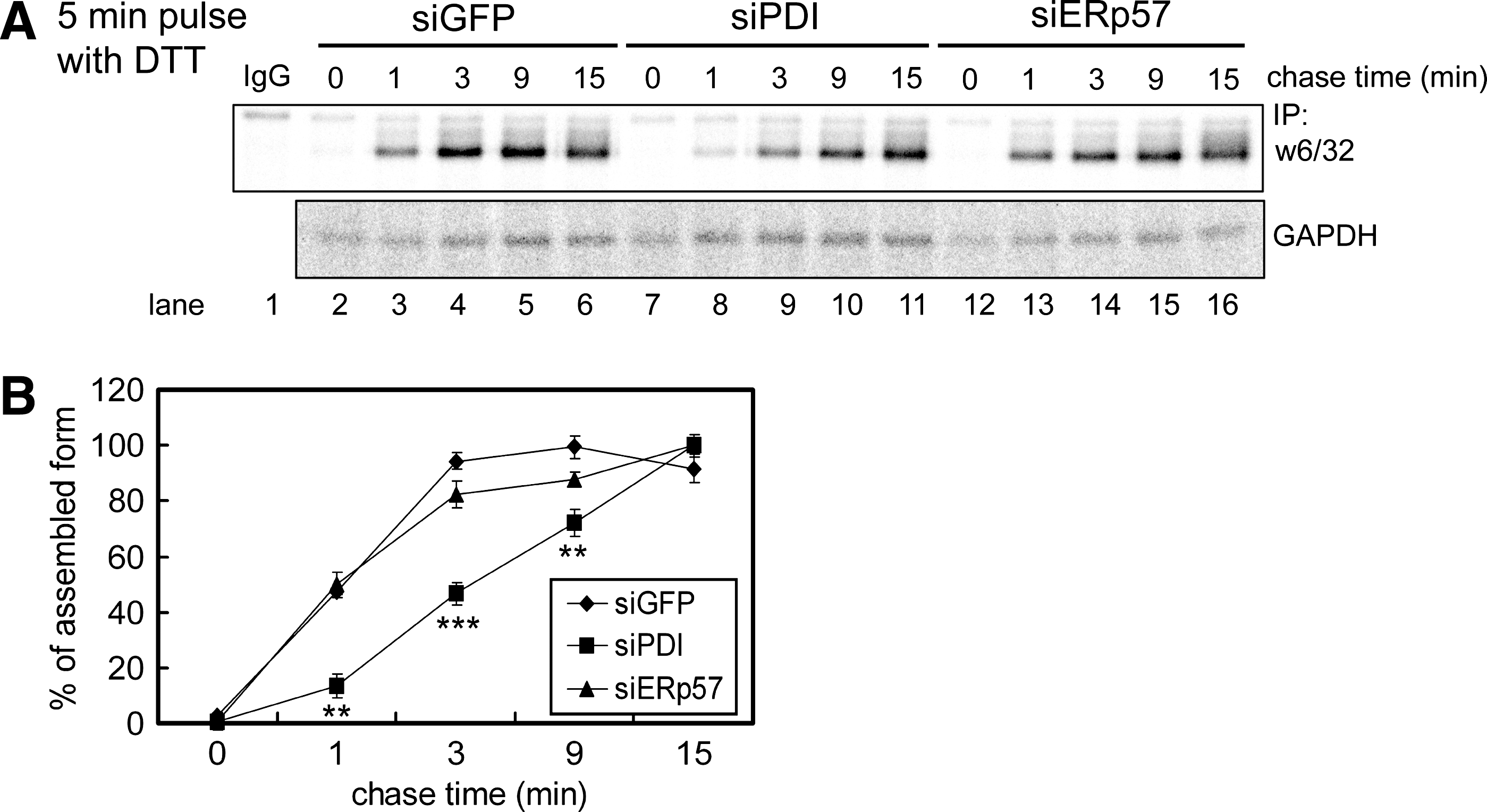
Depletion of PDI delays the assembly of the MHC class I heavy chain–β2m heterodimer
These results suggest that the formation of correct disulfide bonds is required for correct folding and assembly of MHC class I molecules. We examined the effect of PDI or ERp57 depletion on the kinetics of MHC class I HC–β2m heterodimer formation. By using a conformation-specific monoclonal antibody (W6/32) that recognizes only complexed MHC class I HC and β2m, we were able to measure the level of the MHC class I HC–(2m heterodimer. The MHC class I HC‱β2m level was quantitated by using a phosphoimaging device with GAPDH levels as the loading control (Fig. 3A and B). PDI knockdown caused a decrease in the rate of MHC class I HC‱β2m heterodimer formation (Fig. 3A; compare lanes 7 through 11 with 2 through 6), whereas ERp57 knockdown did not affect the kinetics of MHC class I HC‱β2m heterodimer formation (Fig. 3A; compare lanes 12 through 16 with 2 through 6). These results are consistent with the previous report that only fully disulfide-bonded class I molecules efficiently assemble with β2m (30, 39).
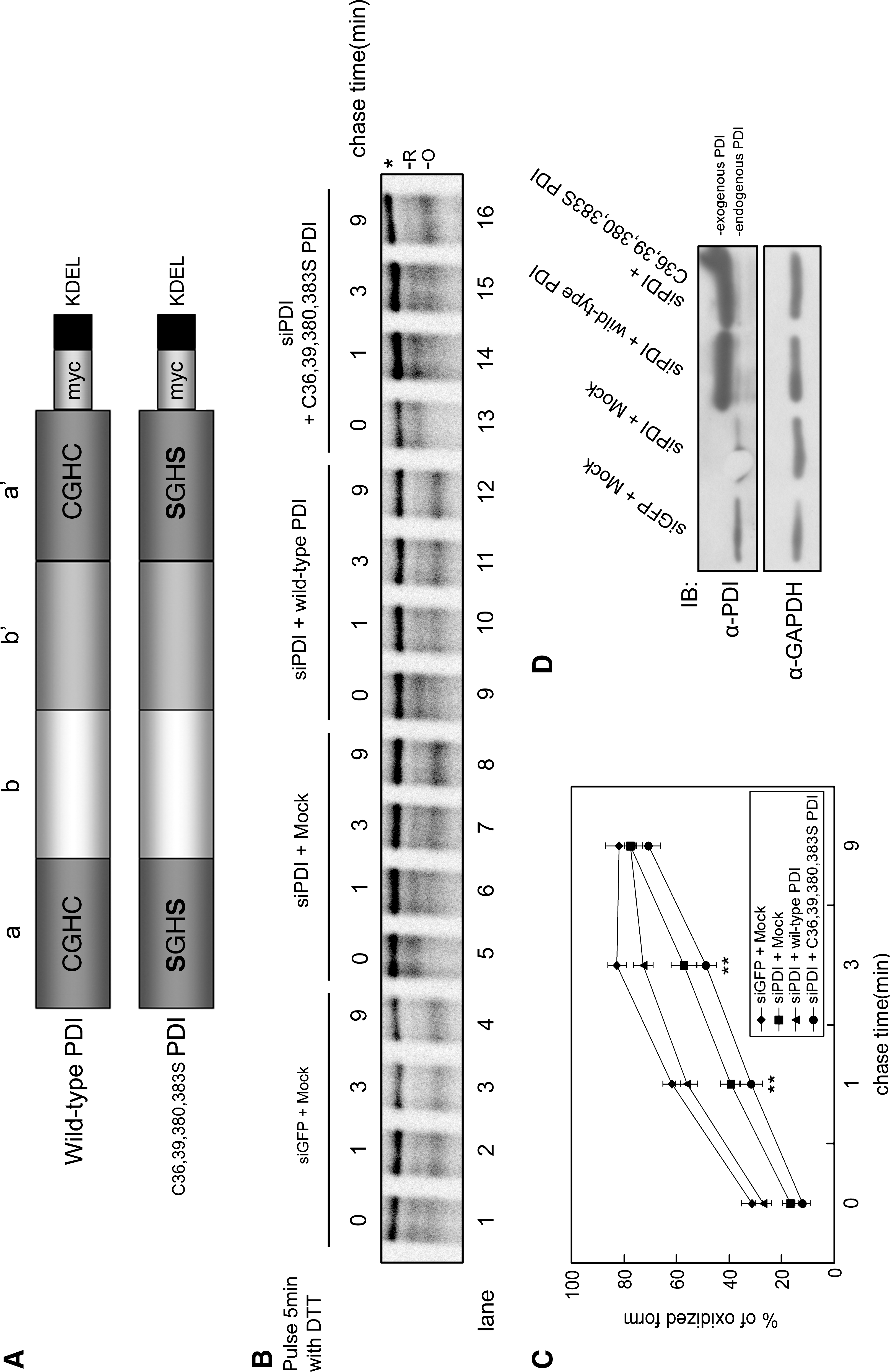
Catalytic activity of PDI is required for folding of MHC class I molecules
The a and a' domains of PDI contain four cysteine residues that are thiol oxidoreductase active sites (CXXC) (9). To confirm that formation of disulfide bonds in MHC class I HC involves the catalytic activity of PDI, we made the PDI C36,39,380,383S mutant, in which the four catalytic cysteine residues of PDI have been replaced by serine (Fig. 4A). HeLa cells expressing siPDI were mock transfected or were transfected with wild-type PDI or mutant PDI before being pulse-chased. Because the PDI siRNA was constructed to target the 3' UTR, we were able to express the full-length PDI in a background depleted of endogenous PDI (Fig. 4D, upper panel). In endogenous PDI-depleted cells, the oxidation rate of MHC class I HC was decreased compared with the rate in control cells (Fig. 4B; compare lanes 1 through 4 and 5 through 8), thereby confirming the result in Fig. 1. Expression of wild-type PDI rescued the oxidation rate of MHC class I HC (compare lanes 5 through 8 and 9 through 12), whereas expression of mutant PDI did not (compare lanes 5 through 8 and 13 through 16). From these data, we conclude that oxidation of free HC in the early assembly necessitates the catalytic activity of PDI.
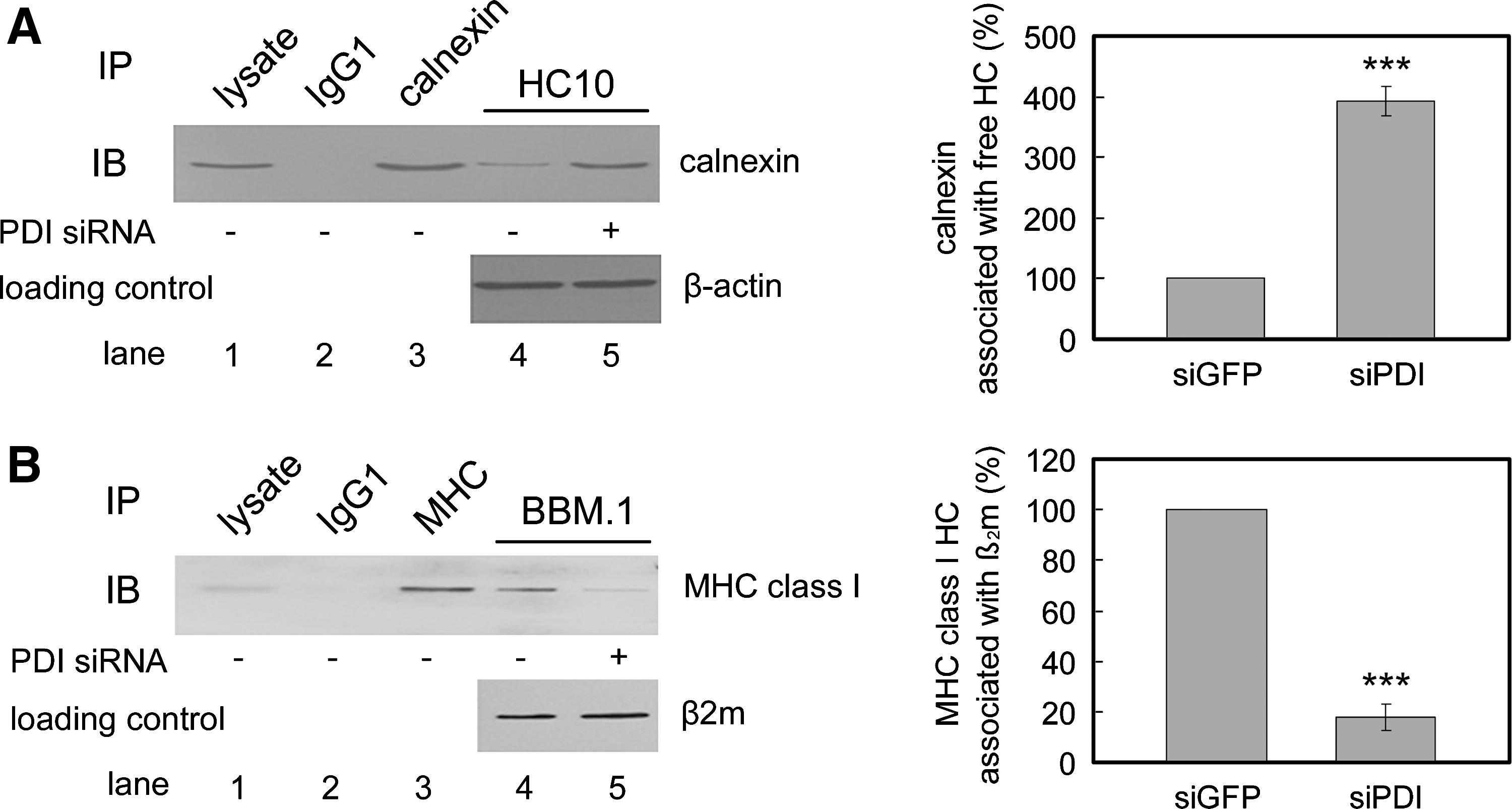
PDI acts at an early stage after the association of MHC class I heavy chain with calnexin but before the MHC class I heavy chain–β2m association
Calnexin is involved in the biogenesis of newly synthesized MHC class I HCs (34, 37) and associates with both reduced and oxidized free HC before the association of MHC class I HC with β2m (35). We addressed the question of at which stage PDI acts in the process of MHC class I assembly. We examined the steady-state levels of both the MHC class I HC–calnexin association and the MHC class I HC–β2m association in PDI-depleted cells. Immunoprecipitation and Western blot analysis revealed that PDI knockdown caused an accumulation of the MHC class I HC–calnexin complex (Fig. 5A; compare lanes 4 and 5). The accumulation was increased fourfold in PDI-depleted cells (Fig. 5A). In the same cells, the steady-state level of MHC class I HC–β2m heterodimers was substantially decreased (Fig. 5B; compare lanes 4 and 5). The filter was reprobed with an anti-β2m antibody (BBM.1) to confirm the equal loading of the protein (Fig. 5B, bottom panel). The quantity of MHC class I HC associated with β2m decreased by 20% in PDI-depleted cells (Fig. 5B). In human cells, calnexin binds to free HC but not to β2m-assembled HC (20, 28). Thus, accumulation of the MHC class I HC–calnexin complex with a concomitant decrease of HC–β2m heterodimers in the absence of PDI demonstrates that PDI-mediated oxidation of class I HC may occur at a stage between the MHC class I HC–calnexin and MHC class I HC–β2m interactions.
Discussion
This study demonstrated that PDI plays a role in the in the early stages of the folding and assembly of MHC class I molecules. We recently reported that PDI forms a disulfide intermediate with MHC class I HC within the PLC and catalyzes the oxidation of the α2 domain in the peptide-binding groove. In this study, we also detected an HC-PDI disulfide intermediate outside the PLC (23). This PDI-associated HC outside the PLC could represent the pool of HC undergoing early oxidative folding. Alternatively, it could represent the HC pool that was en route to the ERAD pathway as parts of ER quality control, because PDI is involved in the retrotranslocation of misfolded proteins (14, 38).
In this study, we focused on the role for PDI in the early stage of MHC class I assembly. To investigate this possibility, we monitored the folding and assembly of MHC class I molecules with short pulse-chase experiments in PDI knockdown cells. Our results showed that depletion of PDI delayed the folding and assembly of MHC class I molecules (Figs. 1 and 3). In addition, newly synthesized free MHC class I HCs interacted with PDI (Fig. 2). Given the previous data that PDI forms a disulfide intermediate with MHC class I HC outside the PLC (23), PDI would directly interact with free HCs through intermolecular disulfide bonds. Furthermore, because calnexin specifically binds to ERp57 but not PDI (21, 27), we conclude that the interaction between PDI and MHC class I HC is direct but not indirect via calnexin. Taken together, these results strongly suggest that PDI is involved in the early stage of MHC class I assembly (Fig. 6). Other PDI family members, such as ERp57, may be involved in the early folding and assembly of MHC class I molecules (8, 41). However, no study demonstrates this. This is the first report showing a function for PDI in the early folding and assembly of MHC class I molecules.
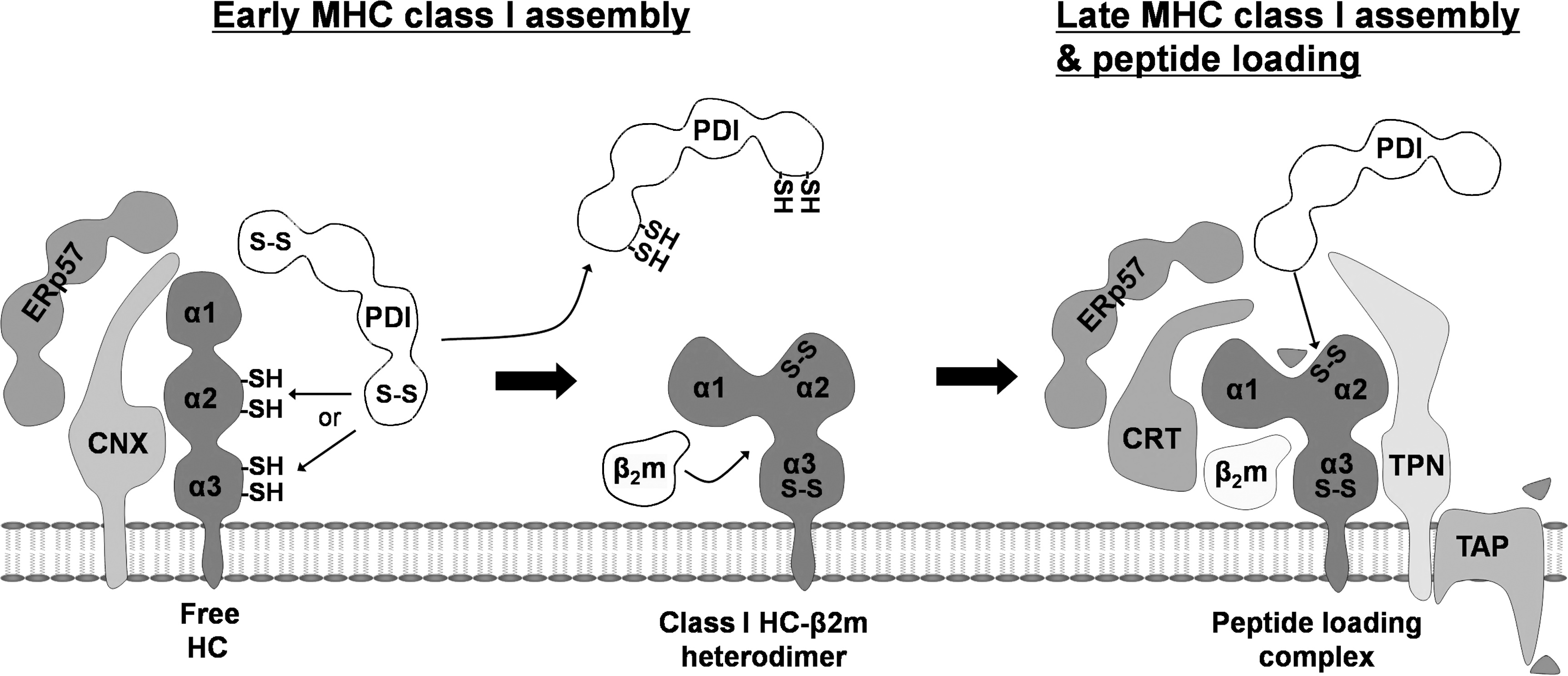
Calnexin binds to free HCs, whereas β2m-assembled MHC class I HCs disassociated from calnexin in human cells (20). Therefore, we can divide the early assembly of MHC class I molecules into two stages: a calnexin-associated stage and a calnexin-disassociated stage. In which stage does PDI affect the folding of MHC class I molecules? Given our results, PDI may participate in the calnexin-associated stage. We showed that the interaction of free HC with calnexin accumulated in the absence of PDI, whereas the interaction of MHC class I HC with β2m was reduced (Fig. 5). Calnexin distinguishes free HC from assembled MHC class I molecules and retains free HC in the ER (28). Hence, the accumulation of free HC with calnexin would result from an increased pool of free HCs in the absence of PDI. This result indicates that PDI catalyzes the oxidation of MHC class I HC at the calnexin-associated stage. In addition, reduced interaction of MHC class I HC with β2m in the absence of PDI implies that the PDI-mediated oxidation of MHC class I HC occurs before its assembly with β2m because fully oxidized MHC class I HCs efficiently assemble only with β2m (30, 39). A mutation that disrupted the intra-disulfide bonds of MHC class I HC strongly inhibited the assembly with β2m (30, 39). Therefore, we conclude that PDI-mediated oxidation of MHC class I HC may occur at the calnexin-associated stage and before assembly with β2m.
An interesting feature of our results is that depletion of ERp57 did not affect the folding of MHC class I molecules in HeLa cells. This result contrasts with that of a previous report. Zhang and colleagues (41) showed that depletion of ERp57 delayed the α3 disulfide formation of MHC class I molecules in mouse cells. Two possibilities might explain this discrepancy: (a) the assembly of MHC class I molecules in human cells differs from that in mouse cells; and (b) PDI is redundant enough to allow the folding of MHC class I molecules without ERp57 in human cells. The first possibility is supported by several reports. York et al. (40) reported a mutant cell with a defect in the early assembly of MHC class I molecules by an unknown factor. Interestingly, the assemblies of primate, but not mouse, MHC class I molecules were impaired in mutant cells. In addition, calnexin interacts with mouse assembled MHC class I, but not with human MHC class I (7, 20). These lines of evidence suggest a difference between human and mouse early assembly of MHC class I molecules. For these reasons, the function of ERp57 in the early assembly of MHC class I molecules may be different between human and mouse cells. The second possibility is that PDI and ERp57 may have redundancies with regard to their function in MHC class I assembly. However, because the expression level of PDI varies in each tissue (4), the study of the effects of depletion of both PDI and ERp57 in various cell lines will determine whether they act redundantly.
In this study, we identified a novel function of PDI in the early folding and assembly of MHC class I molecules. However, unsolved questions remain. First, we could not distinguish which domain PDI targets. PDI could oxidize the α2 domain in the early stages of MHC class I assembly, similar to the way PDI regulates the disulfide bond of the α2 domain in the PLC (23); or PDI could play a role in oxidation of the α3 domain as does ERp57 (41). To study this, antibodies that recognize the different redox states of each domain of MHC class I HCs are required.
The second unknown is the dynamics of the interaction between PDI and MHC class I molecules. It would be interesting to study whether MHC class I HC and PDI maintain their interaction from the early stage to the late stage. These data will provide further insights into the molecular basis of thiol-based redox regulation of MHC class I–restricted antigen presentation.
Footnotes
Acknowledgments
We thank J. Neefjes for providing the HC10 antibody. This work was supported by the Creative Research Initiatives Center for Antigen Presentation of MOST/KOSEF. K. K. and C. O. were supported by the BK21 fellowship.
Author Disclosure Statement
This work was supported by National Creative Research Initiative Program of MOST/KOSEF.