
Abstract
Chronic redox imbalance in erythrocytes of individuals with sickle cell disease (SCD) contributes to oxidative stress and likely underlies common etiologies of hemolysis. We measured the amounts of six antioxidant enzymes—SOD1, catalase, glutathione peroxidase 1 (GPx1), as well as peroxiredoxins (Prxs) I, II, and VI—in red blood cells (RBCs) of SCD patients and control subjects. The amounts of SOD1 and Prx VI were reduced by about 17% and 20%, respectively, in SCD RBCs compared with control cells. The amounts of Prx II and GPx1 did not differ between SCD and normal RBCs. However, about 18% of Prx II was inactivated in SCD RBCs as a result of oxidation to sulfinic Prx II, whereas inactive Prx II was virtually undetectable in control cells. Furthermore, GPx1 activity was reduced by about 33% in SCD RBCs, and the loss of activity was correlated with hemolysis in SCD patients. RBCs from SCD patients taking hydroxyurea demonstrated 90% higher GPx1 activity than did those from untreated SCD patients, with no differences seen for the other catalytic antioxidants. Hydroxyurea induced GPx1 expression in multiple cultured cell lines in a manner dependent on both p53 and NO-cGMP signaling pathways. GPx1 expression represents a previously unrecognized potential benefit of hydroxyurea treatment in SCD patients. Antioxid. Redox Signal. 13, 1–11.
Introduction
Normal RBCs are subject to a high level of oxidative stress as a result of the continuous production of the superoxide anion that accompanies Hb autoxidation, but even more so in SCD (13). The superoxide anion is dismutated to hydrogen peroxide (H2O2), which is further converted to the hydroxyl radical (OH•) through the Fenton reaction in the presence of iron (41). In addition, the instability of Hb in sickled RBCs results in an increase in the amount of iron associated with lipid or protein components of the cell membrane, providing a biologic “Fenton reagent” for the generation of hydroxyl radicals at the membrane and the consequent oxidation of membrane lipids (33). This increased membrane oxidation promotes hemolysis and the associated release of Hb into the plasma. Additional oxidative stress derives from the increased activity of superoxide anion–generating enzymes (NADPH oxidase, xanthine oxidase) apparent in the endothelium, and leukocytes of individuals with SCD can also increase oxidative stress in RBCs (3, 51). This oxidative stress is amplified during cycles of polymerization and depolymerization of sickle Hb, promoting oxidation of RBC cytoskeletal proteins, membrane lipids, and many enzymes, associated with significant depletion of reduced glutathione and NADH (20).
To cope with oxidative stress, RBCs are equipped with Cu- and Zn-dependent SOD (SOD1), catalase, glutathione peroxidase 1 (GPx1), and three isoforms of peroxiredoxin (Prx I, Prx II, and Prx VI) (18, 29, 42). SOD1 converts the superoxide anion to H2O2, which is then removed by catalase and the Prxs. Prx II, the third most abundant protein in RBCs, is responsible for eliminating low concentrations of H2O2, whereas catalase scavenges H2O2 efficiently at high concentrations of the oxidant (18, 29, 42). Mice that lack Prx II thus develop hemolytic anemia (26), whereas RBC-related defects are not apparent in catalase-deficient mice (15). Deficiency of GPx1 renders human RBCs susceptible to oxidant stress (47). The primary physiologic substrate of GPx1 in RBCs is lipid hydroperoxide (19). GPx1 is susceptible to irreversible inactivation by its own substrates, likely as a consequence of the irreversible conversion of the active-site selenocysteine residue to dehydroalanine (DHA) (6). Prx enzymes also are inactivated occasionally during catalysis because the active-site cysteine undergoes oxidation to sulfinic acid (Cys–SO2H) (50). Reactivation of Prx I and Prx II is achieved by reduction of the sulfinic moiety catalyzed by sulfiredoxin (4, 50). No mechanism has been identified for reactivation of the sulfinic form of Prx VI, however (50). Catalase is resistant to inactivation by its own substrate.
Administration of hydroxyurea (HU) in SCD reduces the number of painful vaso-occlusive crises (5) and appears to prolong the life span (46). The effectiveness of HU in the management of SCD is attributed primarily to its ability to increase the synthesis of fetal Hb (HbF), which inhibits the polymerization of sickle Hb (5). HU is oxidized by heme groups to produce NO (11), which activates soluble guanylyl cyclase (sGC) to increase the production of cGMP and the consequent transcription of the HbF (γ subunit) genes (7, 16). HbF expression also has been observed to reduce oxidant stress in the sickle cell mouse (23).
SCD is characterized by chronic oxidative stress caused by an imbalance between ROS production and the activity of antioxidant enzymes. However, the few studies that have examined antioxidant enzymes in RBCs of patients or mice with SCD have yielded contradictory results with respect to RBC levels of SOD, catalase, and GPx (37, 43). The activation status of antioxidant enzymes in RBCs of individuals with SCD and the expression of the more recently discovered Prx enzymes have not been examined. We therefore determined whether SCD might be associated with a loss of the amount or activity of any of six antioxidant enzymes (SOD1, GPx1, catalase, Prx I, Prx II, and Prx VI) expressed in RBCs and explored a potential role for their regulation by treatment with HU.
Materials and Methods
Materials
Nutlin-3, S-nitroso-N-acetyl-
Subjects
Blood specimens were obtained under National Institutes of Health Institutional Review Board–approved protocols from adult patients with SCD or healthy African American adult control subjects, all of whom provided signed informed consent (ClinicalTrials.gov identifier numbers NCT00081523 and/or NCT00542230), with all clinical investigation conducted according to Declaration of Helsinki principles. Baseline patient characteristics are provided in Table 1.
All patients (n = 32) have homozygous sickle cell disease.
SD, standard deviation; TRV, tricuspid regurgitant velocity; NT-proBNP, amino-terminal pro-brain type natriuretic peptide.
ELISA of SOD1 and Prx I
ELISAs were performed as recommended by the manufacturer. Lysates of RBCs in PBS (100 μg of protein), together with various amounts of recombinant enzymes (0.1, 0.2, 0.4, 0.8, 1.6, 3.2, and 6.4 ng for SOD1; 0.78, 1.56, 3.12, 6.25, 12.5, 25, and 50 ng for Prx I), were analyzed in triplicate. Comparison of signal intensities for lysate proteins with those for protein standards yielded estimates for the amounts of SOD1 and Prx I in RBCs. Mean values from five independent measurements were used for statistical analysis.
Quantitative immunoblot analysis of catalase, Prx II, and Prx VI
Lysates of RBCs in PBS (10 μg for catalase, 5 μg for Prx II, and 15 μg for Prx VI) together with various amounts of the corresponding purified enzymes (10, 20, 40, and 80 ng for catalase; 25, 50, 100, and 200 ng for Prx II; 0.38, 0.75, 1.5, and 3 ng for Prx VI) were subjected to SDS-PAGE on a 14% gel. The separated proteins were transferred electrophoretically to a nitrocellulose membrane, which was then incubated consecutively with primary antibodies, HRP-conjugated secondary antibodies, and ECL reagents. To estimate catalase, Prx II, and Prx VI as accurately as possible, exposure to x-ray film was stopped at various times to obtain a linear dose–response curve of the blot intensities with respect to the amount of standard protein. Band intensities on x-ray film were measured with the use of a densitometer. Comparison of band intensities for lysate proteins with those for protein standards yielded estimates for the amount of each enzyme in RBCs. Immunoblot analysis for each antigen was repeated 5 times, and the resulting average values were used for statistical analysis.
Measurement of the activity and abundance of GPx1
Each well of a 96-well plate was incubated first overnight at 4°C with 200 μl of the GPx1 capture Ab (mAb 2A10, 3 μg/ml) in 0.1 M sodium bicarbonate (pH 8.6) and then for 1 h at room temperature with 200 μl of blocking solution, consisting of 5% dried skim milk in PBS-T buffer [10 mM sodium phosphate, 2 mM potassium phosphate (pH 7.4), 140 mM NaCl, 3 mM KCl, and 0.1% Tween 20]. The plate was washed 3 times with PBS-T, after which ELISA Coating Stabilizer (100 μl) was added to each well. After incubation for 1 h at room temperature, the stabilizer solution was removed by aspiration, and the plate was allowed to dry before storage at 4°C. For measurement of GPx1 activity, RBC lysate (100 μg of protein in 200 μl per well) was added to the Ab-coated plate and incubated for 15 h at 4°C. After washing the plate twice with a solution containing 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 0.3% Tween 20, the GPx reaction was performed in 200 μl of a solution containing 0.5 mM EDTA, 200 μM NADPH, 0.2 U of yeast glutathione reductase, 1 mM reduced glutathione, 1 mM t-butyl hydroperoxide, and 100 mM Tris-HCl (pH 7.0). The reaction was initiated by the addition of t-butyl hydroperoxide, and NADPH oxidation was monitored for 30 min at 30°C by measurement of the decrease in absorbance at 340 nm with the use of a microplate reader. The initial rate of the reaction was determined from the linear portion of the time course. After measurement of enzyme activity, the plate was washed 3 times with PBS-T and subjected to quantification of the amount of GPx1 in each well. The detection Ab (50 ng of biotinylated mAb 42C9 in 200 μl of PBS-T) was added to each well and incubated for 1 h at 37°C, after which the plate was washed 3 times with PBS-T, loaded with 200 μl per well of avidin-conjugated HRP (0.5 μg/ml in PBS-T), and incubated again for 1 h at 37°C. The plate was then washed 3 times with PBS-T and allowed to dry, after which 100 μl of 3,3′,5,5′-tetramethyl benzidine solution was added to each well.
After incubation for 15 min at 30°C, the reaction was stopped by the addition of 100 μl of 1N sulfuric acid, and the absorbance of the mixture in each well was measured at 450 nm and 30°C. Four different amounts of recombinant GPx1 were included in the ELISA, and comparison of absorbance values for lysate protein with those for the protein standard yielded estimates for the amount of GPx1 in RBCs. The activity and abundance measurements for GPx1 were repeated 5 times for each RBC sample, and the resulting mean values were used for statistical analysis.
Quantitative immunoblot analysis of the sulfinic form of Prx II
The sulfinic form of Prx II in RBC lysates was measured with immunoblot analysis with rabbit polyclonal Abs that recognize a specific sequence surrounding the cysteine sulfinic acid at the active site (50). To prepare a standard sample for which the extent of sulfinic Prx II was known, we exposed RBCs to a solution containing glucose and glucose oxidase, and the extent of sulfinic Prx II in the cells was determined with two-dimensional gel analysis, as described (50).
Clinical laboratory tests
Clinical laboratory tests, including absolute reticulocyte count, indirect bilirubin, lactate dehydrogenase, and fetal Hb were performed by the Clinical Center Department of Laboratory Medicine, National Institutes of Health, by using standard approved clinical laboratory methods.
Cell culture and chemical treatment
HEL92.1.7 and Jurkat cells were cultured under a humidified atmosphere of 5% CO2 at 37°C in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). HCT116 cells, MEFs, and HepG2 cells were cultured under the same conditions, with the exception that RPMI 1640 was replaced with DMEM. Each cell line (5 × 106 cells in 2 ml per well) was seeded in six-well plates and incubated for 24 h before treatment. The cells were then incubated with the indicated concentrations of HU (0.5 M stock in distilled H2O), nutlin-3 (8 mM stock in DMSO), SNAP (0.2 M stock in DMSO), L-NMA (2 M stock in methanol), BAY 41-2272 (5 mM stock in DMSO), or LY83583 (5 mM stock in ethanol) for various times.
Statistical analysis
Data are presented as the mean ± SEM and were compared with ANOVA and t tests with the use of Sigmastat 3.0 software (Jandel Scientific, Chicago, IL). A p value of <0.05 was considered statistically significant.
Results
Comparison of the amounts of antioxidant enzymes among RBCs from healthy controls and SCD patients treated or not with HU
Although the presence of SOD1, catalase, GPx1, and three Prx isoforms (Prx I, Prx II, and Prx VI) in RBCs has been demonstrated, the amounts of these six enzymes have not previously been measured. We therefore examined the abundance of the six enzymes in lysates of RBCs from three groups of African-American subjects: a control group of healthy individuals (n = 17), SCD patients not previously treated with HU [SCD-HU(–) group; n = 9)], and SCD patients treated with HU [SCD-HU(+) group; n = 13]. The amounts of SOD1 and Prx I were measured with the use of a sandwich ELISA, whereas those of catalase, Prx II, and Prx VI were determined with immunoblot analysis. To estimate catalase, Prx II, and Prx VI as accurately as possible, four different amounts of purified protein were used to obtain a dose-dependent curve for each protein and exposure to x-ray film was stopped at various times to obtain a linear dose–response curve of the blot intensities with respect to the amount of standard protein (Supplemental Fig. 1; see
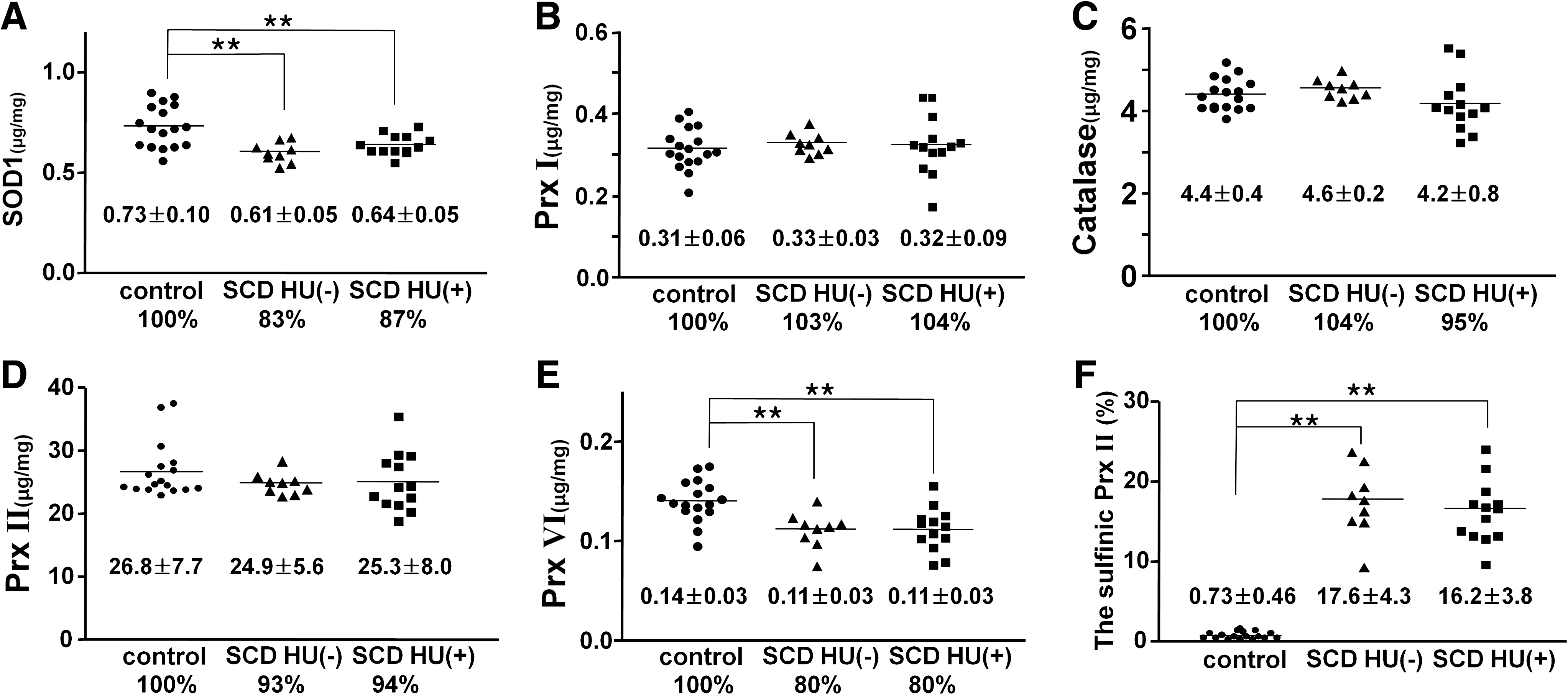
The active-site cysteine of Prxs is readily oxidized to sulfinic acid, resulting in catalytic inactivation. The sulfinic form of Prx proteins can be detected with immunoblot analysis with rabbit polyclonal antibodies that recognize a specific sequence surrounding the cysteine sulfinic acid at the active site (50). Given that the active-site sequence (DFTFVCPTEI) is the same for Prx I and Prx II and that the sizes of Prx I and Prx II are identical, the sulfinic forms of Prx I and Prx II cannot be differentiated with immunoblot analysis. However, because the amount of Prx II in RBCs is about 80 times that of Prx I and because Prx II is more sensitive to oxidation to sulfinic acid than is Prx I (27), the sulfinic Prx II accounts for most of the sulfinic form of the two enzymes detected in these cells. The extent of Prx II oxidation was estimated by including in the immunoblot analysis a standard RBC lysate for which the extent of Prx II oxidation had been measured with two-dimensional electrophoresis (50). Whereas the sulfinic Prx II was essentially not detected in RBCs of the control group of subjects, 17.6 ± 4.3% and 16.2 ± 3.8% of Prx II was oxidized to sulfinic form in those of SCD-HU(–) and SCD-HU(+) groups, respectively (Fig. 1F).
Comparison of the activity and amount of GPx1 among RBCs from healthy controls and SCD patients treated or not with HU
The active-site selenocysteine of GPx1 is more sensitive to oxidative damage than is the active-site cysteine of Prxs. The oxidation of GPx1 selenocysteine to the selenite or selenate state appears to result in loss of the selenium atom and conversion of selenocysteine to DHA (6). It therefore was important to measure both the amount and activity of GPx1 in the three subject groups of the present study. We prepared two mAbs (2A10 and 42C9) that bind independently to human GPx1 and then developed an immunoaffinity capture assay schematically shown in Fig. 2A. By using the assay, we could measure the activity and the amount of GPx1 in the same 96-well plate.
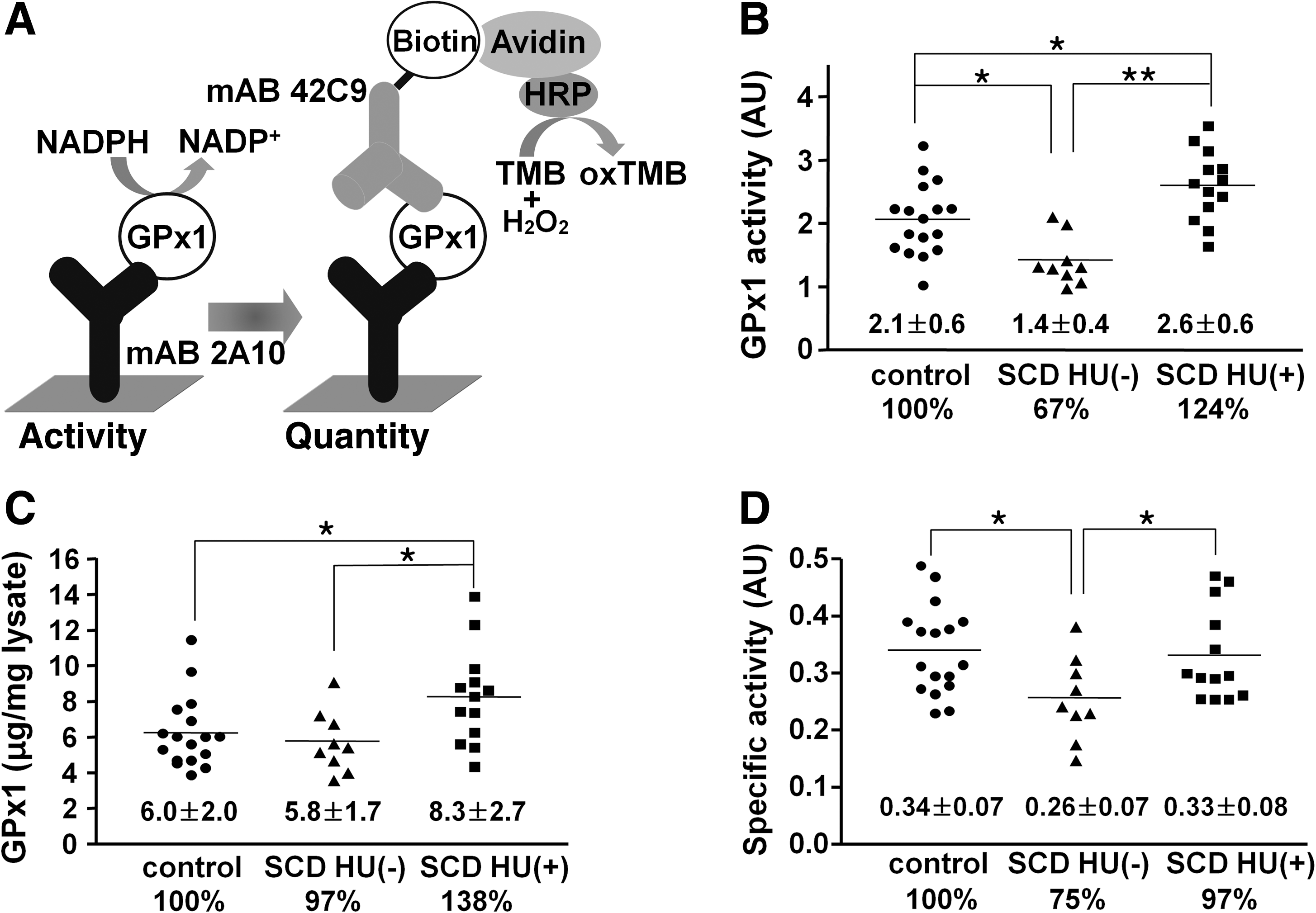
Compared with the control group, the activity of GPx1 in RBCs was reduced by 33% in the SCD-HU(–) group (p < 0.05) and was increased by 24% in the SCD-HU(+) group (p < 0.05) (Fig. 2B). SCD patients receiving HU thus had 90% higher GPx1 activity than did patients not receiving HU (p < 0.001). In contrast, the amount of GPx1 protein in RBCs was similar for the control and SCD-HU(–) groups but was increased by 38% in the SCD-HU(+) group compared with that in the control group (p < 0.05; Fig. 2C). The specific activity (activity/amount) of GPx1 in the SCD-HU(–) group was reduced by 25% compared with that in the control group (p < 0.05; Fig. 2C), indicating that GPx1 was inactivated in SCD patients by the high levels of ROS and that the inactive enzyme accumulated in RBCs. In addition to the activity (Fig. 2B) and amount (Fig. 2C) of GPx1, the specific activity of the enzyme was increased by 29% in the SCD-HU(+) group compared with that in the SCD-HU(–) group (p < 0.05; Fig. 2D). These results suggested that HU therapy both increased GPx1 expression and protected the enzyme against oxidative damage in the RBCs of individuals with SCD.
Relation between GPx1 activity in RBCs and hemolyis in SCD patients
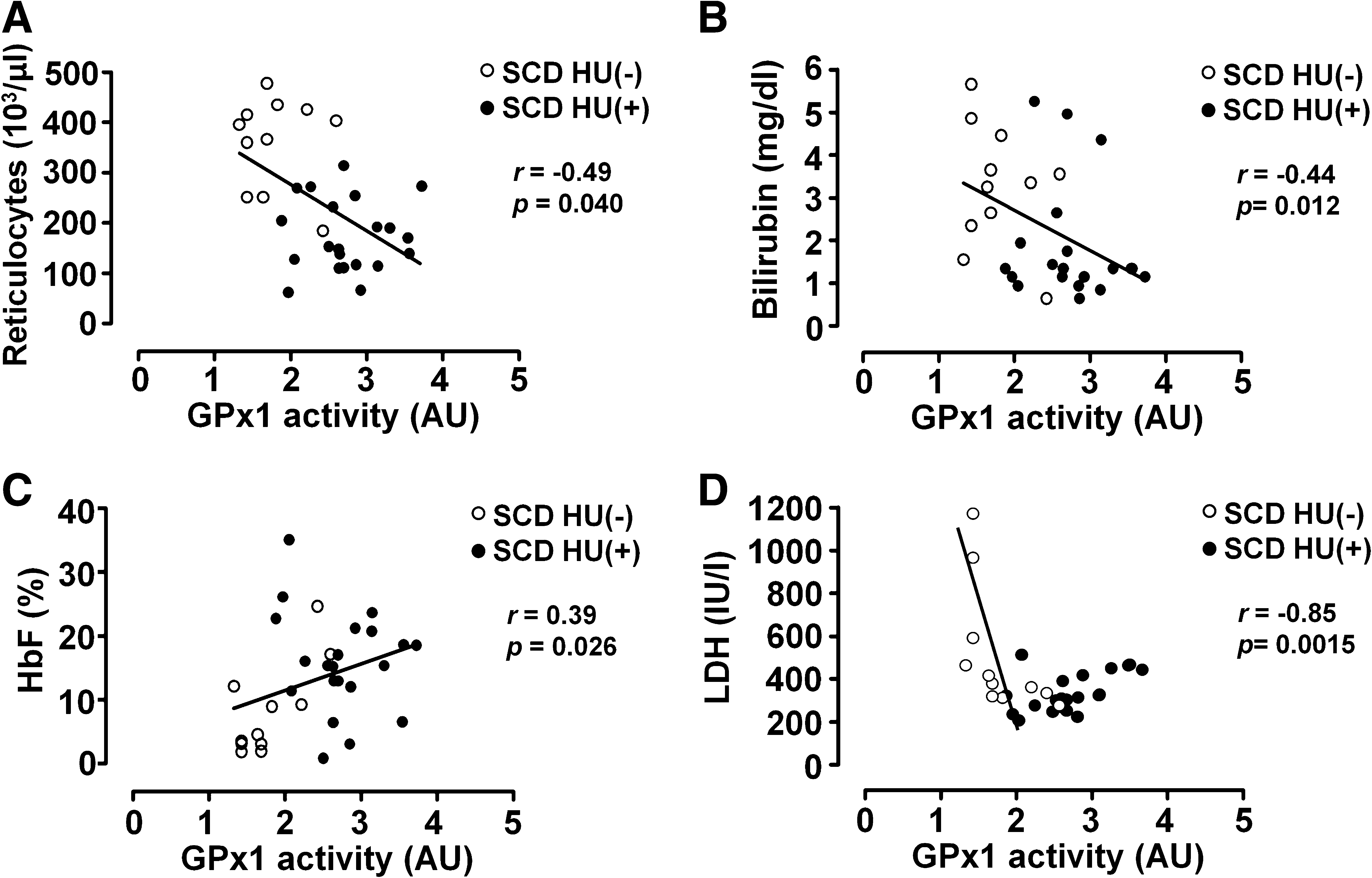
Given that the primary physiologic substrate of GPx1 in RBCs is lipid hydroperoxide (19), membrane oxidation would be expected to be increased in the RBCs of individuals with a low GPx1 activity. In addition, among the antioxidant enzymes, only GPx1 showed significant increase in HU(+) compared with HU(-). To investigate the relation between GPx1 activity in RBCs and hemolysis in individuals with SCD, we analyzed several indicators of hemolysis in SCD patients treated (n = 21) or not (n = 11) with HU. Hemolysis results in the release of Hb and cytosolic lactate dehydrogenase (LDH) into plasma as well as indirectly increases the plasma concentration of bilirubin, a product of catabolism of the released heme. The absolute reticulocyte count in SCD patients is highly elevated as a compensation for the shortened RBC lifetime (about 10 to 20 days) (Table 1). GPx1 activity showed a significant inverse correlation with absolute reticulocyte count (Fig. 3A), as well as with the reticulocyte count adjusted for HbF level (not shown). GPx1 activity also was inversely correlated with the serum levels of bilirubin (Fig. 3B). GPx1 activity increased in parallel with the percentage of HbF (Fig. 3C), the expression of which is induced by HU. Unexpectedly, the relation between serum LDH level and GPx1 activity was confusing when data from all SCD patients were used for analysis (Fig. 3D). A strong inverse relation was apparent, however, when HU-treated patients were excluded from analysis (r = −0.85; p < 0.001), whereas serum LDH levels of HU-treated patients showed an apparent direct correlation with GPx1 activity (r = 0.56; p < 0.01; Fig. 3D). We considered the possibility that HU treatment induces LDH in RBCs. The amounts of LDH in RBC lysates were determined with immunoblot analysis. The amount of LDH in RBCs did not differ between the control and SCD-HU(–) group (Supplemental Fig. 2; see
p53-Dependent GPx1 induction by HU
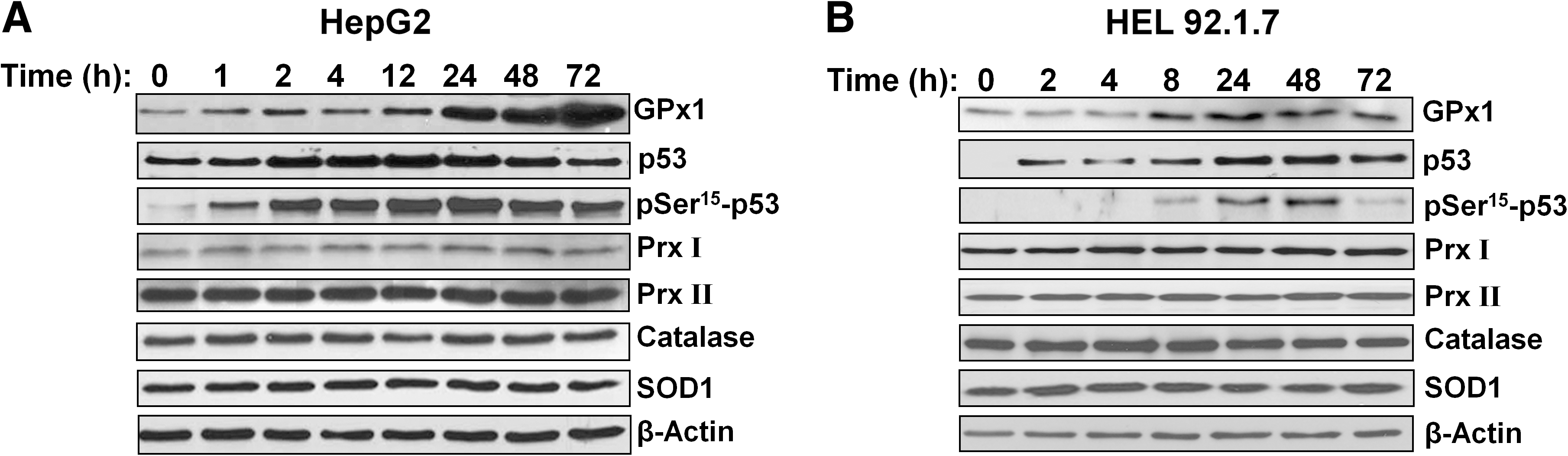
HU increases GPx activity in B16 murine melanoma cells (9). It also induces the accumulation of the transcription factor p53 and its phosphorylation on Ser15 in various cell types (45). The target genes of p53 include those for several antioxidant enzymes, including GPx1, SOD2 (Mn-dependent SOD), and catalase (28). We therefore examined whether HU might induce the expression of GPx1 in HepG2 human hepatocellular carcinoma cells and HEL92.1.7 human erythroleukemia cells and whether such induction might be related to p53 activation. Exposure to 0.5 mM HU increased the amount of GPx1 in a time-dependent manner in both cell lines (Fig. 4A and B). It also induced the accumulation and Ser15 phosphorylation of p53 (Fig. 4A and B). In contrast, the abundance of other antioxidant enzymes, including Prx I, Prx II, catalase, and SOD1, was not affected by HU in either cell line (Fig. 4A and B). The induction of GPx1, as well as the accumulation and phosphorylation of p53 in HEL92.1.7 cells, was dependent on HU concentration (Fig. 4C).
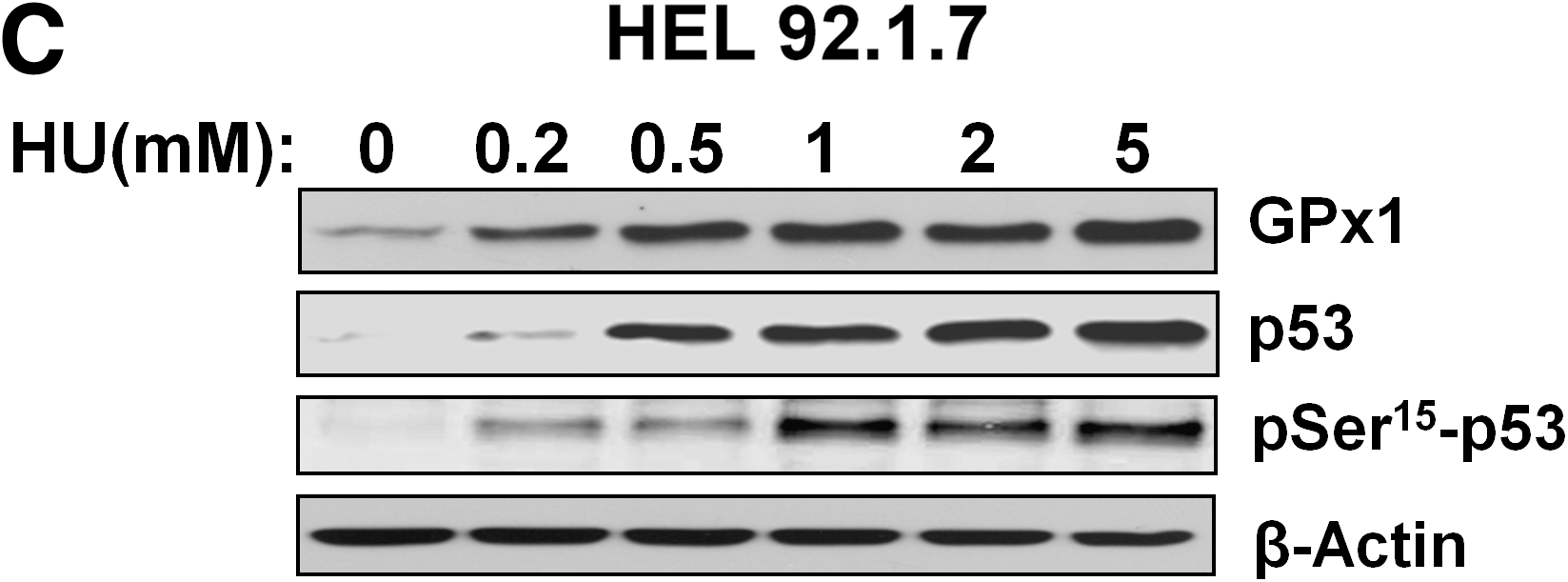
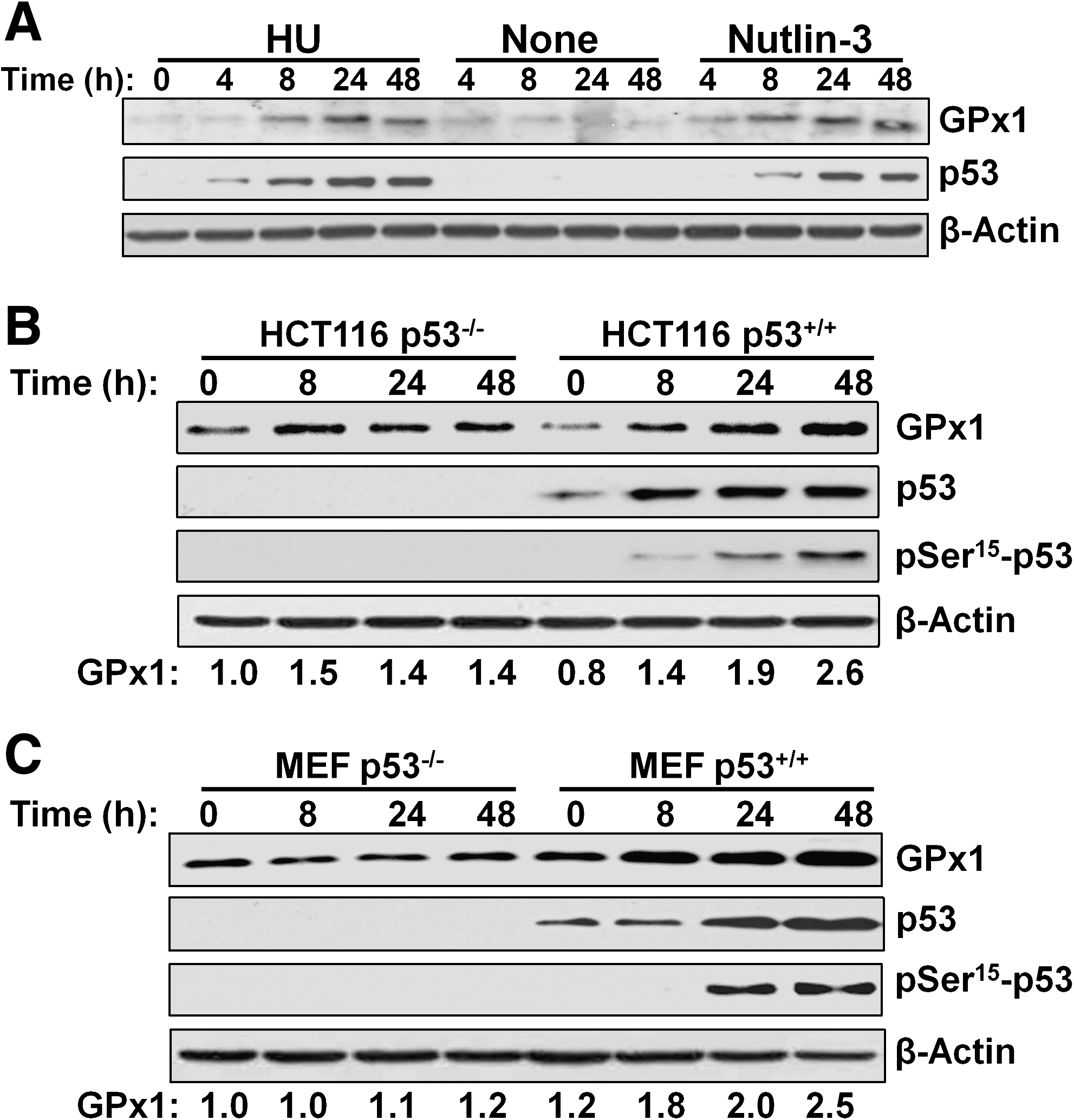
To evaluate further the contribution of p53 to the upregulation of GPx1, we first treated HEL92.1.7 cells with nutlin-3, which binds the ubiquitin ligase MDM2 at its p53-binding pocket and thereby induces the accumulation of p53 by inhibiting its ubiquitin-dependent degradation (49). Nutlin-3 induced GPx1 expression and p53 accumulation, as did HU (Fig. 5A). We also examined p53-null lines of HCT116 human colon cancer cells and mouse embryonic fibroblasts (MEFs). Treatment of the p53-null cells with 0.5 mM HU for 48 h induced 1.2- to 1.4-fold increases in the amount of GPx1, whereas the same treatment induced 2.5- to 2.6-fold increases in GPx1 abundance in the corresponding p53+/+ cells (Fig. 5B and C), suggesting that GPx1 induction by HU is largely, but not entirely, dependent on p53.
NO-dependent GPx1 induction by HU
HU is metabolized to NO by heme groups, and NO then activates sGC to produce cGMP (16). The NO-cGMP signaling pathway targets several transcription factors, including AP-1 and Sp-1, binding sites for both of which are present in the promoter of the human GPx1 gene (35). To examine whether the induction of GPx1 expression by HU might be mediated by the NO-cGMP pathway in addition to the p53 pathway, we exposed HEL92.1.7 cells to the NO donor S-nitroso-N-acetyl penicillamine (SNAP). SNAP increased the abundance of GPx1 in a concentration-dependent manner (Fig. 6A), and the effects of HU and SNAP on GPx1 expression were additive (Fig. 6B).
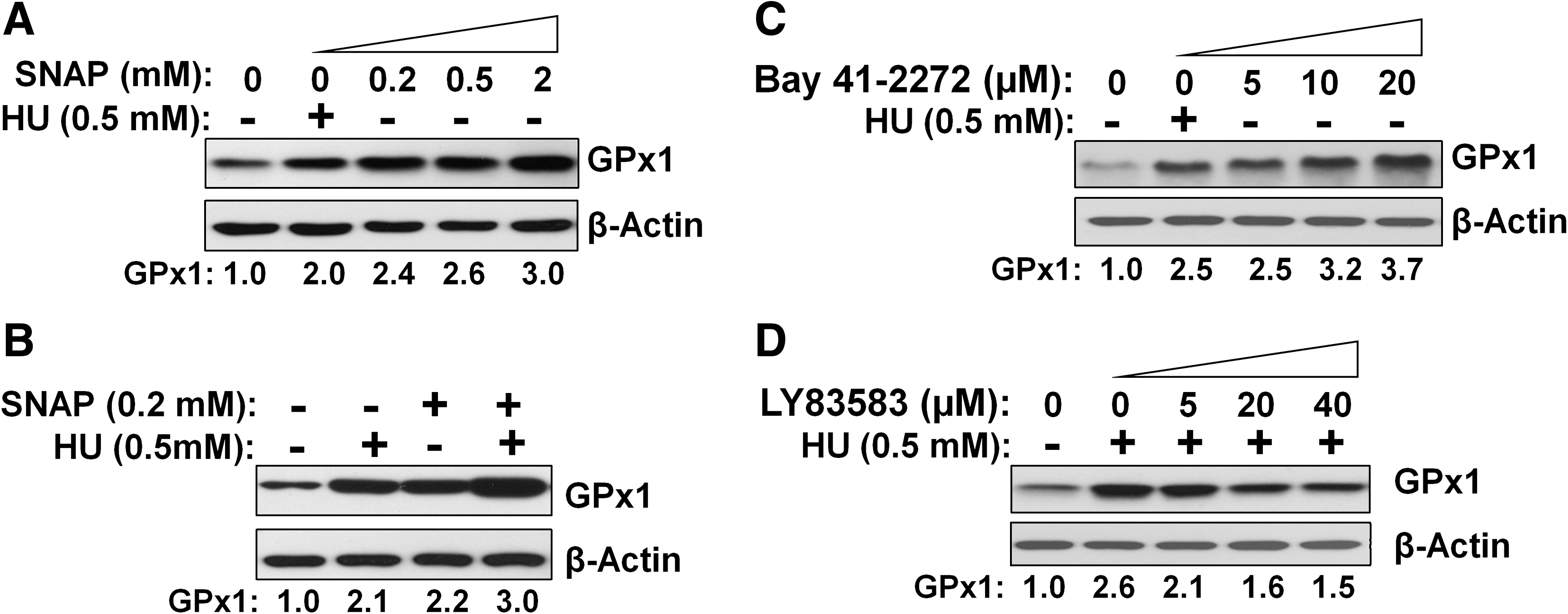
We also tested the effects of activation or inhibition of sGC in HEL92.1.7 cells. BAY 41-2272, an sGC activator, increased GPx1 expression in a concentration-dependent manner (Fig. 6C), whereas the HU-induced expression of GPx1 was inhibited by the sGC inhibitor LY83583, also in a concentration-dependent manner (Fig. 6D). These data thus suggested that HU increases GPx1 expression in part through the NO-cGMP pathway.
Discussion
Mammalian cells express two types of SOD (SOD1, SOD2), one type of catalase, four types of GPx (GPx1–4), and six types of Prx (Prx I–VI) to deal with ROS that include the superoxide anion, H2O2, and lipid hydroperoxides. Six of these enzymes (SOD1, catalase, GPx1, Prx I, Prx II, and Prx VI) have been detected in human RBCs, and we have now measured their concentrations in these cells and found them to predominate according to the rank order Prx II > GPx1 > catalase > SOD1 > Prx I > Prx VI.
The superoxide anion generated as a result of Hb autoxidation in RBCs is rapidly converted to H2O2 by SOD1 in these cells. Otherwise, the superoxide anion depletes NO to produce peroxynitrite (ONOO–) or supports Fenton chemistry to produce the hydroxyl radical. Both peroxynitrite and the hydroxyl radical potently oxidize membrane lipids and other cellular components (39, 41). Our finding that the amount of SOD1 was reduced by 17% in RBCs of SCD patients compared with controls suggests that the sickle RBCs may undergo oxidative damage by the superoxide anion. SCD patients have high plasma levels of the proinflammatory cytokine TNF-α (30), which is known to inhibit the activity of the human SOD1 gene promoter through activation of the JNK–AP-1 signaling pathway (1), possibly accounting for the reduced level of SOD1 expression we observe in sickle RBCs.
Despite the continuous production of H2O2 that results from the combination of Hb autoxidation and the SOD1 reaction, RBCs maintain a low steady-state level (0.05 nM) of H2O2 (18), largely as a result of the activity of Prx II, which, at a concentration of 0.41 mM, is the most abundant antioxidant enzyme in RBCs and efficiently eliminates low concentrations of H2O2 (18, 42). In contrast to Prx II, catalase scavenges H2O2 efficiently only at high concentrations of the oxidant. We detected no significant differences in the amounts of Prx II or catalase among control subjects and SCD patients treated or not with HU. However, immunoblot analysis with antibodies specific for the sulfinic form of Prx enzymes indicated that 16% to 18% of Prx II in RBCs of SCD patients was inactivated as a result of oxidation of the catalytic cysteine to sulfinic acid, not observed in control RBCs. The rate of Prx oxidation to the sulfinic form is proportional to the rate at which Prx eliminates H2O2 (50). The accumulation of the sulfinic form of Prx II in RBCs of SCD patients thus indicates that the rate of H2O2 elimination by Prx II is increased in these cells and that the action of sulfiredoxin is not sufficient to maintain all Prx II molecules in the active state. The amount of Prx I in RBCs was found to be only 1/80 that of Prx II and did not differ between control subjects and SCD patients. Prx VI is unique among Prx isoforms in that it is a bifunctional enzyme with phospholipase A2 activity in addition to peroxidase activity (31). The phospholipase A2 activity of Prx VI, which is not affected by cysteine oxidation, contributes to the repair of oxidized phospholipids by cleaving oxidized fatty acids (31). The observation that catalase and GPx1 could not compensate for an increase in the extent of myocardial ischemia/reperfusion injury induced by ablation of Prx VI suggested that Prx VI has a nonredundant role (36), which might be attributable to its ability to repair cell membranes. Our finding that the amount of Prx VI was reduced by 20% in RBCs of SCD patients thus suggests that membrane repair might be impaired in these cells. The mechanism responsible for downregulation of Prx VI expression in SCD remains unclear.
GPx1 is more vulnerable to inactivation by its own substrates than is Prx II because its catalytic selenocysteine is readily oxidized. Exposure of purified GPx1 to various hydroperoxides thus resulted in its gradual and irreversible inactivation (38). To measure the catalytic activity and abundance of GPx1 simultaneously, we prepared two mAbs to GPx1 and developed microplate-based assays. With the use of these assays, we found that the level of GPx1 activity of SCD patients not treated with HU was 33% lower than that of controls, with no decrease in GPx1 protein abundance. These results suggest that more GPx1 molecules were inactivated in RBCs of SCD patients than in those of control subjects, despite the much shorter life span of sickle RBCs compared with that of normal RBCs. However, HU treatment was associated with a 1.9-fold higher level of GPx1 activity in RBCs of SCD patients, an effect largely due to increased expression of GPx1 protein, but also due to slower inactivation of GPx1 in HU-treated patients.
GPx1 has been suggested to be a minor antioxidant in RBCs on the basis of the findings that RBCs of mice lacking GPx1 appear normal unless stressed with lipid hydroperoxide and that the primary physiologic substrate of GPx1 in these cells is lipid hydroperoxide rather than H2O2 (14, 19). However, humans with deficient RBC GPx1 activity have been reported to demonstrate hemolysis (47), suggesting that GPx1 might be more important in human RBCs. Lipid oxidation is low in normal RBCs because the superoxide anion and H2O2 produced as a result of Hb autoxidation are removed efficiently by SOD1 and Prx II, respectively. The role of GPx1 is thus relatively minor in normal RBCs. In RBCs of SCD patients, however, the rate of Hb autoxidation is increased, the amount of SOD1 is reduced, and Prx II is partially inactivated, all of which may result in promotion of lipid peroxidation. In addition, the amount of membrane-bound iron is greatly increased in RBCs of SCD patients as a result of Hb instability, leading to an increased production of hydroxyl radicals at the cell membrane and lipid peroxidation (17, 41). The observed decrease in the amount of Prx VI in RBCs of SCD patients also likely contributes to the increased level of oxidized lipids, given that Prx VI participates in the repair of oxidized phospholipids. This increased membrane oxidation results in an increased susceptibility to hemolysis.
Exposure to HU was previously shown to increase GPx activity in B16 murine melanoma cells (9). We have now shown that HU induces GPx1 expression in human erythroleukemia and hepatocellular carcinoma cells. Our data show that the induction of GPx1 expression by HU is largely dependent on the p53 pathway. These results suggest that HU-induced upregulation of GPx1 might be a general phenomenon in a wide range of cell types expressing p53. Depending on the cellular context, p53 acts as a pro- or antioxidant effector in redox regulation through direct induction of prooxidant or antioxidant genes or modulation of cellular metabolic pathways (28). The expression of antioxidant proteins, such as GPx1 and SOD2, as well as that of prooxidant proteins, such as BAX and PUMA, is thus induced by p53 (28). Speculatively, antioxidant activity may simultaneously be induced in diverse organs in patients with SCD treated with hydroxyurea, potentially providing resistance to the oxidative stress of the ischemia/reperfusion injury of SCD (22).
Our experiments with p53-null cells indicated that HU also induces GPx1 expression, albeit to a relatively small extent, in a manner independent of p53 and mediated by the NO-cGMP pathway. Although RBCs express a functional NOS (25), the NOS inhibitor N-omega monomethyl-
GPx activity was previously shown to be inversely correlated with membrane fluidity and lipid peroxidation in RBCs of individuals with chronic renal failure treated with continuous ambulatory peritoneal dialysis (32). We have now shown that GPx1 activity is inversely correlated with the extent of hemolysis in SCD patients. The induction of GPx1 expression in RBCs with HU treatment may thus be clinically beneficial in SCD patients. Furthermore, given that HU increases GPx1 expression in many cell types, the associated clinical benefit of HU treatment in SCD may not be restricted to RBCs. For example, in monocytic cells, GPx1 is an efficient inhibitor of 5-lipoxygenase activity, which contributes to the biosynthesis of proinflammatory leukotrienes (48).
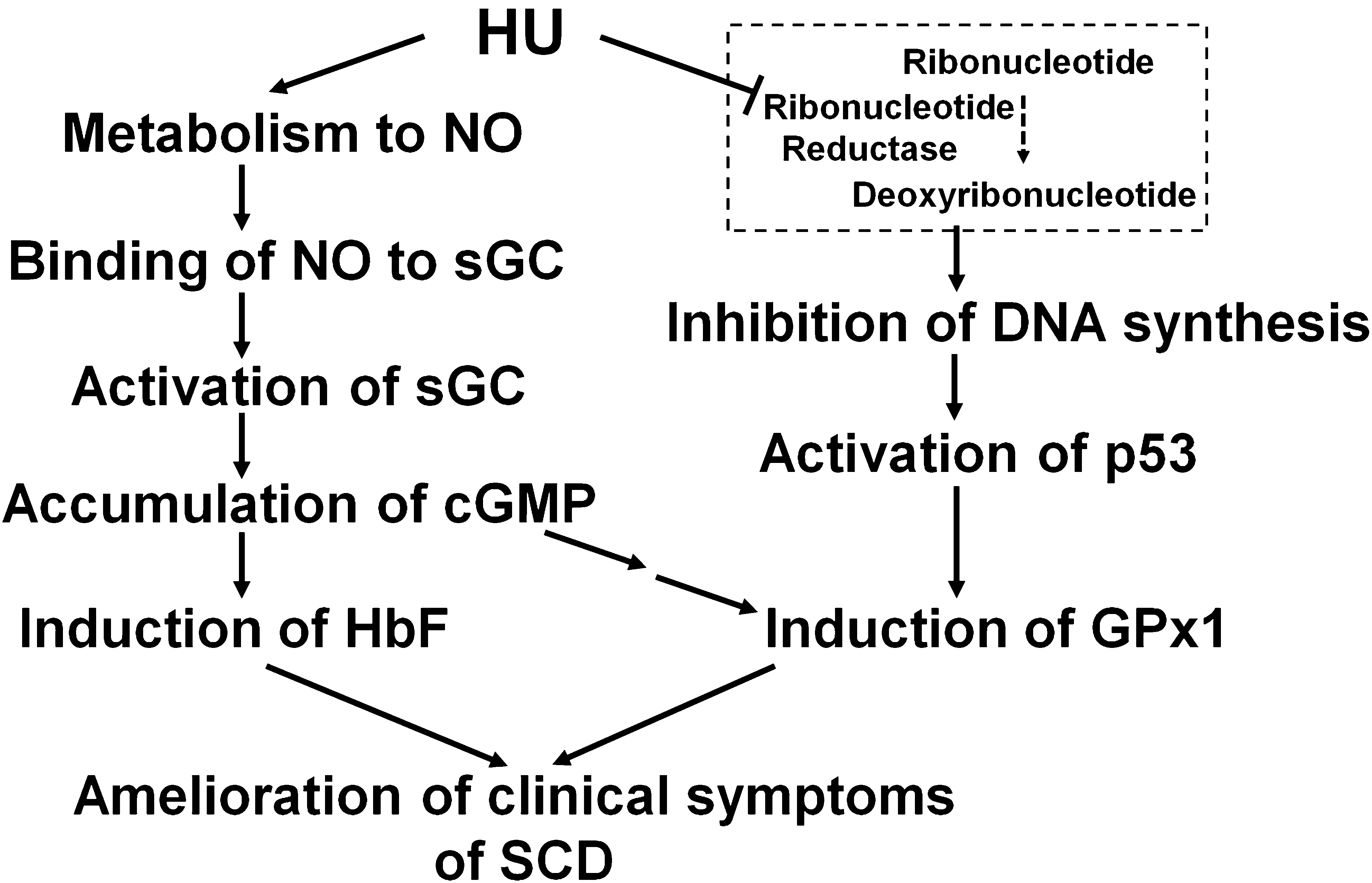
In summary, our findings suggest that the oxidative stress that results from Hb autoxidation is exacerbated in the RBCs of individuals with SCD by a decrease in the catalytic capacity of antioxidant enzymes, including SOD1 and Prx VI. Although the amounts of Prx II and GPx1 in RBCs did not differ between individuals with SCD and control subjects, both of these antioxidant enzymes were found to be partially inactivated in the RBCs of the SCD patients. Treatment of SCD patients with HU is associated with markedly increased GPx1 activity in RBCs, mostly through increased GPx1 protein expression but also in part by decreased inactivation. The increased GPx1 appears to decrease the rate of hemolysis. HU-induced GPx1 expression appears to be mediated by two distinct mechanisms, one involving activation of the p53 pathway as a result of inhibition of DNA synthesis by HU and the other involving activation of the cGMP pathway by NO produced from HU (Fig. 7). This induction of GPx1 expression may thus constitute a previously unrecognized beneficial effect of HU treatment in patients with SCD (Fig. 7).
Footnotes
Acknowledgments
This study was supported by grants from the Korean Science and Engineering Foundation (National Honor Scientist Program grant 2006-05106 and Bio R&D program grant M10642040001-07N4204-00110) to S.G.R. Support was provided to G.J.K. and M.T.W. by the Division of Intramural Research of the National Institutes of Health. We acknowledge the contributions of James Nichols and Laurel Mendelsohn for nursing and technical assistance in specimen procurement and processing. We thank the patients who provided specimens and data for this study.
Author Disclosure Statement
No competing financial interests exist for any of the authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.