
Abstract
This study investigated the time course of heme oxygenase (HO)-1 expression and the role of endogenous HO-1 in hepatic ischemia and reperfusion (I/R). Rats were pretreated with hemin, an HO-1 inducer, and zinc protoporphyrin (ZnPP), an HO-1 inhibitor. Hepatic HO activity increased at 1 h after reperfusion, reaching a maximum at 6 h after reperfusion and then declined. HO-1 mRNA and protein expression in I/R liver were upregulated prior to reperfusion and highly induced again by reperfusion. The ALT level was upregulated at all time points, with a peak at 4–6 h. This increase was augmented by ZnPP but attenuated by hemin. Lipid peroxidation and serum HMGB1 release significantly increased at 1 h after reperfusion and remained elevated throughout the 24 h of reperfusion period, whereas the glutathione content decreased markedly at 4–6 h after reperfusion. These changes were attenuated by hemin but augmented by ZnPP. The levels of serum TNF-α, iNOS, and COX-2 protein and mRNA expressions were upregulated after reperfusion, further enhanced by ZnPP, and suppressed by hemin. HO-1 overexpression protects the liver against I/R injury by modulating oxidative stress and proinflammatory mediators. Antioxid. Redox Signal. 13, 1503–1512.
Introduction
Upregulation of antioxidant/detoxification genes, including HO-1 and glutathione S-transferase, is mediated by antioxidant response element (ARE). The transcription factor nuclear factor erythroid 2-related factor (Nrf )2 plays a pivotal role in the activation of ARE-driven antioxidant gene expression against ROS (1). HO-1 is an endogenous, cytoprotective enzyme that is upregulated under conditions of oxidant stress, and the production of HO-1 ameliorates the liver I/R injury in fat Zucker rats and cirrhotic rats (2, 39). The main activity of HO-1 is to catabolize the oxidative degradation of heme into carbon monoxide (CO), free iron, and biliverdin. Bilirubin/biliverdin is a potent antioxidant that scavenges peroxyl radicals, and CO exerts powerful anti-inflammatory and antiapoptotic effects (27). Based on previous observations, exogenous induction of HO-1 has been proposed as a potentially powerful therapeutic option to protect liver against I/R injury. Despite the protective effects of upregulation of the HO-1 pathway, there is increasing evidence that HO-1 overexpression and activity are not exclusively cytoprotective. Recent studies indicate that the protection might be restricted to a narrow threshold of HO-1 overexpression. Indeed, several studies have shown the excessive overexpression of HO-1 is directly related to increased injury (32, 33). Therefore, the potential beneficial effect of HO-1 during reperfusion injury in liver is not clear.
The aim of this study was to clarify the role of HO-1 in a time sequence during hepatic I/R in the rat. To that end, we conducted this experiment to elucidate: 1) the time course of the induction of HO-1 mRNA, protein and its activity in the liver in response to I/R; and 2) the mechanisms underlying the beneficial effects of hepatic HO-1 during I/R.
Materials and Methods
Liver warm ischemia and reperfusion procedure
All animal protocols were approved by the Animal Care Committee of Sungkyunkwan University. Male Sprague-Dawley rats (body weight 270–300 g, Samtako, Inc., Osan-si, Korea) were fasted for 18 h before the experiments. Briefly, rats were anaesthetized with an intraperitoneal injection of ketamine (60 mg/kg) and xylazine (8 mg/kg). The rats were laparotomized and liver ischemia was induced by clamping the pedicles of the left and median lobes for 60 min. At the end of the ischemic period, the clip around the left branches of the portal vein was removed. Liver tissue and blood samples were taken at 0 (immediately after reperfusion), 1, 2, 4, 6, 8, 12, and 24 h after reperfusion. The liver specimens for histopathological analysis were obtained 24 h after reperfusion. The remaining liver tissues were frozen immediately in liquid nitrogen and kept at −80°C until analyzed.
Drug treatment
Zinc protoporphyrin (ZnPP, 10 μmol/kg) and hemin (30 mg/kg) were prepared under subdued light by dissolving the compound in 1 ml of 0.2 M NaOH, adjusting the pH to 7.4 with 1 M HCl, and diluting the solution to the final volume with 0.9% NaCl (20). Rats were pretreated twice with ZnPP or with hemin at 16 and 3 h prior to ischemia; an equal volume of saline was administered for the controls. The dose and the injection time of ZnPP and hemin treatment were based on previous reports (15, 16) and our preliminary studies. Rats were randomly divided into six groups: (a) vehicle-treated sham; (b) ZnPP-treated sham; (c) hemin-treated sham; (d) vehicle-treated ischemic (I/R); (e) ZnPP-treated I/R; (f ) hemin-treated I/R. Because there were no differences in any of the parameters between ZnPP-, hemin-, or vehicle-treated rats in the sham groups, the results of groups (a), (b), and (c) were pooled and were referred to as sham.
Preparation of liver microsomal fraction and HO enzyme activity
Liver tissues were homogenized on ice in 1.15% (w/v) KCl containing protease inhibitors and centrifuged at 10,000 g for 35 min at 4°C; the supernatant was then centrifuged at 100,000 g for 60 min at 4°C to obtain the microsomal fraction as a pellet. Microsomal fractions were suspended in 0.1 M potassium phosphate buffer (pH 7.4). Microsomal HO activities were assayed by determination of the formation of bilirubin according to the protocol described by Maines (23).
HMGB1 analysis
Blood samples were collected from inferior vena cava and centrifuged at 500 g for 10 min at 4°C. After centrifugation, serum samples were filtered and concentrated through Centricon YM-100 and YM-10 (Millipore, Billerica, MA) with fixed-angle (35°), 7,500 g for 20 min and 5,000 g for 15 min at 4°C, respectively. The concentrated samples were then subjected to SDS-polyacrylamide gel electrophoresis.
Western blot analysis
Microsomal proteins (50 μg per well), concentrated serum proteins (15 μg per well), nuclear extract (15 μg per well), and liver tissue proteins (15 μg per well) were separated on 12% polyacrylamide-SDS gel and transferred to polyvinylidene difluoride membranes. Bands were immunologically detected using polyclonal antibodies against rat HO-1 (1:1000 dilution, Stressgen Co., Victoria, BC, Canada), HMGB1 (1:1000 dilution, Abcam, Cambridge, MA), Nrf2 (1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, CA), iNOS (1:1000 dilution, BD Biosciences, San Jose, CA), and COX-2 (1:200 dilution, Cayman, Ann Arbor, MI). Immunoreactive bands on the membranes were visualized using an ECL detection system (iNtRON Biotechnology Inc., Sungnam, South Korea) according to the manufacturer's instructions. The intensity of the immunoreactive bands was evaluated densitometrically with ImageQuantTM TL software version 2005 (Amersham Biosciences, Piscataway, NJ). Data are expressed as percentage of the sham (fold increase).
Total RNA extraction and reverse transcription-polymerase chain reaction
Total RNA was extracted from the liver tissue using TRIzol® reagent (GibcoBRL, NY) according to the manufacturer's instructions. Polymerase chain reaction (PCR) primers for amplification of each gene are listed in Table 1. All the PCR reactions included an initial denaturation step at 94°C for 5 min and a final extension at 72°C for 5 min in the GeneAmp 2700 thermocycler (Perkin-Elmer, Inc., Waltham, MA). 10 μl samples of amplified products were resolved by electrophoresis in 1.5% agarose gel, stained with ethidium bromide (0.1 μg/ml), and visualized under UV light. The intensity of each PCR product was evaluated semiquantitatively using a digital camera (DC120, Eastman Kodak, Rochester, NY) and a densitometric scanning analysis program (1D Main, Advanced American Biotechnology, Fullerton, CA).
Assessment of serum ALT levels
Serum alanine aminotransferase (ALT) levels were measured by the standard spectrophotometric procedure using INFINITYTM 52-UV kits (Sigma Chemical Co., St. Louis, MO).
Hepatic lipid peroxidation and glutathione content
The steady-state level of malondialdehyde (MDA), the end-product of lipid peroxidation, was determined in the liver tissues by measuring the level of thiobarbituric acid-reactive substances as described by Buege and Aust (6). The total hepatic glutathione level was determined using yeast-glutathione reductase, 5,5′-dithio-bis(2-nitrobenzoic acid), and NADPH in the liver homogenates after precipitation with 1% picric acid. The level of oxidized glutathione (GSSG) was determined by the same method but in the presence of 2-vinylpyridine. The reduced glutathione (GSH) level was calculated as the difference between the total glutathione and the GSSG levels (3).
Measurement of serum TNF-α level
The serum TNF-α level was quantified by enzyme-linked immunosorbent assay (ELISA) with a commercial kit (eBioscience, San Diego, CA) according to the manufacturer's instructions.
Histological analysis of tissue
The fresh liver tissue samples were fixed in 10% neutral buffered formalin and embedded in paraffin; 5-μm sections were then cut and stained with hematoxylin and eosin (H&E). H&E-stained sections were evaluated at 200× magnification with an Olympus CKX 41 microscope (Olympus Optical CO., Tokyo, Japan).
Statistical analysis
The overall significance of the results was examined using two-way analysis of variance (ANOVA). The differences between the groups were considered statistically significant at a P value 0.05 with the appropriate Bonferroni correction made for multiple comparisons. The results are presented as the mean ± SEM.
Results
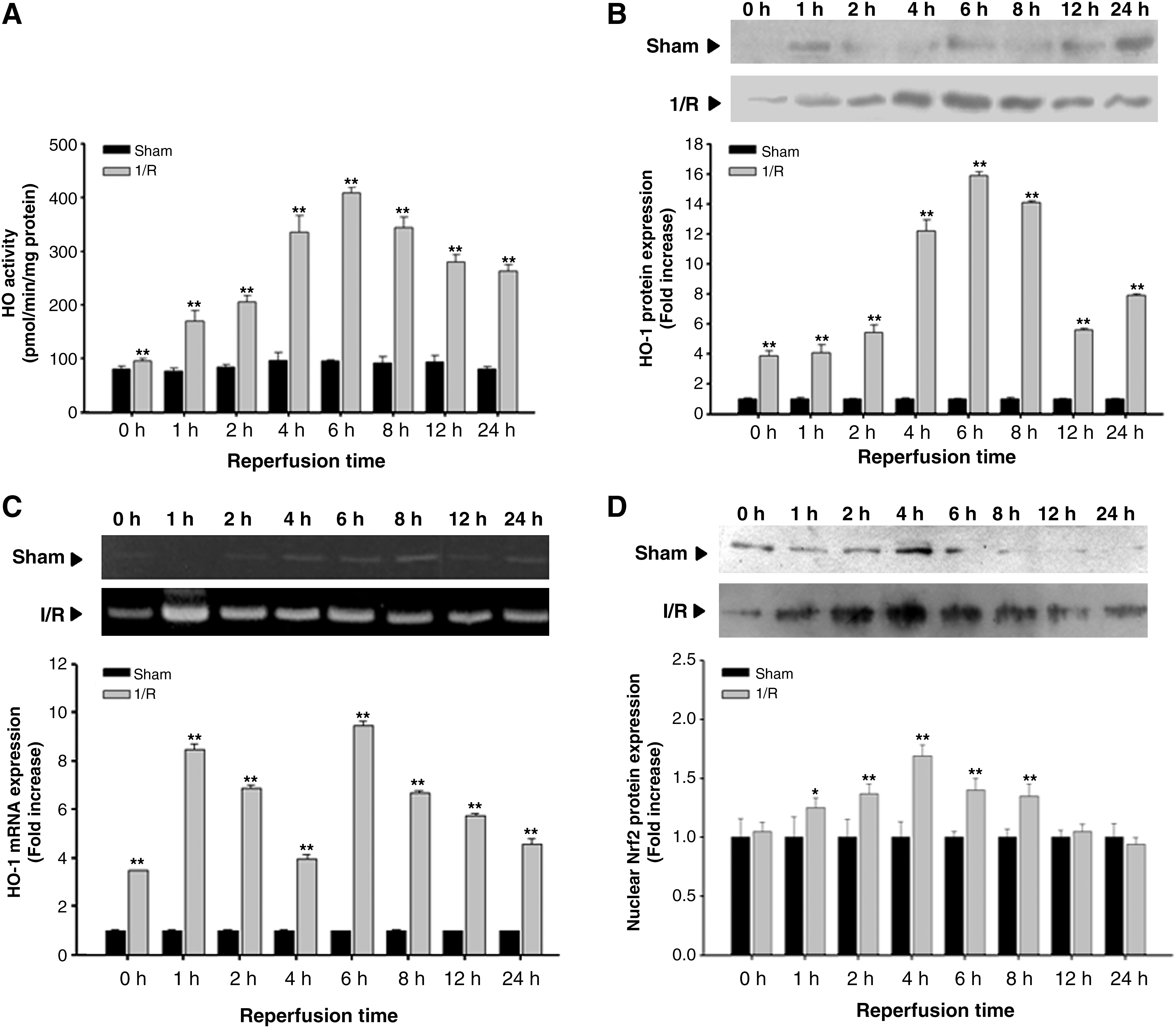
Time course of changes in HO-1 activity, protein, mRNA, and nuclear Nrf2 expression in the liver following hepatic I/R
Figure 1A shows that the hepatic microsomal HO activity in sham-operated rats remained at 76.9 ± 5.3–93.8 ± 11.8 pmol/min/mg protein throughout the experiment. While there was no significant change in HO activity immediately upon reperfusion, the activity began to increase 1 h after reperfusion, was maximal at 6 h after reperfusion, and gradually decreased until 24 h after reperfusion. As shown in Figure 1B, the level of hepatic HO-1 protein expression began to increase immediately after reperfusion, and peaked at 6 h after reperfusion. Thereafter, the level of hepatic HO-1 protein expression began to decrease gradually. The level of hepatic HO-1 mRNA expression increased immediately after reperfusion, dramatically increased by 1 h after reperfusion, moderately at 2 and 4 h after reperfusion, and increased again at 6 h after reperfusion (Fig. 1C). The Nrf2 translocalization into the nucleus significantly increased at 1, 2, 4, 6, and 8 h after reperfusion (Fig. 1D).
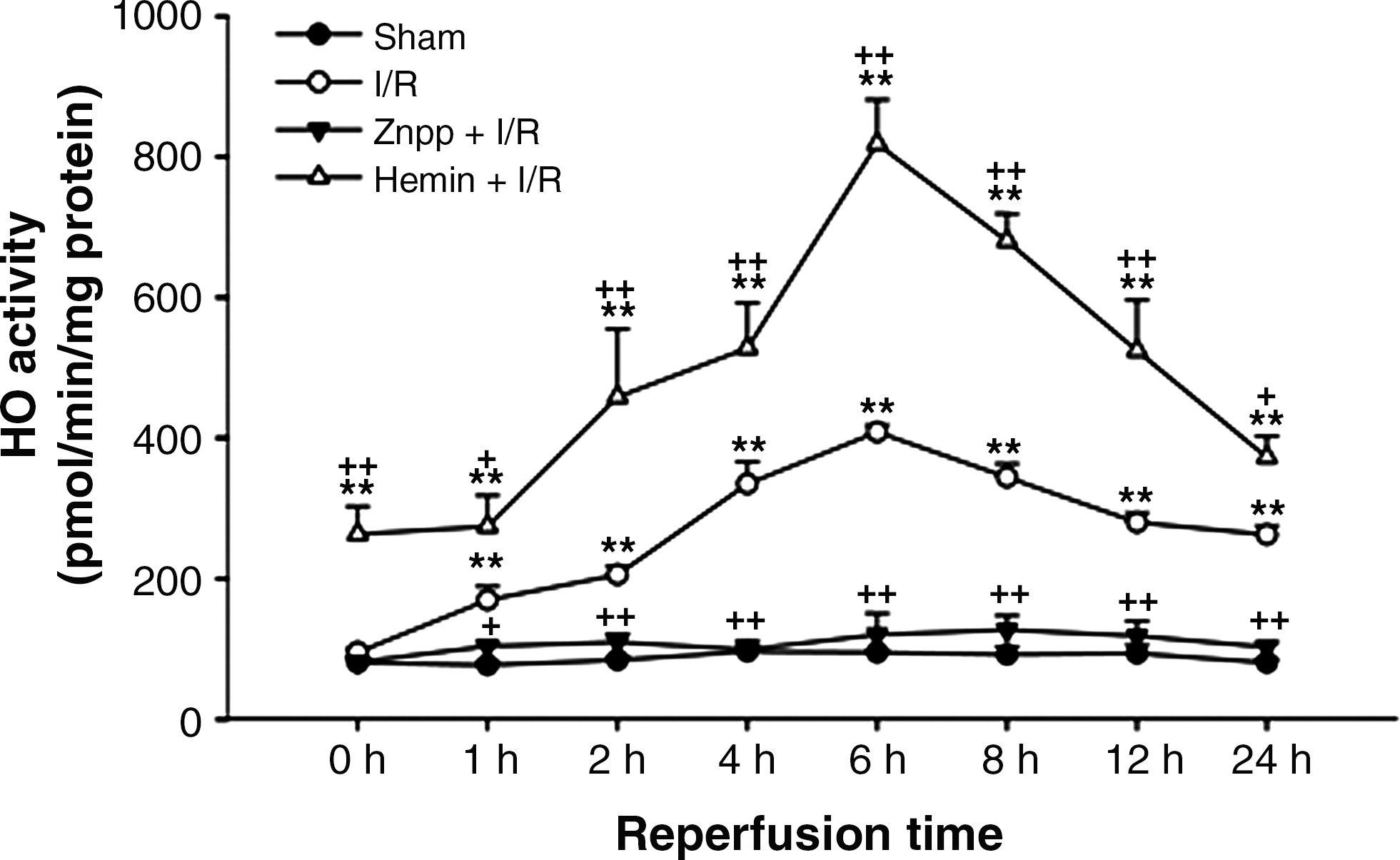
The role of HO-1 in the liver damage caused by hepatic I/R
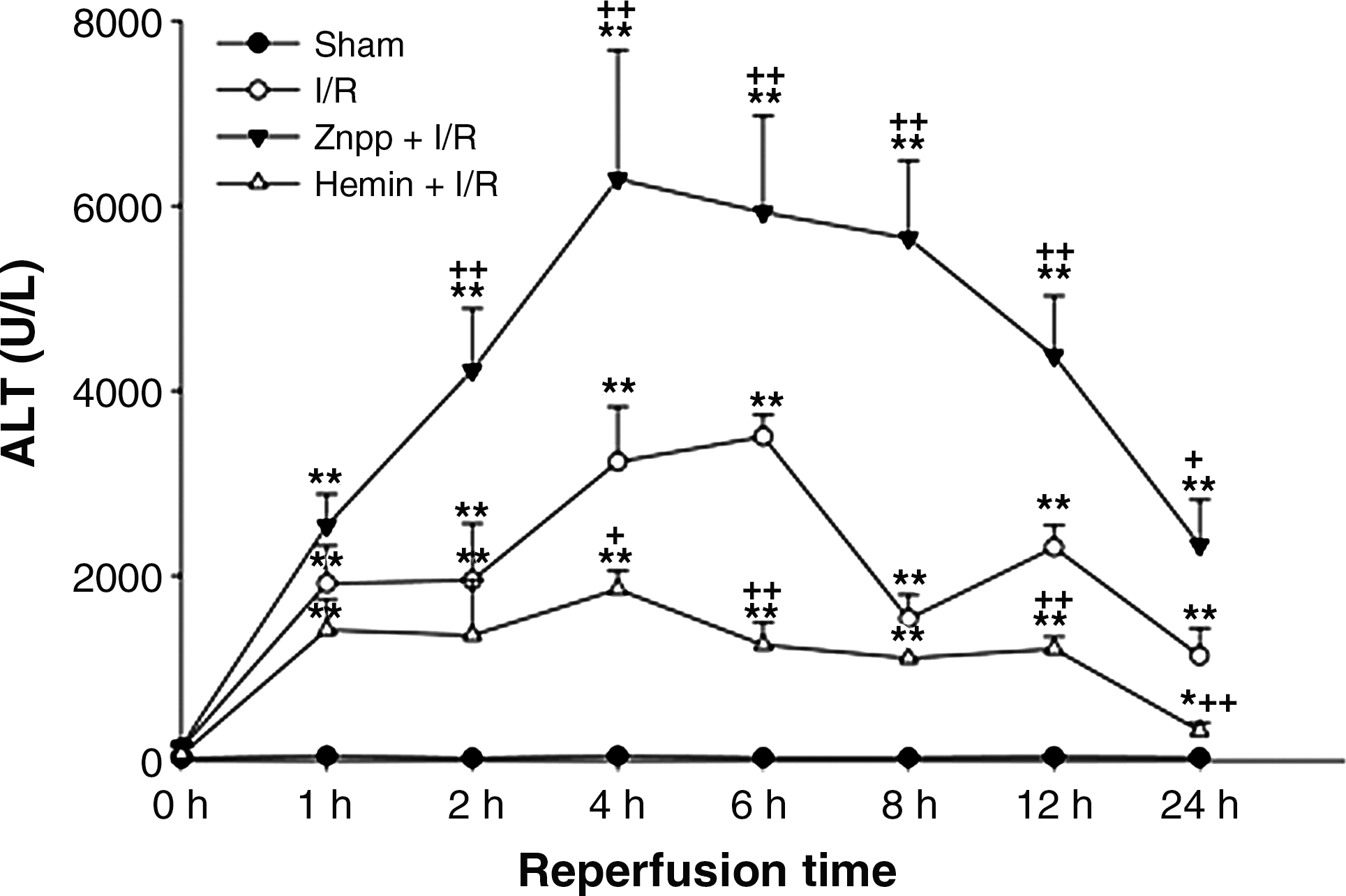
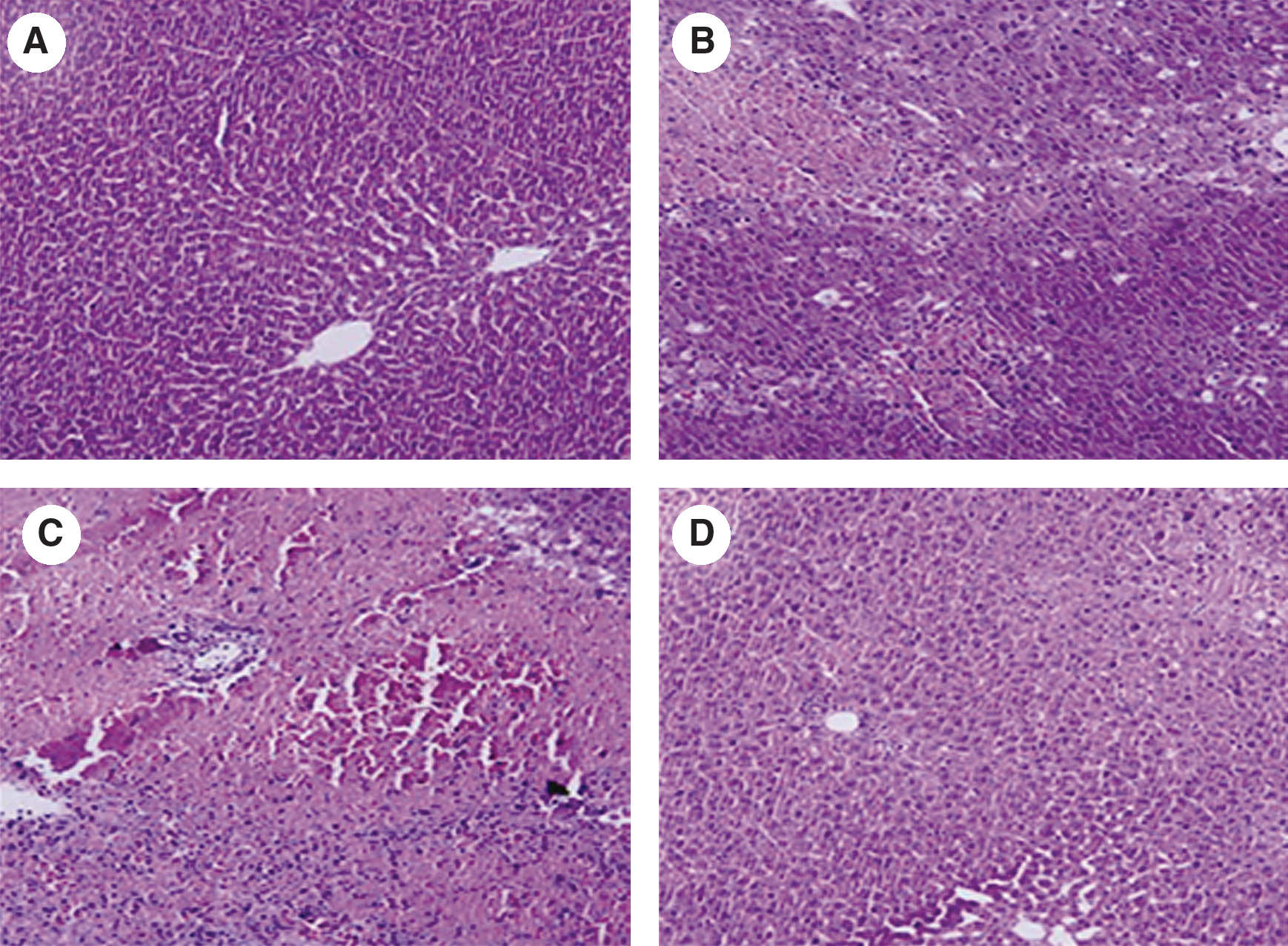
In the livers of rats pretreated with ZnPP and subjected to I/R, HO activity was completely inhibited and remained at the basal level throughout the entire reperfusion. In contrast, HO activity dramatically increased in the hemin-treated I/R group (Fig. 2). The serum ALT activity averaged 39.2 ± 8.1 U/l in the sham-operated group. While there was minimal increase in serum ALT activity immediately upon reperfusion, the ALT activity increased dramatically by 1 h after reperfusion, peaked at 6 h after reperfusion, and decreased gradually thereafter. However, the serum ALT level did not recover completely to the normal basal level even after 24 h of reperfusion. The increase in serum ALT activity was markedly augmented by ZnPP, but attenuated by hemin (Fig. 3). Liver sections isolated after 24 h of reperfusion showed multiple and extensive areas of portal inflammation and hepatocellular necrosis that were distributed randomly throughout the parenchyma, as well as a moderate increase in inflammatory cell infiltration and congestion. Hemin attenuated these pathological changes but ZnPP resulted in more severe hepatocellular damage (Fig. 4).
The role of HO-1 in oxidative stress following hepatic I/R
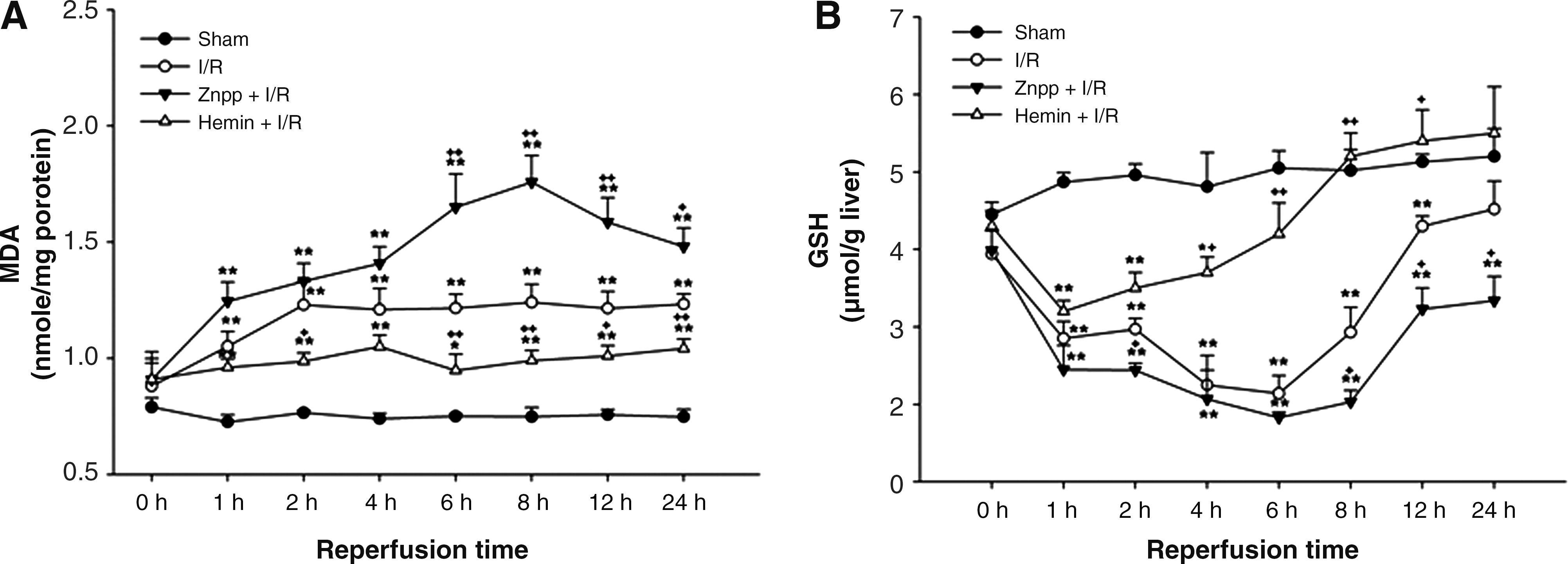
The level of MDA started to increase at 1 h after reperfusion and this elevation persisted until 24 h after reperfusion. This elevation in the level of MDA at all time points was augmented by ZnPP but attenuated by hemin treatment (Fig. 5A). In contrast, the hepatic GSH content started to decrease 1 h after reperfusion and decreased markedly 4 and 6 h after reperfusion. After 24 h of reperfusion, this content returned to normal levels. These decreases in GSH content were augmented by ZnPP but attenuated by hemin treatment (Fig. 5B).
The role of HO-1 in the regulation of TNF-α activity and HMGB1 release
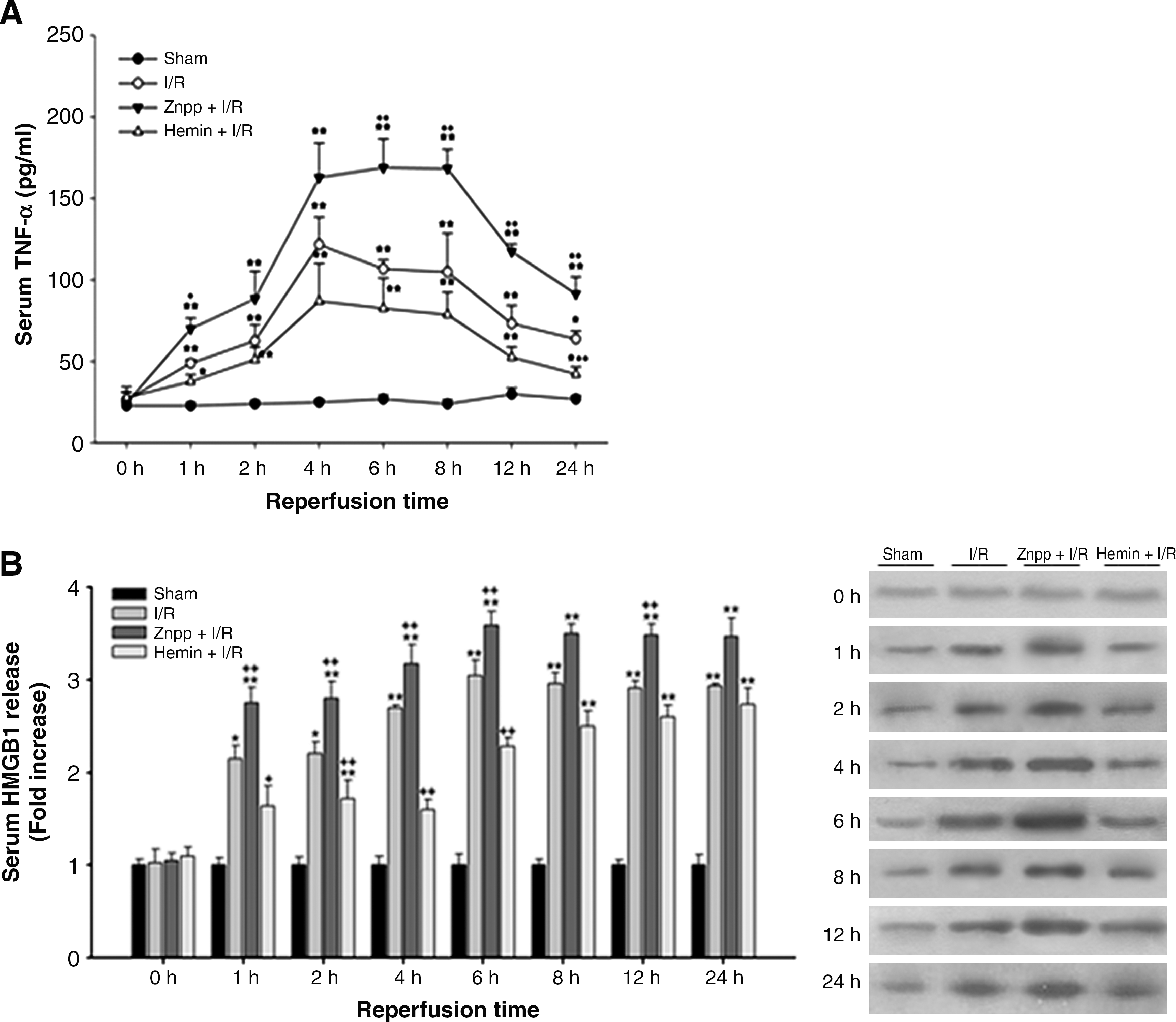
As shown in Figure 6A, the serum level of TNF-α started to increase 1 h after reperfusion, peaked between 4–8 h after reperfusion and thereafter gradually declined. The increase in TNF-α level was augmented by ZnPP but tended to attenuate by hemin treatment. In rats subjected to I/R, serum HMGB1 release markedly increased at 1 h after reperfusion, was maximal at 6 h after reperfusion. This increase remained elevated even at 24 h after reperfusion. The increases occurred at 1, 2, 4, and 6 h after reperfusion were markedly augmented by ZnPP and attenuated by hemin pretreatment (Fig. 6B).
The role of HO-1 in the regulation of hepatic iNOS and COX-2 expression following hepatic I/R
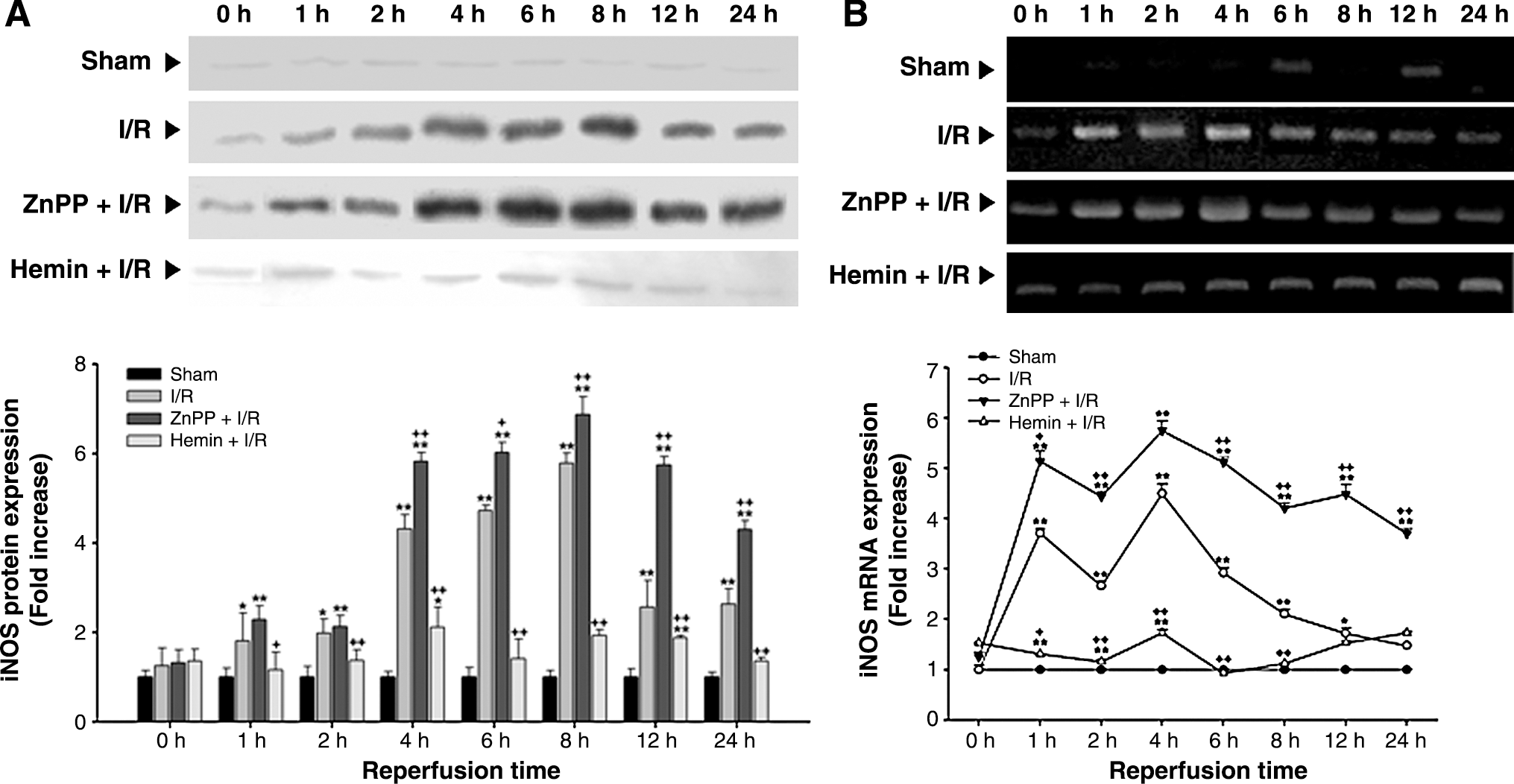
The level of iNOS protein expression in the liver subjected to I/R significantly increased at 1 h after reperfusion, peaked at 8 h after reperfusion. The level of iNOS mRNA expression in the liver significantly increased at 1 h after reperfusion and peaked at 4 h after reperfusion. Thereafter, the level of iNOS mRNA expression decreased gradually, reaching the normal level at 24 h after reperfusion. The increases in iNOS protein and mRNA expression were augmented by ZnPP but attenuated by hemin treatment (Fig. 7).
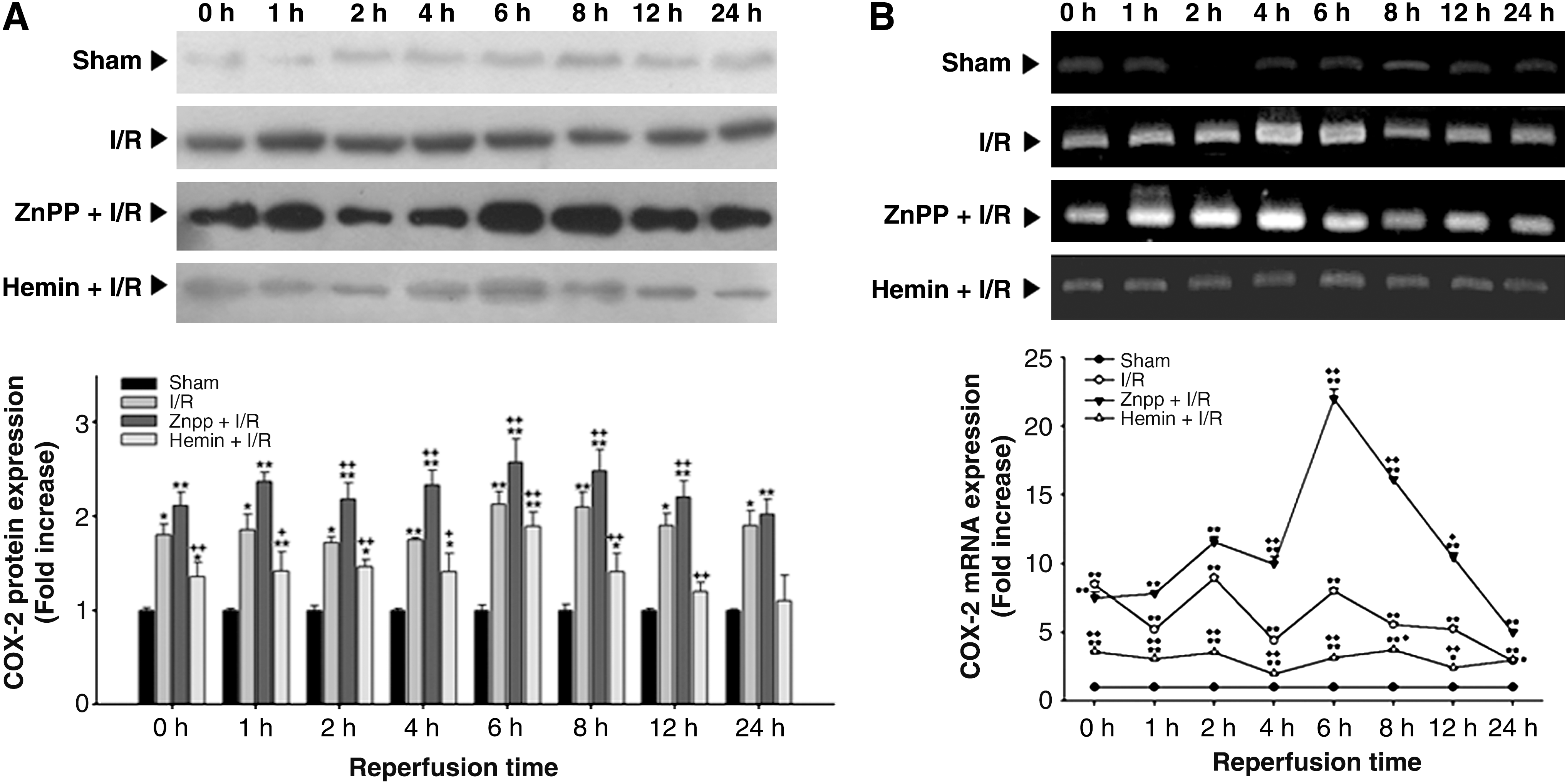
As shown in Figure 8, the level of COX-2 protein expression increased immediately upon reperfusion, and remained elevated even at 24 h after reperfusion. These increases were augmented by ZnPP at 2, 4, 6, 8, and 12 h after reperfusion. Hemin pretreatment significantly reduced the elevations in COX-2 protein expression at all time points measured. In parallel with COX-2 protein expression, the level of COX-2 mRNA also showed significant increase immediately after reperfusion and persisted until 24 h of reperfusion. The increases in COX-2 mRNA expression were augmented by ZnPP pretreatment at 4, 6, 8, and 12 h after reperfusion. Hemin pretreatment attenuated the elevations in COX-2 mRNA expression at all time points measured.
Discussion
HO-1 has been implicated in cytoprotection against oxidative stress in vitro and in vivo. Despite the large number of studies supporting the cytoprotective effect of HO-1, there have been several reports showed contradictory results (10, 32, 33, 40). Moreover, little if any is known about how local HO-1 expression might function in the most basic and well-established rat model of hepatic warm ischemia followed by reperfusion. Thus, in this study, we report the time course of changes in hepatic HO-1 activity, protein, and gene expression in response to in vivo hepatic I/R model, and their relation to various events during the reperfusion period in a time sequence.
In the present study, HO-1 activity increased 1 h after reperfusion, peaked 4–6 h after reperfusion, and then gradually decreased. ALT activity also increased considerably immediately after reperfusion, peaked 4–6 h after reperfusion, and then declined gradually. Thus our data show a temporal association between increased HO-1 activity and hepatocellular injury. Moreover, pretreatment with ZnPP, a potent HO-1 inhibitor, augmented ALT release; in contrast, pretreatment with hemin, a naturally occurring HO-1 substrate, attenuated ALT activity at various reperfusion time points. This phenomenon was also supported by histological examinations. The present findings indicate that induction of HO-1 protects against hepatocellular damage induced by I/R.
It has long been assumed that the oxidative stress, cytokine production, or their interconnection, are responsible for the induction of initial cellular injury leading to necrosis. Many studies suggest that ROS generated by Kupffer cells at the early phase of reperfusion and additional ROS produced from late phase of reperfusion could destroy the cell membrane through lipid peroxidation (18). In corroboration of previous reports, MDA concentrations in liver samples were measured and a significant increase in hepatic lipid peroxidation throughout the entire reperfusion period was observed. Interestingly, this increase was inhibited by hemin but augmented by ZnPP treatment. GSH plays an important role as a free radical scavenger. It has been reported that HO-1 is induced by agents that are known to interact with or modify cellular GSH levels (4). Furthermore, prior induction of HO-1 with glutathione depletor ameliorated the renal I/R injury in the rat (14). In our data, hepatic GSH began to decrease at 1 h after reperfusion and further decreased 4–6 h after reperfusion. The decreased hepatic GSH content began to be restored at 8 h after reperfusion. This decrease was also inhibited by hemin treatment. Our findings indicate that the induced HO-1 expression by changing the intracellular redox state may protect hepatic cells against I/R injury. In particular, biliverdin/bilirubin and CO, the end products of heme metabolism, are thought to be responsible for modulating intracellular redox state with their potent antioxidant properties (13, 26).
It has been reported that the intracellular production of ROS is implicated in the activation of signal transduction cascades and in the regulation of gene expression. In our study, HO-1 mRNA was induced in a biphasic pattern: during ischemia, and then highly induced again during reperfusion. Hypoxia-inducible factor (HIF )-1 is a key regulator of the expression of numerous genes during hypoxic stress such as ischemia. In rats, widespread induction of HIF-1α was observed after exposure to systemic hypoxia, whereas, after coronary occlusion, localized expression occurred primarily at the border of infarcted tissue and persisted for 4 weeks. Upregulation of HIF-dependent proteins such as HO-1 was also detected in the peri-infarct zone (19). Here, we attempted Western blot analysis of I/R livers using mouse anti-HIF-1α antibody. Unfortunately, we could not identify any bands equivalent to HIF-1α (120 kDa; data not shown), presumably because HIF-1α is present in very low levels in the liver, even when induced by hypoxia (41).
One cellular defense mechanism for coping with oxidative stress is enhancing the expression of a selected set of genes that encode antioxidant enzymes via activation of several cytoplasmic redox-sensitive transcription factors such as NF-κB, AP-1, and Nrf2 (7, 12). This leads to enhanced production of the GSH and NO needed for rapid scavenging of ROS. However, NO is continuously overproduced following induction of iNOS, and this NO can scavenge O2·- and generate large amounts of ONOO-. This ONOO- can then rapidly oxidize GSH, causing nitrosative stress and, in response, Nrf2 can become activated (25). Nrf2 plays a central role in the transcriptional regulation of antioxidant enzymes such as glutathione transferase, quinone reductase, and HO-1 that provide additional cytoprotective activities and is considered one of the major transcription factors for the ARE. Upon activation, Nrf2 enters the nucleus where it binds to the ARE in the HO-1 promoter to trigger gene expression (1). A recent study demonstrated that HO-1 is induced via Nrf2 to protect the kidney from remote organ injury after hepatic I/R (34). In the present study, a second phase of hepatic HO-1 mRNA induction occurred after reperfusion (6 h of reperfusion) following the overinduction of iNOS and the decrease in GSH content (4 h of reperfusion). Interestingly, in our data, nuclear translocation of Nrf2 began to increase at 1 h of reperfusion and peaked at 4 h of reperfusion, then gradually decreased. These results indicate that Nrf2 may be activated by oxidative/nitrosative stress, triggering the second phase of HO-1 mRNA expression and GSH biosynthesis. Consistent with the present results, induction of HO-1 was caused by overproduction of NO·, resulting from LPS-derived iNOS induction in cultured RAW264.7 cells. When this iNOS-derived delivery of NO· was combined with prior depletion of GSH, HO-1 induction was potentiated (30). Besides, pretreatment of hemin resulted in reversal of iNOS expression and GSH content. Toda et al. (35) showed that pretreatment of SnCl2, a HO-1 inducer, significantly decreased microsomal heme content, which serves as the prosthetic group of NOS.
Previous studies have demonstrated that TNF-α plays a pivotal role in I/R-induced liver injury. HO-1 overexpression in the allograft rat pancreas was accompanied by significant decreases in TNF-α, IL-2, IL-6, and interferon-γ (5). Scott et al. (28) observed a significant increase in remote intestinal TNF-α expression following hind limb I/R that was greatly attenuated with inhaled CO throughout reperfusion. As expected, in our study, the serum TNF-α concentration was greatly increased in I/R animals. Furthermore, TNF-α levels in I/R rats coincided with the increase in HO activity after reperfusion. The inhibition of HO activity by ZnPP significantly increased serum TNF-α levels.
COX-2, the inducible isoform of cyclooxygenase, is capable of producing large amounts of prostaglandins and is of particular interest due to its potential role in I/R injury. Reperfusion causes cellular injury via activation of arachidonic acid cascade followed by induction of COX-2, which promotes release of TNF-α, generation of ROS, neutrophil infiltration, and lipid peroxidation (8). Csiki et al. (9) reported that COX-2 is upregulated in hypoxic lung cancer cells in an HIF-1-dependent manner. We found here, in response to hypoxia, hepatic COX-2 expression is enhanced immediately after reperfusion, suggesting that hypoxia may contribute to COX-2 overexpression in the early stages of I/R. Furthermore, our data indicate that the release of pro-/anti-inflammatory cytokines and mediators during reperfusion is preceded by the induction of early response genes such as COX-2 and HO-1 during the ischemic phase. Hemin reduced COX-2 expression in I/R liver and these effects were reversed by ZnPP treatment. Li Volti et al. (21) reported that induction of HO-1 led to heme depletion and consequently decreased expression of other important heme proteins, such as COX-2 and NADPH oxidase, and Suh et al. (31) found that CO binds to promoter elements of the COX-2 gene to decrease its transcription. These results support negative regulation of COX-2 by HO-1.
HMGB1 is a nuclear protein released from stressed or damaged cells that serves as a signal for inflammation. Izuishi et al. (17) have recently shown that whereas HMGB1 is a late mediator of systemic inflammation, it can also play a role as an early mediator following acute local organ injury. In previous reports, HMGB1 levels are increased in liver subjected to I/R and anti-HMGB1 antibody decreases hepatic cytokine expression and hepatocellular damage through toll-like receptor-4 dependent signaling (38). In contrast to the proinflammatory role of HMGB1 released post insult, preconditioning with HMGB1 results in protection from inflammation and organ injury following hepatic I/R (17). HO-1 derived CO reduces HMGB1 released in LPS-activated cells and LPS- or CLP-induced animal model of sepsis (36). However, despite the critical role of HMGB1 in early response to hepatic I/R injury, there is no report on the interconnection between HO-1 upregulation and HMGB1 release in warm hepatic I/R model. In our data, the HMGB1 release began to increase at 1 h after reperfusion, peaked 6 h after reperfusion, and remained elevated even at 24 h after reperfusion. Pretreatment of hemin significantly abolished the HMGB1 release into plasma at 1, 2, 4, and 6 h after reperfusion and ZnPP augmented it. Although the HMGB1 release at 8, 12, and 24 h after reperfusion was significantly elevated, either hemin or ZnPP did not affect the HMGB1 release. Our data indicate that HO-1 regulates HMGB1 release in the early phase of reperfusion.
In summary, we have demonstrated that HO-1 expression was upregulated in the livers of rats subjected to I/R. Inhibition of the enhanced HO activity by ZnPP administration, which thus blocked the degradation of free heme and the production of CO and bilirubin, enhanced liver damage. Conversely, prior induction of endogenous HO and provision of substrate for the elevated HO activity through hemin administration attenuated liver damage caused by I/R. Thus, upregulation of HO-1 expression is one of the key adaptive survival responses that occur in liver undergoing sequential hypoxic and oxidative stresses caused by hepatic I/R. Our results further indicate that prior induction of HO-1 expression may provide a new strategy to protect the liver against injury caused by I/R that occurs during liver transplantation.
Footnotes
Acknowledgment
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2006-521-E00023).
Author Disclosure Statement
No competing financial interests exist.