
Abstract
The development of an embryo constitutes a complex choreography of regulatory events that underlies precise temporal and spatial control. Throughout this process the embryo encounters ever changing environments, which challenge its metabolism. Oxygen is required for embryogenesis but it also poses a potential hazard via formation of reactive oxygen and reactive nitrogen species (ROS/RNS). These metabolites are capable of modifying macromolecules (lipids, proteins, nucleic acids) and altering their biological functions. On one hand, such modifications may have deleterious consequences and must be counteracted by antioxidant defense systems. On the other hand, ROS/RNS function as essential signal transducers regulating the cellular phenotype. In this context the combined maternal/embryonic redox homeostasis is of major importance and dysregulations in the equilibrium of pro- and antioxidative processes retard embryo development, leading to organ malformation and embryo lethality. Silencing the in vivo expression of pro- and antioxidative enzymes provided deeper insights into the role of the embryonic redox equilibrium. Moreover, novel mechanisms linking the cellular redox homeostasis to gene expression regulation have recently been discovered (oxygen sensing DNA demethylases and protein phosphatases, redox-sensitive microRNAs and transcription factors, moonlighting enzymes of the cellular redox homeostasis) and their contribution to embryo development is critically reviewed. Antioxid. Redox Signal. 13, 833–875.
In Vivo Gene Inactivation Affects Embryonic Redox Homeostasis (Knockout Mice)
Redox-Dependent Epigenetic Control of Embryonic Gene Expression
Redox-Dependent Transcriptional Control of Embryonic Gene Expression
Redox-Dependent Post-Transcriptional Control Mechanisms of Gene Expression
Clinical Relevance of Redox Imbalance in Embryo and Fetal Development
I. Introduction
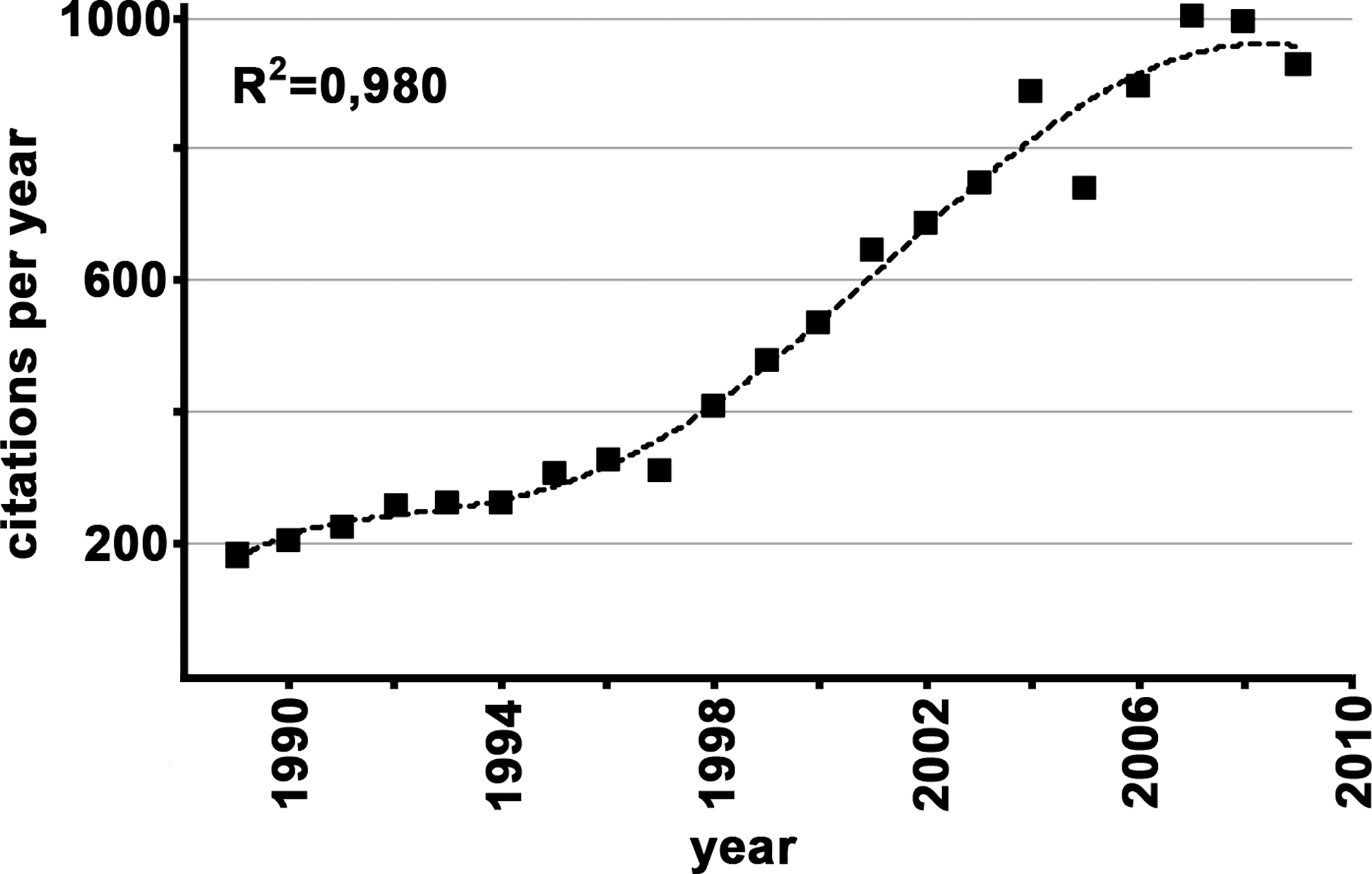
The biological role of redox chemistry is currently a busy area in developmental research. A PubMed search using the key words “redox” and “development” revealed some 13,000 entries. More than 90% of these articles have been published during the last 20 years (1989–2009). Since 2004 (the last 6 years), an averaged yearly output of some 900 articles was calculated (Fig. 1). During the late 1980s and early 1990s, there was a fairly constant output of about 220 articles per year, but between 1996 and 2006 an exponential growth was observed. More recently, the number of published articles leveled at about 990 per year. Because of these high numbers, it was not possible to consider all publications for this review. In fact, although we cited more than 400 references they only represent less than 5% of the reports published in the field. To adhere to the space limitations set by the publisher, we were forced to make selections, being aware of the fact that such selections are always a personal matter. Moreover, because of space limitations, we were unable to include redox control mechanisms in prokaryotes, yeast, and lower animals. However, since many redox-controlled processes have first been explored in lower model organisms, we will occasionally address nonmammalian studies.
II. Mammalian Redox-Homeostasis
The redox state of mammalian cells is defined by the equilibrium between pro- and antioxidative processes. Pro-oxidative reactions deliver reactive oxygen species (ROS) and/or reactive nitrogen species (RNS), which may be toxic to the cell when produced excessively. In contrast, antioxidative processes detoxify ROS/RNS and/or prevent their formation. Both sites of the redox equilibrium involve enzymatic and nonenzymatic processes and their proper balance is of major biological importance.
A. Pro-oxidative mechanisms
Molecular dioxygen has a fairly stable electron configuration and, thus, is not particularly reactive. This is also the case for its 4 electron reduction product, water. However, if dioxygen is not completely reduced (Fig. 2A) but only takes up one, two, or three electrons, it is activated to superoxide (•O2
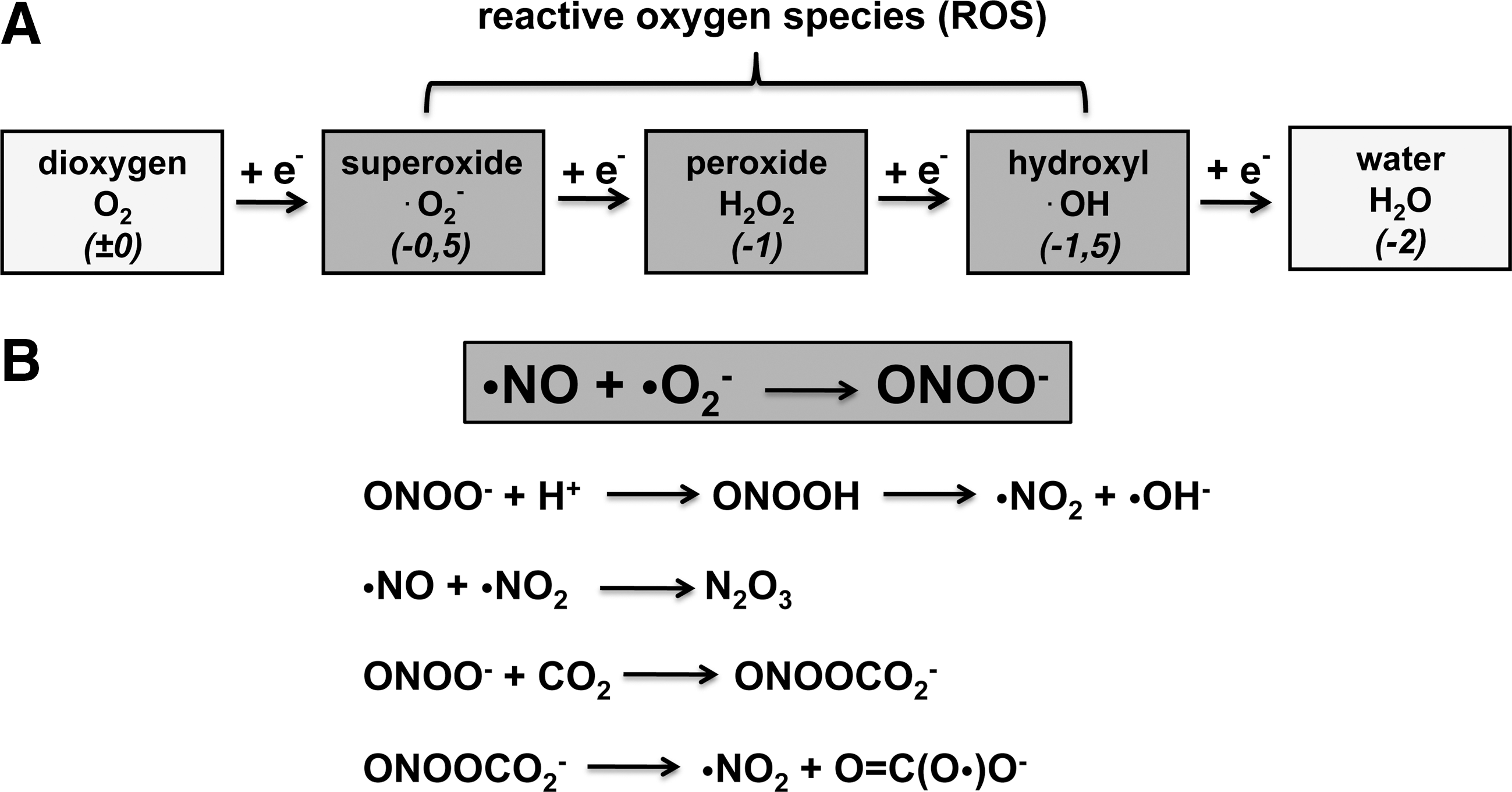
The major origin of RNS is nitric oxide (•NO). It constitutes a stable radical with a biological half-life of 1–10 s. It functions as a vasodilator by activating soluble guanylate cyclase in vascular smooth muscle cells but also as a neurotransmitter. It is biosynthesized from arginine by the catalytic activity of three nitric oxide synthase (NOS) isoforms (neuronal, endothelial, inducible), which are encoded for by three different genes and show cell-specific expression patterns (240). During inflammation, large amounts of superoxide (NADPH-oxidase) and nitric oxide (iNOS) are simultaneously formed in inflamed tissues. These redox mediators react with each other (Fig. 2B) to form peroxinitrite (OONO-), which is highly reactive and can directly attack various cellular components (224). Peroxinitrite can be protonated to form peroxynitrous acid, which decomposes to yield nitrogen dioxide (•NO2) and hydroxyl radical (•OH), two highly reactive compounds capable of modifying biomolecules. Nitric oxide and nitrogen dioxide can recombine to form dinitrogen trioxide (N2O3) that preferentially reacts with protein thiols to form covalent protein adducts. Alternatively, peroxynitrite can react with carbon dioxide (CO2) to form nitrosoperoxycarbonate (ONOOCO2 -), which can decompose (Fig. 2B) to yield nitrogen dioxide (•NO2) and the carbonate radical [O = C(O•)O-].
B. Antioxidative defense mechanisms
To avoid excessive ROS/RNS formation, a complex antioxidative defense system has evolved and several reviews have recently been published characterizing the mammalian antioxidative defense system under different pathological conditions (122, 133, 303). In brief, it consists of nonenzymatic antioxidants (ascorbic acid, tocopherols, and bilirubin) and antioxidative enzymes such as superoxide dismutases, catalase, the glutathione (GSH) system (GSH peroxidases, GSH reductase), peroxiredoxins, thioredoxins, and others. The oxidative pentose monophosphate shunt and its rate-limiting enzyme, glucose-6-phosphate dehydrogenase (G6PD), play an important role in the antioxidative defense system since they provide NADPH required as reductant. G6PD-deficiency induces major malfunctions of the antioxidative defense system and leads to hemolytic anemia and developmental retardation. An alternative source of cellular NADPH is the cytosolic malic enzyme that catalyzes decarboxylation of malate forming pyruvate (138).
C. Developmental consequences of redox alterations
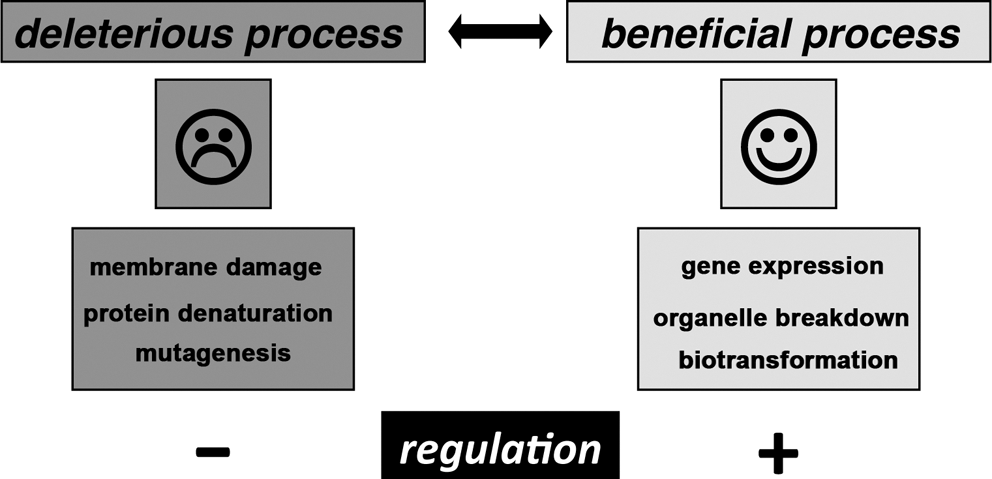
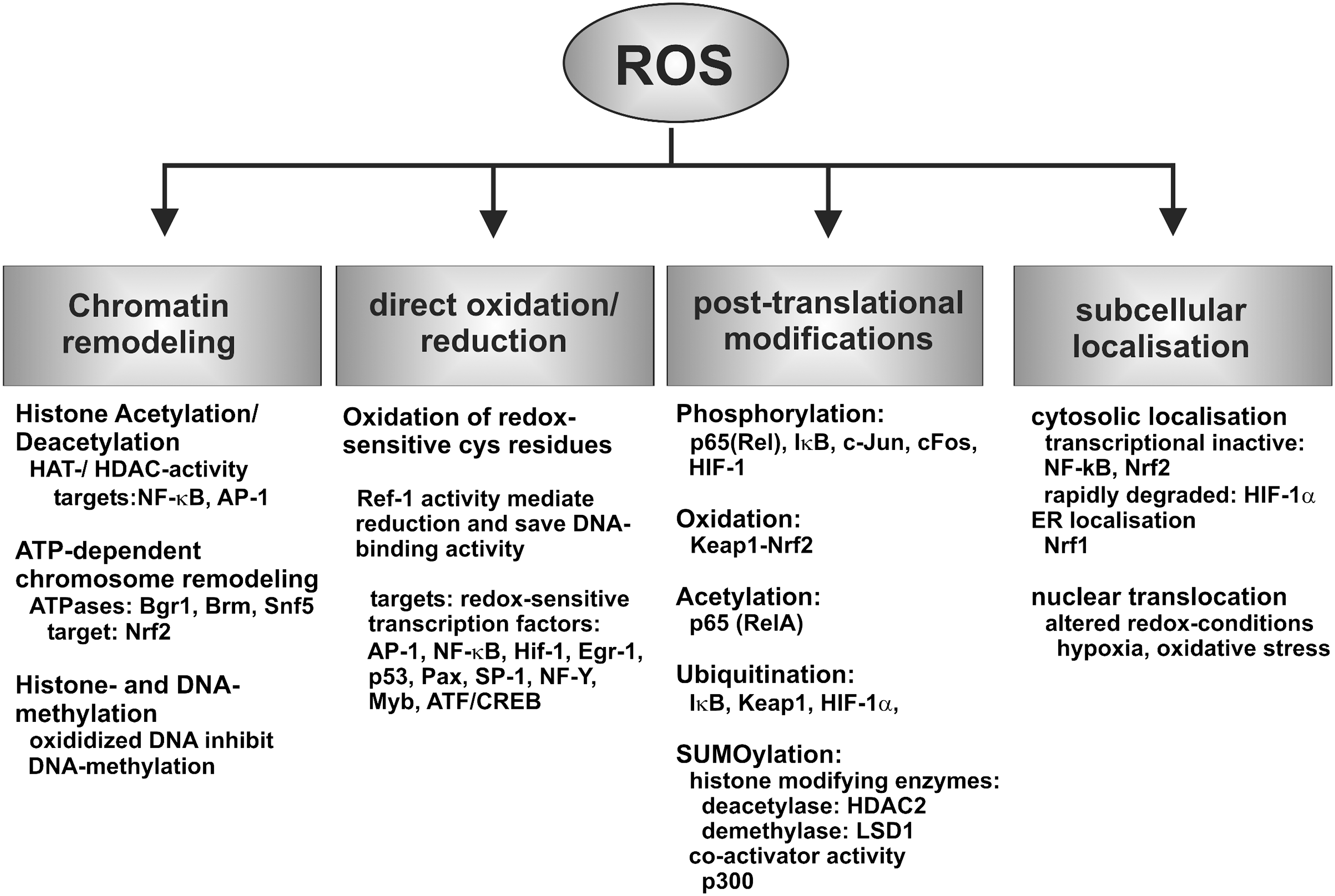
Because of their high chemical reactivity, ROS and RNS are capable of modifying biomolecules such as lipids, proteins, nucleic acids, and even carbohydrates. These modifications (Fig. 3) often lead to cellular dysfunction (detrimental activity). However, in recent years it has been well established that ROS/RNS at lower concentrations also exhibit beneficial effects (Fig. 3). The cellular redox state is an important regulator of the cellular gene expression cascade, and the activity state of a variety of transcription factors is altered by redox changes. In other words, a certain level of ROS/RNS is essential for normal cell function. For instance, hydrogen peroxide is not only considered a deleterious oxidant but also an intracellular signal transducer, which is frequently generated in a controlled manner and leads to the selective post-translational modification of cysteine residues of certain target proteins (80). Reversible thiol oxidation, in particular disulfide bond formation, changes the functional properties of affected proteins and alters their functional properties. One prominent example is the transient inactivation of protein tyrosine phosphatases in receptor tyrosine kinase signaling (393). Thus, precise control of the cellular redox homeostasis is essential for regular cell function, and this is of particular importance for complex developmental processes such as embryogenesis.
Starting at fertilization, the embryo encounters variable levels of ROS/RNS during its development, and these reactive intermediates originate from the developing embryo itself but also from maternal processes. For instance, hydrogen peroxide formed as systemic signal transducer (121) by the mother can penetrate the placental barrier to modify embryonic processes. To control the systemic redox state, ROS/RNS concentrations must be sensed so that the organism can adequately respond in case of disturbance. The consequences of redox alterations in biological systems are rather complex but they mainly affect cellular energy metabolism and the gene expression cascade. Since changes in gene expression regulation also alter energy homeostasis, the two major consequences are interrelated. This review is focused on redox-sensitive mechanisms regulating embryonic gene expression and there is no space for discussing in detail the metabolic consequences. The interested reader is referred to the primary reports cited herein.
Regulation of gene expression takes place at various levels of the expression cascade, and epigenetic, transcriptional, and post-transcriptional mechanisms have been reported. Redox-sensitive transcription factors such as HIF-1α (269), PPARs (374), or NF-κB (137) are important players in transcriptional regulation, and their biological roles have been characterized in the past. Oxidative stress affects embryo development, and redox-dependent transcriptional mechanisms have previously been summarized (83). Unfortunately, our knowledge on epigenetic and post-transcriptional mechanisms in the regulation of embryonic gene expression is less advanced. For a long time post-transcriptional mechanisms have been focused on iron-regulatory proteins (296), but in recent years other translation factors (309, 399) and miRNA-based mechanisms (51) have been studied in more detail. On post-translational levels, proteasomal degradation (416) of redox-sensitive trans-regulatory proteins plays a role in embryogenesis. Moreover, intracellular signaling cascades, which involve reversible protein tyrosine phosphorylation, appear to be redox dependent (393) and thus, alterations in the cellular redox homeostasis alter intracellular signaling.
III. Redox Control During Embryo Development
A. Normal mouse embryo development
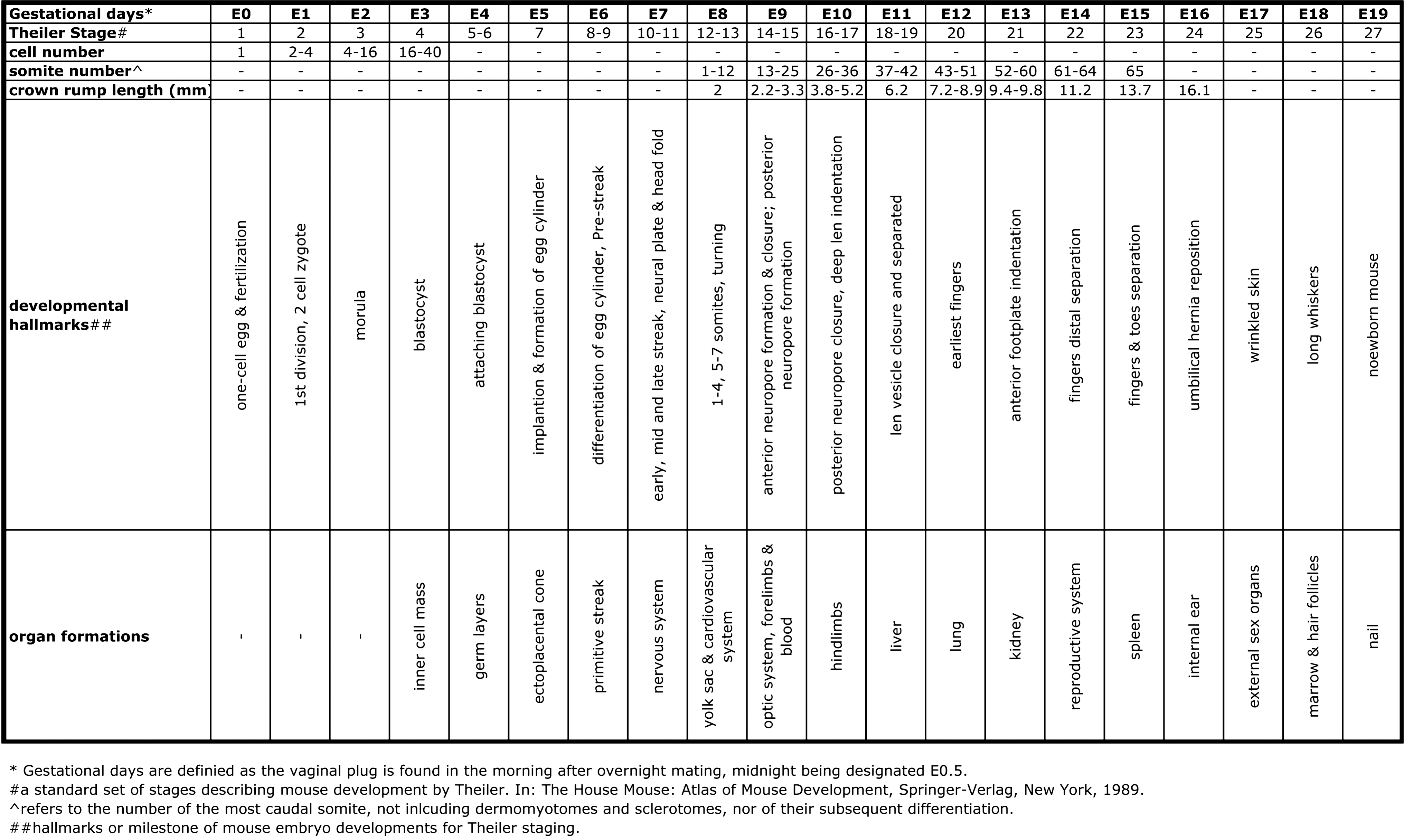
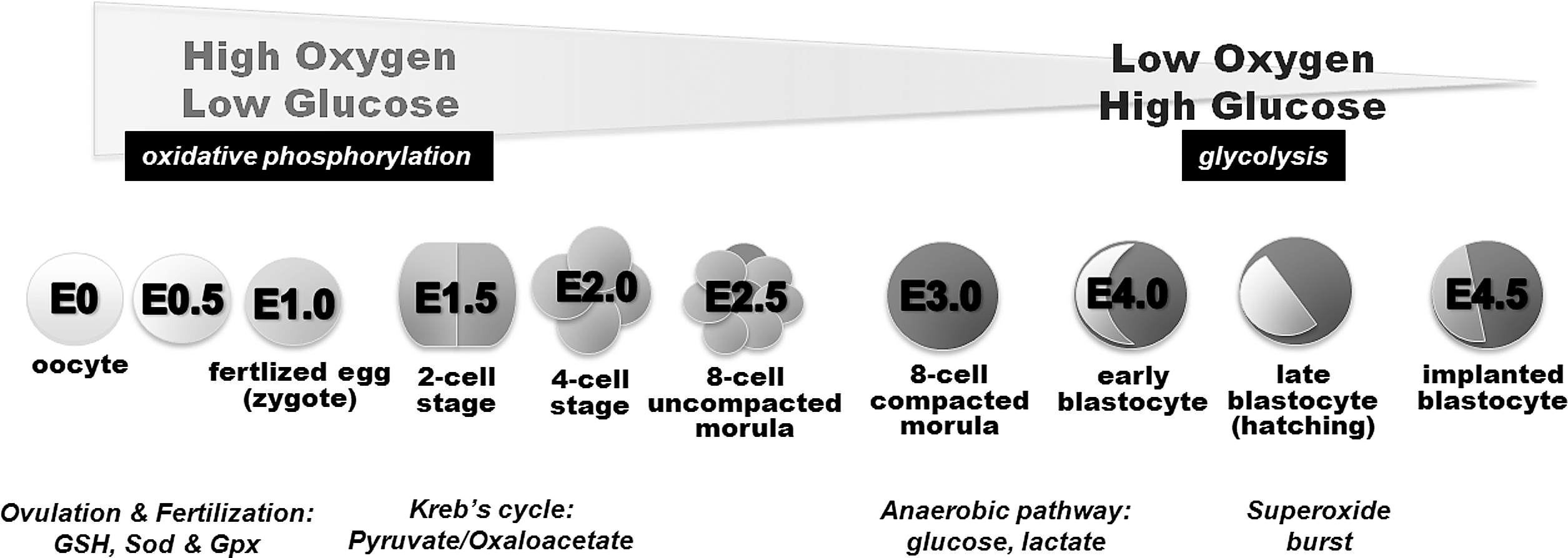
Embryogenesis is initiated by fertilization of the oocyte (gestational day 0, E0) and lasts 18 to 19 days (gestational day 18 to 19, E18 to E19). Normally, it leads to fully functional newborns and various developmental stages can be separated. In Figure 4 a time course of murine embryogenesis is depicted and important growth parameters and major developmental milestones are summarized. Development of the central nervous system starts early at E6–7 and continues throughout the entire life. Development of the cardiovascular system and erythropoiesis begins around E8 but organogenesis of liver, lung, kidney, and the reproductive system is initiated at later developmental stages (98).
B. Embryonic antioxidative capacity
A tightly controlled redox system is essential for normal embryogenesis, and dysregulation of the redox equilibrium in either direction severely hampers embryo development. Reductive stress is just as dangerous as oxidative challenge (42) and thus, the equilibrium between oxidative and reductive forces is a key factor for proper embryogenesis. At certain developmental periods (early postimplantation stage, completion of embryonic circulation) the embryo is particularly susceptible to redox alterations and at these stages a functional antioxidative defense system is of particular importance. As for the adult organism, low molecular weight antioxidants (GSH, vitamin C, and vitamin E), metal chelators (desferoxamine), and antioxidative enzymes play an important role in protecting developing embryos from oxidative damage. Unfortunately, the enzymatic activity of many antioxidative enzymes, including GPx, GSH-reductase, SOD and CAT, is much lower than that of the adult organisms (301, 431) and thus embryos are particularly sensitive to oxidative damage. A known exception is embryonic G6PD, which is expressed at similar levels as in adult individuals (280). However, in some cases an overdose of antioxidants deteriorates the redox state and may induce teratogenic alterations (407). Owing to the high complexity of the redox network more research is needed to understand the involved regulatory mechanisms. A better understanding of embryonic redox network is vital for optimizing in vitro embryogenesis and for ensuring normal development in vivo (83).
C. Oxygen requirement and energy metabolism during early embryogenesis
Oxygen and glucose are essential for normal embryo development. Hypoxia (52) and hypoglycemia (333) are major risk factors for embryonic lethality, and oxidative stress is a mechanism in teratogenesis (129). However, the relative importance of oxygen at different stages of embryogenesis is variable, depends on anatomic preconditions, and the developing embryo is able to cope with these challenges by adapting its metabolism depending on the substrates available (Fig. 5). In this context, embryonic mitochondria play a vital role in controlling the relative production of ROS and/or providing reducing equivalents for the cell to cope with ROS (89). Oviductal oxygen levels are lower than atmospheric oxygen concentrations (215). In the uterus, where implantation takes place, even lower oxygen levels have been determined (101). Thus, following fertilization in the oviduct, the embryo encounters a decreasing oxygen gradient when moving down the reproductive tract. In fact, during early implantation the embryo is confronted with an almost anoxic environment (215). The glucose concentration in oviductal fluid is relatively low when compared with corresponding levels in uterine fluid. These differences in substrate concentrations severely alter the energy metabolism of the developing embryos. In the oviduct, where oxygen is present at saturating conditions, but glucose (major energy supplying nutrient for early embryos) appears to be rate-limiting (low glucose, high oxygen), oxidative ATP production via glycolysis, citrate cycle, and oxidative phosphorylation are the major energy sources (Fig. 5). However, at later developmental stages, when the embryo reaches the uterus (high glucose, low oxygen), glucose supply may not be critical any more so that less efficient anaerobic metabolic pathways (lactate formation) may contribute higher shares to systemic ATP production (15). In brief, the energy metabolism of early mammalian embryos (one-cell stage) proceeds via oxidative phosphorylation, whereas at blastocyst stage anaerobic glycolysis prevails (388). The switch between aerobic and anaerobic energy supply is balanced by two major regulatory elements: i) expression regulation of enzymes involved in energy metabolism, which is directly related to oxygen sensing, and ii) NAD(P)+/NAD(P)H + H+ ratio, which reflects the cellular redox state in early embryogenesis (134).
Molecular oxygen sensors play an important role in regulation of embryonic energy metabolism. They adjust the cellular influx and efflux of energy supplying substrates through different metabolic pathways and thus, they are crucial for effective ATP production (6, 419). Mitochondria are of major importance for the cellular redox state and contribute to oxygen sensing. They are a major source of ROS (432), which control the activity state of various transcription factors. Mitochondria are not symmetrically distributed within early embryos, and in some species this asymmetric distribution has been related to the specification of the developmental axis (70). The activity of the glycolytic pathway is affected by both oxygen availability via stabilization of hypoxia-inducible factor 1 (HIF-1) and indirectly through the redox state via the NAD(P)+/NAD(P)H ratio (134).
D. ROS-mediated teratogenesis
Dysregulation of embryogenesis induces developmental retardations, organ malformation, and functional deficiencies commonly termed teratogenesis. In teratogenesis, the redox equilibrium plays an important role (134) and endogenous (embryonic) or exogenous (maternal) ROS formation have been implicated. The processes regulating embryonic redox homeostasis are important determinants of teratological risk. It is well known that maternal and embryonic metabolism of xenobiotics is frequently accompanied by ROS formation, but the individual risk of ROS-mediated teratogenesis depends on both genetic and/or environmental factors (424). During certain time windows of embryogenesis, the embryo is more susceptible to oxidative stress and excessive ROS formation (129). Many drugs and environmental chemicals, which are capable of inducing oxidative stress in the embryo, are eliminated or metabolized during maternal circulation before reaching the embryo. Others cannot penetrate the placental barrier and thus may not be dangerous for the embryo (424). A number of xenobiotics (proteratogens), that are relatively nontoxic for both the embryo and the mother, are bioactivated during maternal metabolism to reactive radicals (425), which may then be transferred to the embryo.
Many xenobiotics and their primary biotransformation products are conjugated with glucuronic acid or sulfate to increase water solubility so that they can be effectively secreted in the urine. A deficiency in maternal conjugating pathways will increase oxidative challenge of the embryo (425). Another mechanism by which maternal and/or other extra-embryonic pathways may modulate ROS-mediated teratogenesis is the production of diffusible factors that can cross the placental barrier. This is exemplified by hydrogen peroxide and the peroxynitrite pathways (250).
E. Expression of ROS/RNS producing enzymes during murine embryogenesis
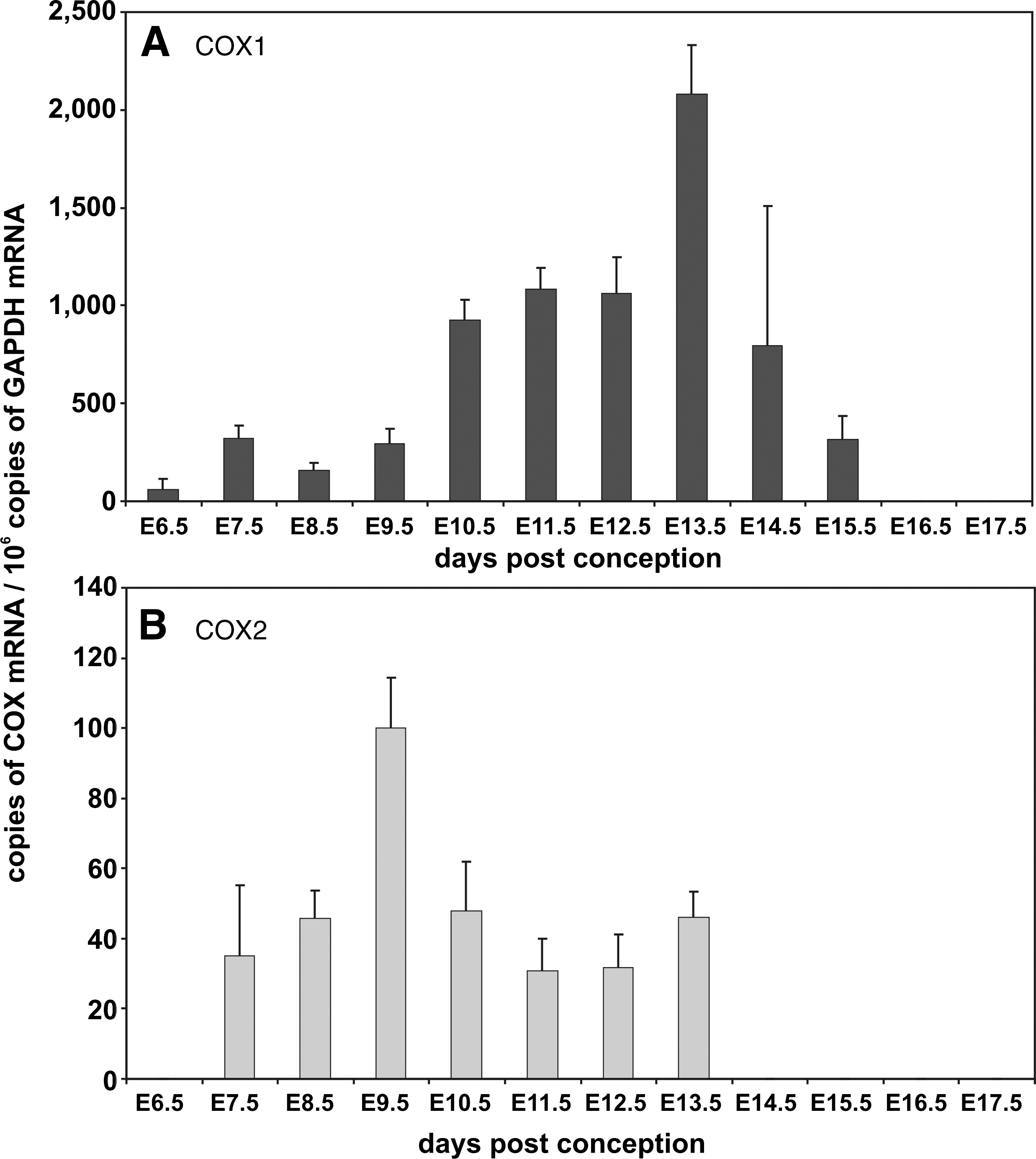
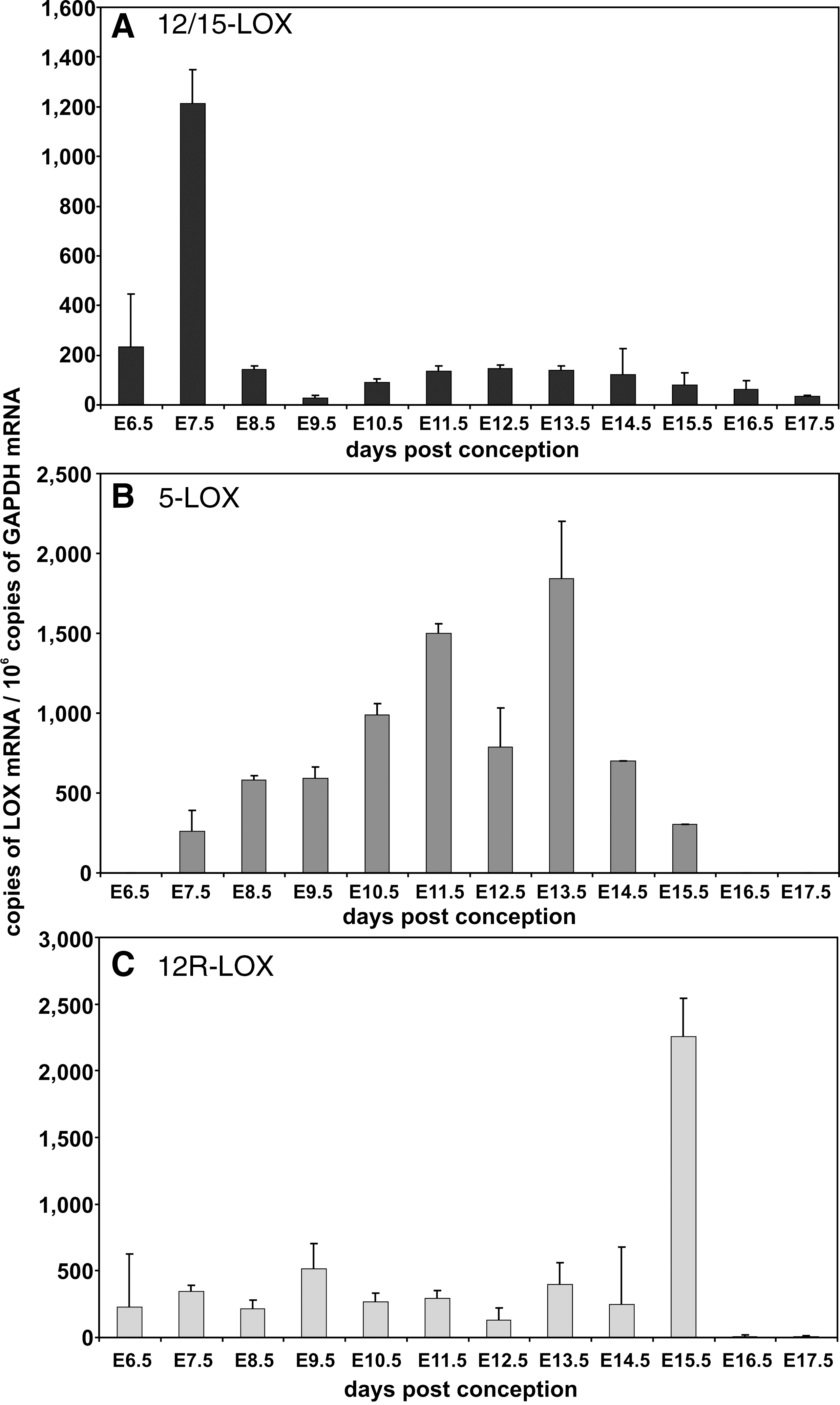
During mouse embryo development, most cytochrome P450 (CYP) isoforms involved in xenobiotic metabolism are not expressed at high levels (173). In humans, most CYP isoforms are also expressed at low levels but at later developmental stages, some CYP isozymes (CYP1B1, CYP3A7, and CYP2E1) can be detected in higher quantities (425). Lipoxygenase (LOX) and cyclooxygenase (COX) isoforms have also been implicated in embryonic ROS formation (Fig. 6). COX1 (430) and COX2 (300) are constitutively expressed in mouse embryos. We detected COX1 mRNA expression throughout embryo development and the mRNA copy number varied between 20 and 2500 copies per 106 GAPDH mRNA copies depending on the developmental stages of the embryo (Fig. 7A). It can be seen that COX1 mRNA increases from E6.5 to E13.5 but then declines when the embryo approaches birth. In contrast, COX2 expression is significantly lower, ranging between only 40 to 100 copies per 106 GAPDH mRNA copies. It peaks early at E 9.5 but then declines similar to COX1 to undetectable levels. Among LOX isoforms, 12/15-LOX is high level expressed during early embryogenesis. At E7.5 we quantified 1200 12/15-LOX mRNA copies per 106 GAPDH mRNA copies. At later developmental stages, 12/15-LOX mRNA expression declines dropping below the threshold value of our assay system when the embryo approaches birth (Fig. 8A). 5-LOX expression is detectable at E7.5 and peaks at E13.5 with about 2000 copies per 106 GAPDH mRNA copies (Fig. 8B). In contrast, 12R-LOX (Fig. 8C) is expressed at lower levels (less that 500 copies per 106 GAPDH mRNA copies) throughout embryo development with a sharp expression peak of more than 2000 copies per 106 GAPDH mRNA copies at E15.5. Between E16.5 and E17.5, neither 5-LOX nor 12R-LOX expression was observed.
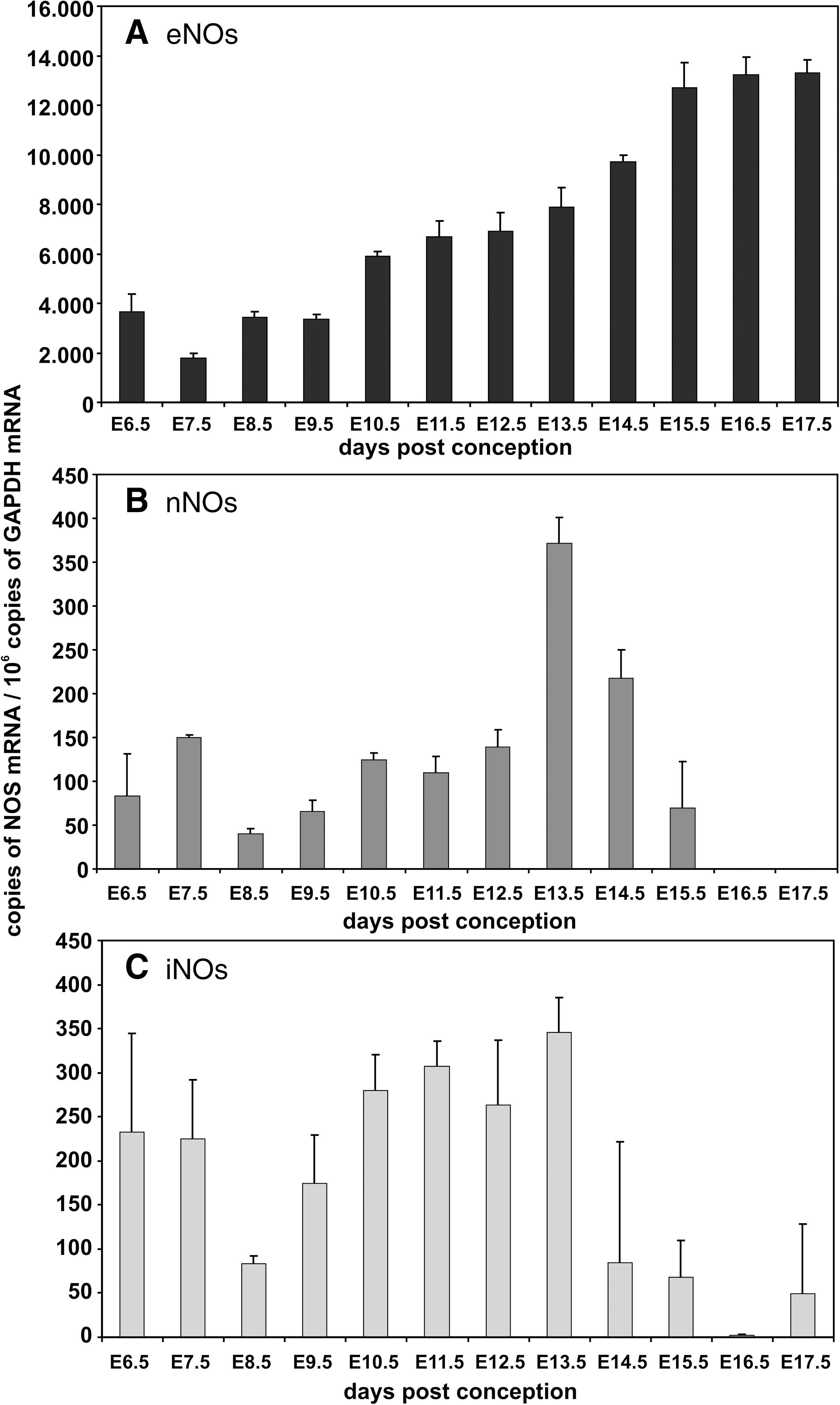
Nitric oxide plays an important role for the formation of RNS. We quantified expression of the three nitric oxide synthase isoforms (eNOS, nNOS, iNOS) during the time course of murine embryogenesis and found that eNOS was expressed at rather high levels (2,000–13,000 copies of eNOS mRNA per 106 GAPDH mRNA copies). During early embryogenesis (E6.5–E11.5) lower mRNA levels (2000–4000 eNOS mRNA copies per 106 GAPDH mRNA copies) were quantified, but at a later stage the mRNA copy number was upregulated (Fig. 9A). In contrast, mRNA levels of both nNOS and iNOS were expressed at lower quantities of less than 400 copies per 106 copies GAPDH mRNA (Figs. 9B and 9C).
F. Gamete formation, fertilization, and organogenesis
Germ cells are sensitive to changes in redox conditions. The high concentrations of polyunsaturated fatty acids in spermatoid cells make them susceptible to ROS-induced lipid peroxidation, which alters the functional characteristics of the membranes (2). Oxidative modification of membrane phospholipids induces changes in sperm motility and morphology, which finally leads to reduced efficiency of oocyte fertilization (152). To avoid such functional deficits, sperm and their cellular precursors are protected by an excess of antioxidative enzymes (316). In mammalian oocytes, SOD1, SOD2, and GPx isoforms are expressed during various stages of oogenesis (93) and peroxiredoxins, particularly Prx6, are upregulated during in vitro oocyte maturation (218).
GSH appears to play an important role in the preparation of a mature oocyte to receive a sperm and in early zygote development. During oocyte maturation, an increase in GSH was reported (238) and high GSH concentrations are maintained during first cell divisions. However, when the late blastocyst prepares for implantation, significantly lower GSH levels have been reported (108). Different superoxide scavengers prevent the blastomere from hatching from the zona pellucida (387) and these data suggest an important role of superoxide in blastocyst formation. Unfortunately, the detailed mechanisms of this effect have not been explored so far. After in vitro fertilization, embryo development is arrested at the two-cell stage, which coincides with the onset of embryonic gene expression. For mouse embryos this developmental arrest can be overcome by treatment with antioxidants (271). Interestingly, expression of peroxiredoxins is sustained during the first cell division and it further declines to the 16-cell stage. However, during blastocyst formation peroxiredoxin expression picks up again (218).
During the early post-implantation period, rodent embryos are most susceptible to oxidative challenge since they have adapted to hypoxic conditions in the uterus (101). Thus, for in vitro embryogenesis, usually low oxygen concentrations (5%–10%) are adjusted. The molecular basis for the reduced oxygen requirement in this developmental stage remains unclear, but if explanted and cultured in vitro under normoxic conditions the embryos develop structural abnormalities (52, 262, 276).
The yolk sac plays an important role for early embryo development since it delivers oxygen and nutrients for the embryo during the early post-implantation period. Later on, the yolk sac regresses and the allantoic placenta takes over its function. These sudden alterations in uteroplacental circulation expose the embryos acutely to higher oxygen concentrations (276). Once the uteroplacental and embryonic circulation systems are developed (E9.5–10.5) the embryo becomes less sensitive for maternal redox alterations, instead endogenous ROS/RNS formation becomes a more disturbing process.
G. Redox control of cellular processes during embryogenesis
The cellular redox equilibrium affects a variety of basic cellular functions such as energy supply, proliferation, differentiation/maturation, and apoptosis (334). On the other hand, these cellular functions may also alter the redox equilibrium, suggesting the existence of multiple regulatory networks that are in part feedback-controlled (76). In embryogenesis, the basic functions of each cell are controlled within the frame of a heterogeneous four-dimensional space-time continuum and this requires intense intercellular cross-talk.
When proliferating mammalian cells are oxidatively challenged, they exhibit a broad spectrum of responses and these responses depend on the cell type, on their metabolic state, on the experimental conditions, and on intensity and duration of the stimulus. Low levels of ROS (hydrogen peroxide, superoxide) stimulate proliferation of cells (330) and overexpression of antioxidant enzymes appears to reverse this effect (358). These data support the hypothesis that ROS may act as signals to maintain cell proliferation and cell growth. On the other hand, higher concentrations of hydrogen peroxide temporarily induce cell growth arrest (82). After temporary growth arrest, many cells exhibit a transient adaptive response, in which genes for oxidant protection and DNA repair are preferentially expressed. At even higher hydrogen peroxide concentrations, mammalian cells are not able to adapt, but instead enter into a permanently growth-arrested state, in which they appear to perform most normal cell functions but cease to divide, a state resembling cellular replicative senescence (82). If oxidative stress is further increased, cells may die in an organized manner (apoptosis).
Cell differentiation and maturation is an integral part of embryogenesis and complete differential arrest in early embryogenesis is lethal (18). After priming, embryonic stem cells differentiate within a certain lineage according to a biological program that has been optimized during evolution. These differentiation processes are characterized by major metabolic alterations and finally result in the formation of highly specialized cell types with limited functionality. Many of these functional alterations are mediated by the intracellular redox equilibrium (38, 211, 219, 336). Spontaneous differentiation of human embryonic stem cells is paralleled by ROS formation. Overexpression of various antioxidant enzymes, including SOD, CAT, and peroxiredoxins, induced marked alterations in the differentiation pattern (63). Continuous exposure of embryonic stem cells to ROS results in an inhibition of cardiomyogenesis and vasculogenesis. On the other hand, a low-level ROS pulse enhances differentiation along some lineages (219, 337).
Apoptosis is an essential process in embryo development and regular organogenesis involves balanced apoptosis as key event (161). During embryonic development of the central nervous system, neuronal apoptosis frequently occurs and this process affects all areas of the brain in a time-dependent manner. In mouse embryos, there appears to be a spatial correlation between developmental apoptosis and ROS formation. In fact, at midgestation a high degree of apoptotic alterations can usually be seen in areas with enhanced ROS formation (334). Another obvious example for this correlation is embryonic limb development. The interdigital regions of primitive limbs are characterized by pronounced apoptotic alterations, and simultaneously a high degree of ROS formation can be detected in these areas (342). Expression of antioxidative enzymes is limited to the developing digits and downregulated in the interdigital regions, thereby establishing a mechanism enhancing the effects of ROS activity and apoptosis in areas where they are needed and to limit ROS formation in neighboring tissues (342, 352). For the in vivo situation, it is difficult to tell whether apoptosis induces secondary ROS formation or whether ROS formation triggers developmental apoptosis, and mechanistic studies in cellular in vitro systems cannot conclusively answer this question. Retinoic acid is known as an inducer of developmental processes, and during embryogenesis retinoic acid-induced signaling has been implicated in the control of apoptosis in many regions of the embryo (77). Retinoic acid induces apoptosis in the interdigital regions, which is paralleled by increased ROS formation but impaired peroxidase activity (342). These data suggest that an increased ROS formation is the primary process required for induction of apoptosis at this developmental period.
IV. In Vivo Gene Inactivation Alters Embryonic Redox Homeostasis (Knockout Mice)
A. Knockout of pro-oxidative enzymes
The innate immune response towards pathogen challenge involves targeted ROS formation (119). Phagocytes (neutrophils, macrophages) bind pathogens at pattern recognition receptors (286) and internalize the microbes via phagocytosis. In phagolysosomes, the pathogens are killed by a concerted action of oxidative and nonoxidative reactions, and NADPH oxidase plays a major role. This multimeric enzyme is localized in the membrane of the phagolysosomes and is activated upon pathogen challenge. During activation several cytosolic and membrane-bound proteins including p47phox and the flavoprotein gp91phox assemble at the phagolysosome membrane and catalyze electron transfer from NADPH+H+ to molecular dioxygen forming superoxide (92). In humans, a defective p47phox gene leads to chronic granulomatous disease characterized by severe recurrent bacterial and fungal infections (9). Similarly, targeted knockout of the p47phox gene in mice resulted in viable animals that frequently suffer from bacterial infections (160). Disruption of the X-linked gp91phox gene led to viable animals that exhibited an increased susceptibility to bacterial infections (311). However, embryonic development of these animals appears not to be affected (Table 1). Myeloperoxidase (MPO) generates highly reactive hypochlorous acid (HOCl), which is employed by phagocytes for oxidative microbial killing. MPO knockout mice develop normally but show impaired host defense when challenged with pathogens (5).
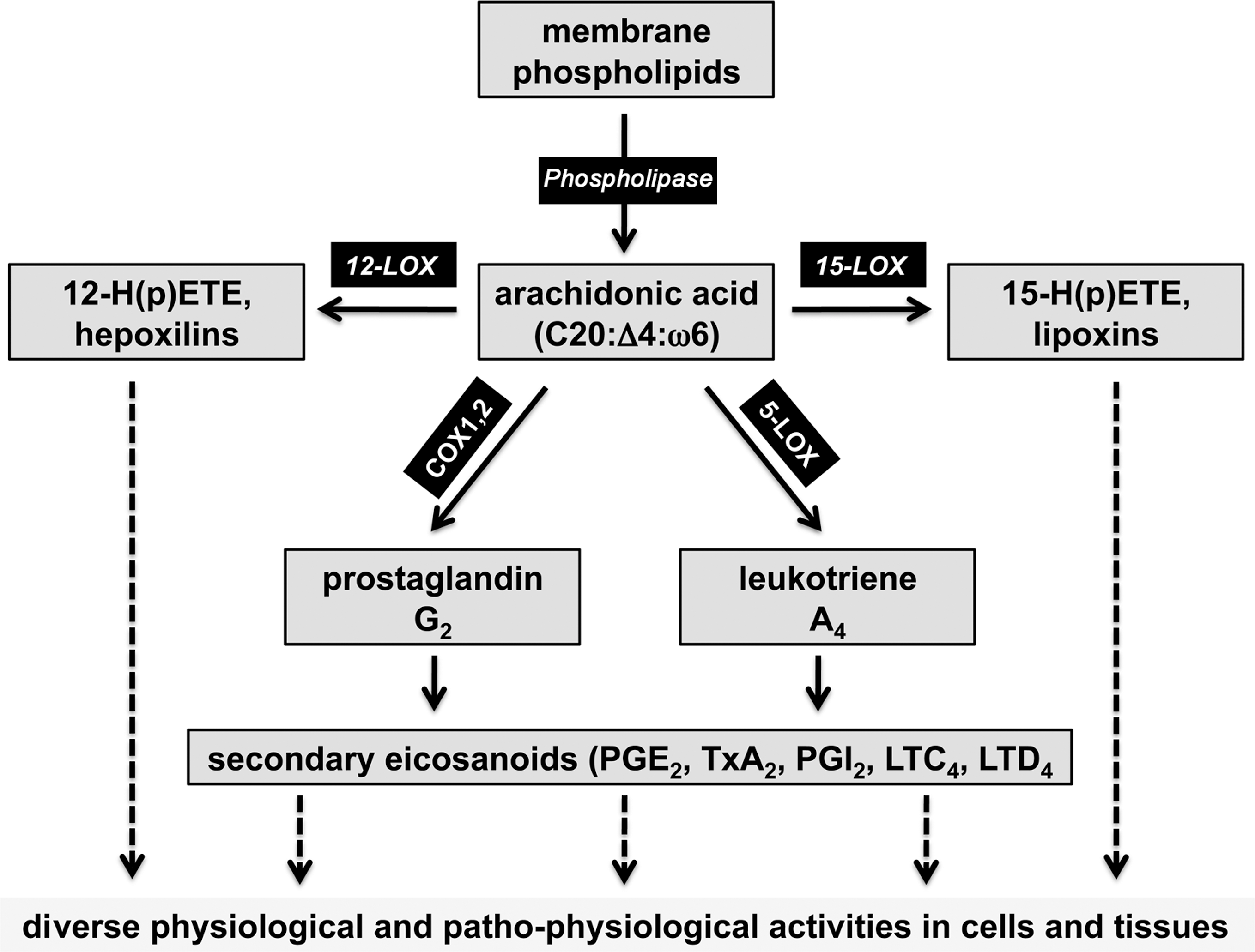
Eicosanoids (prostaglandins, leukotrienes, and lipoxins) form a heterogeneous family of lipid mediators, which have been implicated in the regulation of physiological and patho-physiological processes (202). Eicosanoid biosynthesis involves direct oxygenation of arachidonic acid via the lipoxygenase (LOX) or cyclooxygenase (COX) pathway (Fig. 6), and COX inhibitors are available for prescription as nonsteroidal anti-inflammatory drugs. In mice and humans there are two COX isoforms that originate from separate genes. Disruption of COX1 gene did not dramatically alter embryo development but adult animals exhibited an impaired inflammatory response in selected inflammation models (207). Selective COX2 knockout induced defective postnatal kidney developmental and myocardial fibrosis. In addition, an increased rate of neonatal death was frequently observed (85, 260). Female COX2-/- mice were virtually infertile, which was related to abnormal ovaries lacking the corpora lutea. Adult survivors exhibited impaired inflammatory response in certain inflammation models (85). COX1/COX2 double knockouts (233) develop normally until birth but fail to induce closure of the ductus arteriosus and this leads to premature death in about one-third of the newborns. This data contrasts previous observations suggesting that COX inhibitors stimulate postnatal closure of ductus arteriosus (45). When COX1 expression is put under the control of COX2 regulatory elements (COX1COX2) in a COX2-deficient background, the patent ductus arteriosus phenotype is fully rescued (446). Unfortunately, no offspring with COX1null/COX1COX2 genotype could be generated, suggesting that COX1 and COX2 exhibit discriminate isoform functionality during embryo development (446). Expression of an inactive COX2 mutant rescues the incidence of patent ductus arteriosus in COX2-/- mice when COX1 is expressed, but fails to do so when COX1 is not present (445). Consequently, it has been suggested that COX-derived prostaglandin signaling may be dependent on COX1/COX2 heterodimerization (446). The LOX pathway of the arachidonic acid cascade leads to the formation of other lipid mediators (leukotrienes, lipoxins) but LOX isoforms have also been implicated in cell differentiation (402) and skin development (94). In mice, there are seven functional LOX genes (12/15-LOX, 5-LOX, platelet-type-12SLOX, 12R-LOX, 8-LOX, eLOX3, and epidermis-type 12S-LOX), but in humans the epidermis-type 12S-LOX gene is a functionless pseudogene (106). Functional inactivation of the 12/15-LOX gene, platelet-type 12-LOX gene, and 5-LOX gene did not lead to major phenotypic alterations and there is no obvious impact on embryogenesis (106). However, 5-LOX knockouts are less susceptible to antigen challenge in murine asthma models, have a reduced response for inflammatory stimuli (106), and develop aortic aneurysms when fed a lipid-rich diet (450). 12/15-LOX knockout mice are somewhat smaller than wild-type controls (statistic trend, but not significant) and gain significantly less weight when fed a lipid diet (346). Moreover, they are less susceptible to atherosclerosis (449), but there appear to be gender-specific differences (310). 12/15-LOX + 5-LOX double knockouts develop normally but female individuals are more resistant to atherosclerosis when bred into an apolipoprotein E-deficient background (310).
Monoamine oxidases (MAO-A and MAO-B) are key enzymes in the metabolism of biogenic amines and are located at the outer mitochondrial membrane. They catalyze oxidative substrate deamination and contribute to the cellular redox homeostasis by producing stoichiometric amounts of hydrogen peroxide. Although both isoforms are expressed during mouse embryonic development (Fig. 7) knockout embryos show no severe developmental abnormalities until birth (40, 120). However, brains of MAO-A-/- mice show subtle changes during early postnatal development, which have been related to elevated serotonin concentrations (41). A spontaneous point mutation in the MAO-B gene of MAO-A-/- mice gave rise to MAO-A/B double knockout mice but these animals do not show major developmental defects either (55). Forebrain-specific MAO-A expression on an MAO-A-deficient background restores the wild-type somatosensory cortex barrel field structure and ameliorates the aggressive MAO-A-/- phenotype (54).
As indicated above, elevated concentrations of nitric oxide may induce the formation of RNS and thus, nitric oxide synthase isoforms (eNOS, iNOS, nNOS) are classified pro-oxidative enzymes. Knockout mice for the three NOS-isoforms have been created but none of them showed major abnormalities during embryo development (114, 123, 147, 208). However, nNOS knockout mice develop a hypogonadism and are infertile owing to disturbances of the hypothalamo-pituitary axis (123). NOS triple knockouts (261) are viable and develop normally. This data suggests that targeted NO formation may not be essential for embryo development.
Xanthine oxidoreductase (XOR) is a key enzyme in the breakdown of purine nucleotides and catalyzes the conversion of hypoxanthine to xanthine and its further transformation to uric acid. In mammals, it is present as homodimer and each subunit contains two iron–sulfur clusters, a FAD, and molybtopterin, which are involved in electron transfer (304). Under normoxic conditions, the enzyme works as dehydrogenase transferring electrons from substrates to NAD+. Under hypoxic conditions, it functions as oxidase using molecular dioxygen as electron acceptor generating large amounts of superoxide (338). Thus, under hypoxic conditions, xanthine oxidase significantly contributes to ROS formation. XOR-/- mice are viable, fertile and do not show major signs of embryonic retardation (288). However, in adult individuals, kidney alterations have been described that resemble those reported for COX2-/- mice.
Heme oxygenases (HO) are key enzymes in heme degradation and HO-1 expression is regulated by the redox-sensitive transcription factor Nrf2 (377). The reaction yields equimolar amounts of biliverdin, iron, and carbon monoxide. Two HO isoforms exist, the stress-induced HO-1 and the constitutive HO-2. HO-1 has been suggested to play an antioxidant role since it is involved in the biosynthesis of the antioxidant bilirubin. In contrast, the major function of HO-2 appears to be the production of carbon monoxide as a signaling molecule. HO-1-deficient mice are viable and develop normally (313). However, significant reduction of litter size was observed, but the underlying mechanisms are not clear. Fibroblasts derived from HO-1-/- mice were more susceptible to stress induced by hydrogen peroxide (314). Reduction of HO-1 expression impairs heart function, increases the severity of heart infarction, and HO-1-/- cardiomyocytes are more susceptible to hypoxia (440, 443). Genetic ablation of HO-2 does not induce significant alterations of the embryonic phenotype (312).
Several hundreds of cytochrome P450 isoforms have been identified in mammals and their functional importance has not always been clarified (272). An important step in the cytochrome-P450 cycle is its reduction by cytochrome P450 reductase (CPR). The important role of CPR for embryonic development is indicated by severe phenotypic alterations induced by CPR knockout (291). CPR-/- embryos do not turn, fail to induce neural tube closure, have a shortened anteroposterior axis, and show severe abnormalities in the development of the heart, limbs, and the forebrain (291). Retarded vascularization in CPR-/- animals coincides with strongly reduced expression of HIF-α, which is considered to be important for vascularization and erythropoiesis (291). Complete CPR knockout leads to embryonic lethality before day E10.5. When the CPR membrane binding domain, which is thought to be vital for CPR function, was deleted, a less severe phenotype was observed affecting mostly neural tube and heart development (357).
B. Knockout of antioxidative enzymes
The first line of defense against superoxide is formed by superoxide dismutases (SODs). In mice, there are three major SOD isoforms (cytosolic Cu/Zn-SOD, mitochondrial Mn-SOD, and extracellular SOD) originating from different genes. Homozygous Cu/Zn-SOD-/- mice show developmental defects and are more sensitive to neuronal damage (Table 1) (319). Breeding of these animals is problematic since litter size is reduced and about 75% of the embryos undergo premature intrauterine death (142). Unfortunately, the underlying mechanisms are not well understood. Mn-SOD knockout embryos develop normally with little structural defects at birth (221). After birth, Mn-SOD-/- pups are hypotonic, hypothermic, and develop dilated cardiomyopathy. Moreover, lipid accumulations in liver and skeletal muscles were observed and the animals develop a metabolic acidosis. The knockout mice do not survive for longer than 10 days. Depending on the genetic background, dilated cardiomyopathy can already occur prenatally (148). Knockout of the extracellular SOD has no measurable impact on embryo development (34). However, when stressed by hyperoxia (>99% oxygen) survival rate of these animals is reduced. Hydrogen peroxide is an intracellular oxidant and its excessive accumulation is prevented by catalase (CAT). Catalase knockout mice do not exhibit major developmental abnormalities and are not more susceptible to oxidative stress induced with hyperoxia than wild-type controls (144). However, cortical injury of CAT-/- mice induces more severe impairment of brain mitochondrial function (144).
In addition to SOD and CAT, the glutathione system is one of the major antioxidative defense systems. It strongly depends on the availability of reduced GSH, which is constantly supplied by reduction of its oxidized counterpart GSSG. This reduction requires GSH reductase and NADPH+H+, which mainly originates from the oxidative hexose monophosphate shunt. As indicated above, G6PD is the rate-limiting enzyme for this pathway and thus, it constitutes a key player in the GSH-dependent antioxidative defense system. G6PD-deficient stem cells are extremely sensitive to oxidative stress (294). After implantation, G6PD-deficient embryos develop normally until E7.5. However, at E8.5 they stop growing and die before E11.5 (234). This phenotypic alteration has been related to the onset of erythropoiesis and the development of the circulation system. Because of the early death of G6PD-/- embryos, it is difficult to study the impact of G6PD deficiency in later developmental stages. However, using G6PD-deficient embryonic stem cells, it has been shown that G6PD is dispensable for the differentiation of many cell types (293). In contrast, during erythropoiesis, G6PD-deficient cells undergo apoptosis after the onset of hemoglobin biosynthesis. In a different in vivo model, reduced expression of G6PD renders the mutant mice more susceptible to teratogenic drugs (280). De novo synthesis of glutathione from the precursor amino acids requires the activity of the γ-glutamylcysteine synthase (γGCS). Mice deficient for the heavy subunit (γGCS-HS) of the γGCS show developmental abnormalities. They fail to gastrulate and die before E8.5 owing to excessive developmental apoptosis. Cell lines derived from γGCS-HS deficient animals can regain their proliferation capability by adding GSH into the culture medium (360). Glutathione reductase (GR) is a key enzyme of the GSH-dependent antioxidative defense system, but to the best of our knowledge no targeted GR knockout has been created so far. A GR hypomorphic mouse line that exhibits strongly reduced GR levels and resembles a GR knockout line is more sensitive to diquat-induced oxidative damage. However, considering the key function of GR in antioxidative defense these phenotypic alterations are rather subtle (315, 326, 327).
Glutathione peroxidases (GPx) are also important constituents of the GSH-dependent antioxidative defense system. In mice, there are seven different GPx isoforms (GPx1–GPx7) originating from different genes. In addition, in various mouse strains there are GPx pseudogenes some of which appear to be functional (27). Targeted knockouts for GPx1, GPx2, and GPx5 generate viable offspring and no major developmental retardations have been described under physiological conditions (46, 60, 97). However, when stressed with diquat, GPx1-/- mice are more vulnerable to oxidative damage (104). In contrast to other GPx-isoforms, homozygous GPx4 knockout mice are not viable. In 2003, two groups independently showed that GPx4-/- embryos die at midgestation (E8.5) and undergo intrauterine resorption (150, 439). Interestingly, heterozygous knockout mice are fully viable and breed normally. Until E6.5, GPx4-/- embryos were morphologically indistinguishable from wild-type controls. At E7.5, the embryos still resembled pre-gastrulation individuals. Thus, similar to γGCS-HS knockout mice, GPx4-/- animals fail to gastrulate and suffer from increased apoptotic cell death. Embryonic fibroblasts derived from GPx4+/- were more sensitive to oxidative challenge and radiation (439). GPx4 is expressed in three different isoforms (short [sGPx4], long [lGPx4], and nuclear [nGPx4]), which originate from a single GPx4 gene localized on murine chromosome 10. The lethal phenotype of GPx4-/- mice can be rescued by introducing a mutated human GPx4 gene that encodes for cGPx4 but not by a gene construct encoding lGPx4 (223). The cGPx4 transgene protects GPx4-/- mice from diquat-induced apoptotic cell death. Isoform-specific abrogation of lGPx4 and nGPX4 expression leads to embryos with no aberrant embryonic development (74, 343). However, lGPX4-/- mice are infertile due to the pivotal role of the GPx4 protein as a structural component for the architecture of the sperm mid piece (74, 343). On the other hand, siRNA-mediated knockdown of lGPx4 during in vitro embryogenesis strongly affected embryo development (26). Here, impaired segmentation of the lower rhombomeres was observed (26), which was related to an increased level of neuronal apoptosis (26, 399). A similar phenotype was observed when expression of Grsf1, a translational activator of lGPx4, was silenced (225, 399). These findings are consistent with a protective role of lGPx4 in mitochondria-mediated apoptosis (151).
Peroxiredoxins form a heterogeneous family of small antioxidant proteins that reduce peroxides at the expense of reduced thioredoxins or glutathione. Disruption of the various peroxiredoxin genes does not induce major developmental abnormalities. This may be explained by the fact that other isoforms may take over the function of the defective gene. However, the knockout animals are more susceptible to oxidative challenge (154, 212, 220, 275, 413). Thioredoxins (Trx) are small antioxidant proteins that function as oxidoreductases in their reduced state. When reacting with oxidants they are converted to their oxidized state and must be rereduced to catalyze the next cycle. This reduction depends on the presence of the thioredoxin reductases (TrxR). Two thioredoxin isoforms (cytosolic Trx1 and mitochondrial Trx2) are known and both isoforms appear to be of outstanding importance for embryo development. The Trx1 knockout induces embryonic death around implantation (E3.5) owing to reduced proliferation of the inner cell mass (251). In contrast, Trx2-/- embryos look normal until E8.5. At E10.5, homozygous Trx2 knockout embryos show a massive increase of apoptotic cell death with an open anterior neural tube and undergo intrauterine resorption by E12.5 (282). In mammals, three thioredoxin reductase isoforms (cytosolic TrxR1, mitochondrial TrxR2, and testis-specific TrxR3) are known, and functional inactivation of the corresponding genes induced obvious phenotypic alterations. TrxR1 knockouts lacking exon 15 of the TrxR1 gene that harbors the TrxR1 C-terminal active site exhibited severely retarded development in various embryo regions (164) with particular emphasis on the head and the parts caudal to the heart. These changes, which have been related to a failure of proper cell proliferation, start at E8.5 and at E10.5, the embryos do not turn properly. Interestingly, heart and blood vessel development are not affected in these knockout mice. Indeed, a heart-specific knockout of TrxR1 failed to induce major phenotypic alterations (164). However, a different knockout strategy compromising exons 1 and 2 of the TrxR1 gene leads to an even more pronounced phenotype (24). These TrxR1 knockout embryos show severe disturbances of differentiation and fail to gastrulate. Whereas Jakupoglu and coworkers only remove the TrxR1 active site (164), Bondareva's group removes the translational start sites thereby abrogating all potential TrxR1 protein translation (24). Therefore it is tempting to speculate that the different phenotypes reflect the functionalities of TrxR1 protein domains. However, both knockout strategies result in a sufficiently destabilized messenger RNA and neither TrxR1 transcripts nor TrxR1 protein is detectable. The reasons for the two different phenotypes require further investigations. Disruption of TrxR2 expression is lethal at E13. TrxR2-/- embryos appear anemic, which may be related to impaired hepatic hematopoiesis, and show signs of abnormal heart development. A heart-specific TrxR2 knockout induces dilated cardiomyopathy that leads to death shortly after birth (73). To our knowledge, a knockout of the only recently discovered TrxR3 is not available yet (372).
Glutaredoxins (Glrx) belong to the thioredoxin superfamily and there are two Glrx isoforms (cytosolic Glrx1 and mitochondrial Glrx2). A recently published Glrx1 knockout did neither sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia nor induce major developmental alterations (143). A transgenic mouse model overexpressing Glrx1 did similarly not affect the phenotype but suggested a cardioprotective role of the enzyme (246).
V. Redox-Dependent Epigenetic Control of Embryonic Gene Expression
Expression regulation of mammalian genes involves genetic and epigenetic regulatory elements (Fig. 10). In eukaryotic nuclei, the DNA is packaged in highly organized chromatin structures and this packing determines the accessibility of DNA for DNA-binding proteins to regulate transcription efficiency (298). Several highly conserved mechanisms for altering the chromatin structure have been described and these processes include reversible modifications of nuclear histones (phosphorylation, methylation, acetylation, and ubiquitination) (359, 370), reversible DNA methylation (241, 396), and ATP-dependent chromosome restructuring (206, 447). Under certain conditions, oxidative stress affects the efficiency of epigenetic regulatory processes by limiting the availability of S-adenosylmethionine, the essential cofactor required for DNA and histone methyltransferases. Moreover, the recent discovery of new classes of histone demethylases [LSD1 (lysine specific demethylase 1) and JHDM1 (JMJC domain-containing histone demethylase 1)], which require oxygen as a cofactor, directly links epigenetic expression regulation to oxygen gradients during development (193, 359).
A. Histone acetylation and deacetylation
Reversible acetylation of lysine residues at the N-terminal tails of nuclear histone proteins induces uncoiling of nuclear DNA, which increases the accessibility of transcription factors. Thus, the degree of histone acetylation is crucial for transcriptional regulation of gene expression. Histone acetylation is catalyzed by a group of specific acetyltransferases (HATs). The counterbalancing process, histone deacetylation (370) is mediated by deacetylases (HDACs) and the equilibrium of HATs and HDACs is decisive for reversible chromatin restructuring. In addition to HATs, nuclear coactivators such as CBP/p300 or ATF-2 exhibit intrinsic HAT activity (287) and thus, may contribute to establishing a certain histone acetylation pattern. Oxidative stress frequently activates transcription factors that recruit coactivators such as CBP/p300 or ATF-2. Employing their intrinsic HAT activity, the coactivators catalyze acetylation of specific core histones. This acetylation results in chromatin remodeling, making the promoters of target genes localized in the remodeled region more accessible for transcription factor binding (317). On the other hand, regulatory complexes of transcription are dismantled or disrupted by HDACs, which are sensitive to ROS-dependent post-translational modifications. Thus, acetylation of RelA/p65 and chromatin modifications leads to sustained pro-inflammatory gene transcription (318).
B. DNA and histone methylation
The second important mechanism involved in chromatin remodeling is reversible DNA methylation. DNA methyltransferases (DNMTs) catalyze the introduction of a methyl residue at the 5-position of cytosine leading to the formation of 5-methylcytosine (m5C). DNA methylation is associated with maintaining a stable and condensed chromatin organization that represses eukaryotic transcription. DNMT activity is particularly important for methylating different stretches of genomic DNA during gametogenesis, embryo development, and differentiation of somatic cells (396). Methylation as regulatory mechanism is not just restricted to DNA but also contributes to chromatin remodeling via modification of nuclear histones. Several histone methyltransferases (EHMT2, SUV39H) involved in histone 3 lysine 9 (H3K9) methylation are essential for embryo survival and expression regulation of pluripotency genes. The H3K9 demethylase JMJD2C is stage-specifically expressed in preimplantation mouse embryos and is important for embryonic expression of the transcription factors Myc and Klf4, which are required for cell proliferation. Klf4 in turn transactivates iNOS expression and interacts with the transcriptional co-activator CBP/p300 (410). Depletion of JMJD2C in early embryos caused a developmental arrest before the blastocyst stage and showed a significant downregulation of pluripotency-related genes, including transcription factors Myc and Klf4 (410). In many cases, oxidative challenge of mammalian cells alters the methylation pattern of genomic DNA (397, 421) and thus, affects gene expression. For instance, DNA methylation at the osteocalcin gene locus induces chromatin condensation, which represses eukaryotic transcription. In contrast, oxidative stress impairs DNA methylation and thus, activates transcription of the corresponding gene (405).
C. ATP-dependent chromosome remodeling
In addition to histone modification and DNA methylation, ATP-dependent chromatin remodeling alters the chromatin structure and alters gene expression (222). ATP-dependent chromosome remodeling is conferred by chromatin structure remodeling complexes (RSC). These heteromultimeric proteins exhibit ATPase and helicase activity to disrupt histone–DNA interaction. Brg, which represents a catalytical subunit of such chromatin remodeling complexes, interacts with the redox-sensitive transcription factor Nrf2 to induce expression of heme oxygenase in response to oxidative stress (447). Knockout mice deficient for various RSC ATPase subunits have been created and their embryonic growth characteristics indicated Brg1, Brm, Chd4, Chd2, p400, Etl1, Snf5, and Bptf are required for normal embryo development, hematopoiesis, or regular postnatal survival (206). Inactivation of Snf5 in mouse embryonic fibroblasts impairs cell growth and survival. Snf5-deficient cells are hypersensitive to oxidative stress, show signs of defective mitosis, and exhibit impaired response to apoptotic stimuli (192).
VI. Redox-Dependent Transcriptional Control of Embryonic Gene Expression
ROS/RNS alter the activity state of redox-sensitive transcription factors that regulate cellular gene expression. A variety of transcription factors is responsive to alterations of the redox homeostasis (153, 257, 347) and it would exceed the frame of this review to address all of them. In this review, we will focus on selected transcription factors that have previously been related to embryogenesis and their role during different developmental stages as indicated in Figure 11.
A. Nuclear factor κB
The nuclear factor κB (NF-κB) family of transcription factors consists of five members, p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), c-Rel, and RelB. All members are characterized by the presence of the Rel homology domain (RHD), which is essential for DNA binding and dimerization but also for the interaction of NF-κB with its endogenous protein inhibitor IκB (137). The most common species of NF-κB is the heterodimer p50/p65. Research about these subunits provided the first insight into the mechanisms of the NF-κB pathway and led to the discovery of a huge number of NF-κB target genes (10). Such NF-κB dependent genes include those encoding for pro-inflammatory cytokines (TNFα, IL1, IL2, IL6, and IL8), growth factors (G-CSF), adhesion molecules (VCAM-1, ICAM, E-and selectin), redox-regulated enzymes (COX2, iNOS, SOD, 12-LOX, and GSH-synthase), regulators of apoptosis (Bcl-XL and c-IAPs), acute phase response proteins, and other transcription factors (53). Under resting conditions, NF-κB is present in most cells as inactive cytoplasmic complex associated with its endogenous inhibitor IκB. This complex is retained in the cytosol and requires activation to become functionally relevant. A variety of stimuli are capable to activate NF-κB and these include increased ROS/RNS formation (12, 257) and hypoxia (196). ROS/RNS activate redox-sensitive protein kinases, including IκB-kinase (IKK) that activate NF-κB via phosphorylation of its inhibitor IκB. This phosphorylation prompts ubiquitination of IκB, its dissociation from the IκB–NF-κB-complex, and degradation of IκB by the proteosome (295). The free NF-κB dimerizes, translocates into the nucleus, binds to consensus sequences in the promoter region of NF-κB-sensitive genes, and activates their transcription.
In the nucleus, redox related modifications of NF-κB, such as oxidation of its cysteine residues, phosphorylation or acetylation of the p65 subunit affects its regulatory activity. Oxidation of Cys61 of the p50 subunit impairs DNA binding capabilities of NF-κB (259, 390). On the other hand, phosphorylation of the NH2-terminal amino acid of the p65 subunit by protein kinase A stimulates transcriptional activity by promoting interaction with the transcriptional coactivator CBP/p300 (451). In addition, nuclear p65 is subject to reversible acetylation, which induces nuclear export of the activated NF-κB complex and thereby terminates NF-κB-mediated signaling (56). ROS/RNS can either activate or inhibit NF-κB activity, depending on the level of ROS. Moderate increase of ROS often leads to activation, whereas excessive ROS/RNS results in oxidation of the redox-sensitive Cys62 of the p50 subunit, which impairs DNA binding activity (252, 259).
Embryonic expression of p65, p50 and p52 (204, 265, 281) indicated developmental stage- and cell type-specific expression of the major NF-κB family members. For instance, RelB expression was described at E14 and this subunit mainly occurs in lymphoid tissues, particularly in interdigitating dendritic cells (36). Transcripts of c-Rel (37) were also detected during late embryogenesis (E13), but expression of this subunit is restricted to erythroid precursor cells and to the lymphoid linage (B and T cells). These expression patterns suggest that c-Rel and RelB may not be essential for early embryo development. Using NF-κB-driven LacZ reporter gene constructs, it has been shown that NF-κB activity is first detected in brain and thymus between E12.5–E13 (341). More recent analysis of NF-κB expression during oogenesis and during the preimplantation period of fertilized oocytes indicated that transcription factors containing the Rel homology region (RHR) are already expressed at high level in 2-cell stage and in blastocyst embryos (174). To explore the functional role of NF-κB subunits during embryonic development, a number of knockout and transgenic mice were created (113). Disruption of the genes encoding for individual NF-κB subunits (p50, p52, c-Rel, and RelB) yield viable embryos but postnatal development of various organ systems is significantly impaired (Table 2). However, p65 knockout leads to intrauterine death at E15, owing to extensively increased liver apoptosis that strongly impairs fetal hematopoiesis (Table 2). Taken together, NF-κB subunits play a vital role in the development of various organ systems, but their impact appears to be mostly related to postnatal development.
B. Activator protein-1
Activator protein-1 (AP1) is a redox-sensitive, heterodimeric transcription factor, which is composed of proteins belonging to the c-Fos, c-Jun, ATF, and JDP families. It regulates gene expression in response to a variety of stimuli including cytokines, growth factors, stress, and pathogens, and controls a number of cellular processes including differentiation, proliferation, and apoptosis (354, 355). AP1 upregulates transcription of genes containing the consensus cis-regulatory sequence 5′-TGAG/CTCA-3′ in their promoters and interacts with the acidic phosphodiester backbone of double-stranded DNA via a basic amino acid region. Its dimeric structure is stabilized by a leucine zipper, a super-secondary structural motif consisting of two crossing helices (Fig. 12), which are kept together by interacting leucine side chains. A major AP1 family member is c-Jun, which plays an important role in ROS-induced apoptosis (435, 436). Although the molecular mechanisms for this involvement are not completely understood, experimental data suggested the involvement of the cellular redox-equilibrium in c-Jun signaling. Intracellular concentrations of active AP1 are controlled by redox-dependent mechanisms on transcriptional as well as post-translational levels. The three major classes of MAP-kinases (ERK, p38, and JNK) are involved in the regulation of intracellular AP1 activity and oxidative stress activates c-Jun and ATF2 via phosphorylation by these MAP-kinases, in particular JNK and p38 (177). In addition, expression of c-Jun can be impaired by histone deacetylation (HDAC) or by proteasomal degradation initiated by MEKK1-dependent ubiquitination (435, 436).
Jun proteins are expressed at variable levels during fetal growth and organogenesis of mice and show a tissue-specific expression pattern. C-Jun transcripts were detected in cell populations of developing cartilage, gut, and central nervous system, but JunB is restricted to differentiating epidermal cells and endodermal gut epithelium between E14.5 and E17.5 (428). Expression of JunD family members was shown to start around E7.5. Beginning at E9.5, JunD was detected in the heart, in vessels of the developing cardiovascular system, and later on in the developing central nervous system and musculature (386). cFos expression was observed in the developing nervous system starting at E12 (43). Fra2 expression was shown in the epithelium, in developing cartilage, and in the central nervous system at later stages of organogenesis (35).
Loss of function studies, particularly the creation of knockout mice shed light on the in vivo functionality of the different members of the AP-1 system (Table 2). Disruption of c-Jun and JunB genes resulted in embryonic lethality. Embryos lacking c-jun die at midgestation (E12.5) because of impaired hepatogenesis resulting in fetal liver erythropoiesis as well as generalized edema (140, 170). Lack of JunB (344) causes early embryonic lethality (E8.5–E10) with multiple defects in extra-embryonic tissues (yolk sac, placenta). Inactivation of the Fra1 gene also results in embryonic lethality. Defects in extra-embryonic tissues induce embryonic growth retardations, leading to intrauterine death at E10.0 (345). Fra2 knockouts are viable but ablation of the corresponding gene revealed a function of Fra2 in skeletogenesis and chondrocyte differentiation (128). Functional inactivation of the JunD gene also produced viable mice. However, homozygous individuals exhibit postnatal growth retardations and males exhibited reduced fertility (386). cFos-/- mice are viable (169, 415) but growth-retarded, develop osteopetrosis, and suffer from impaired hematopoiesis (Table 2). FosB knockout mice are profoundly deficient in their ability to nurse their newborns. This behavioral defect is likely due to the absence of FosB in the preoptic area, a region of the hypothalamus that is critical for nurturing (29). In summary, these knockout data indicate distinct phenotypes for inactivation of single genes of the AP1 family. This reflects the universal role of AP1 transcription factors as well as the specific involvement of certain AP1 family members in different developmental processes. It also becomes obvious that AP1 transcription factors are not interchangeable and the knockout of one family member cannot simply be compensated for by another one.
C. Nrf-ARE system
Oxidative stress is frequently induced by pro-inflammatory stimuli via activation of redox-sensitive transcriptional factors such as NF-κB and AP1 and protein kinase signaling (58). To maintain a physiological equilibrium, these pro-oxidative stimuli are met by antioxidative mechanisms. One functional counterpart of these pro-oxidative mechanisms is the Nrf2-ARE system, which contributes to the upregulation of antioxidative defense mechanisms. Thus, modulation of Nrf2 signaling has profound impact on NF-κB and AP1 (190). Coordinated expression of antioxidant enzymes during oxidative stress is mediated by antioxidant response elements (AREs) in the promoter for ROS-sensitive genes (187). The nuclear factor erythroid-derived 2-related factors 1 and 2 (Nrf1, Nrf2) are transcription factors, which bind with high affinity to AREs and thus, are of major importance for expression regulation of gene products containing this cis-regulatory element in their promoters. Nrf1 and Nrf2 are members of the basic leucine zipper family (Fig. 12) of transcription factors (bZIP) and require heterodimerization with small Maf proteins for DNA binding (264). It has been proposed that under resting conditions Nrf2 is localized in the cytoplasm (153), where it is bound to the actin-binding protein Keap1 (Kelch-like ECH-associating protein 1). Following oxidative challenge, Nrf2 is activated by thiol oxidation of multiple reactive cysteine residues of Keap1 (86) and/or by phosphorylation of Nrf2 by protein kinases, such as PCK or PERK (79, 145). These post-translational modifications induce the dissociation of the Nrf2/Keap1 complex, which allows free Nrf2 to translocate into the nucleus. Nrf2 associates in the nucleus with Maf proteins and is now capable of binding to ARE (181, 182).
However, the mechanism by which Keap1 acts to repress Nrf2 activity is a topic of ongoing discussion (278). It is well established that Nrf2 activity is controlled by Keap1, which in turn promotes Nrf2 ubiquitinylation and subsequent proteolytic degradation (254, 279, 406). In contrast to the proposed cytosolic co-localization of Nrf2 with Keap1, it has been suggested that Nrf2 is predominantly localized in the nucleus in the absence of stress signals (279). Therefore, the activation of Nfr2 target genes is limited by the interaction with Keap1 following proteolytic degradation. Under stress conditions, stabilization of Nrf2 is a result of prevented or reduced access of Keap1 to Nrf2 induced by post-translational modifications of Keap1 such as thiol oxidation or phosphorylation (278). Genes containing AREs in their promoter regions include NAD(P)H:quinone oxidoreductase, glutathione S-transferase, heme oxgenase-1, glutathione peroxidase 3, glutamate cysteine ligase, and peroxiredoxin 1 (84, 163). Although Nrf1 and Nrf2 are structurally related, Nrf1 activity is differently regulated and is not controlled by Keap1. In the resting state, Nrf1 is localized at endoplasmic membranes and is liberated by endoplasmic stress (412). This different intracellular localization suggests distinct functionalities for the two family members (289). Nevertheless, both Nrf isoforms play overlapping roles in the regulation of basal expression of ARE-containing genes.
During the time course of murine embryogenesis, Nrf1 is expressed starting at E7–7.5 in both embryonic and extra-embryonic tissues. Around E9, increased levels of Nrf1 were observed in heart, midbrain, head mesenchyme, and migrating neural crest cells (266). Nrf2 mRNA expression was shown between E7–E17 in various organs such as the liver, lung, kidney, digestive tract, and central nervous system (49). Functional disruption of the Nrf1 gene in mice results in embryonic lethality. Nrf1-/- mice (48) suffer from embryonic anemia due to abnormal fetal liver erythropoiesis and die in utero at late gestation (E12.5–E18). Fetal livers explanted from Nrf1 knockout embryos showed signs of increased apoptosis. Moreover, markers of oxidative stress were significantly enhanced and expression of antioxidant enzymes was impaired (57). Fibroblasts derived from Nrf1-deficient embryos showed enhanced sensitivity to oxidative challenge (203). Embryos of Nrf2 null mice are viable but develop multi-organ autoimmune inflammation, show enhanced lymphocyte proliferation, and exhibit a reduced tolerance for oxidative challenge (244). These data suggest that Nrf2 may be involved in regulation of peripheral lymphocyte homeostasis and autoimmune surveillance (244). Nrf1-/-/Nrf2-/- double knockout embryos die at midgestation (E9–E10). This is significantly earlier than single Nrf1-knockout embryos, suggesting that in Nrf1 knockouts Nrf2 may partially compensate the defect (216).
D. Transcriptional activities of hypoxia inducible factors
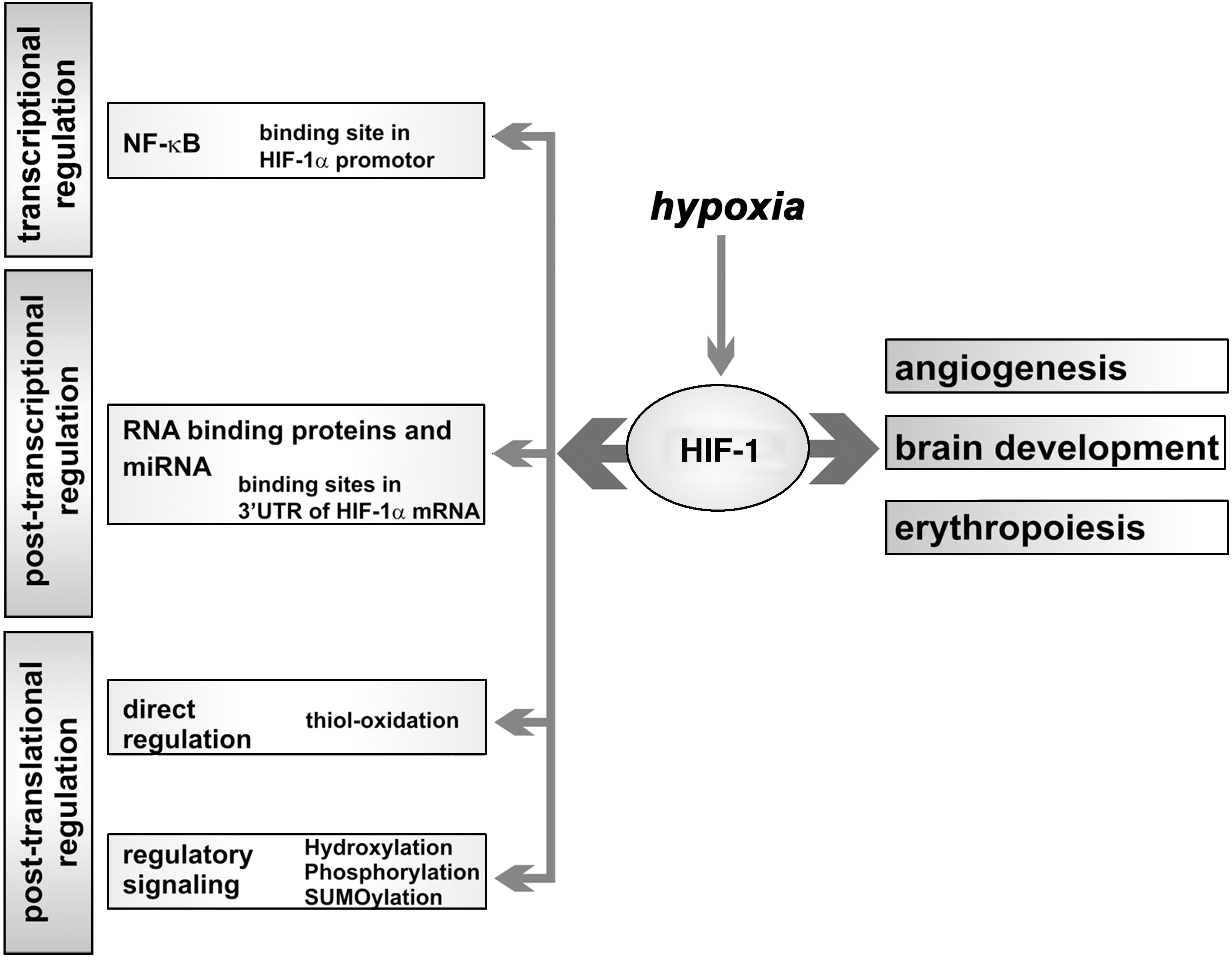
Hypoxia inducible factors (HIF) is a heterodimeric DNA-binding complex composed of two helix-loop-helix proteins, which regulate gene expression in an oxygen-dependent manner (408). The HIF complex consists of an α- (three different genes: 1α, 2α, 3α) and a ß-subunit (three genes: ARNT1ß, ARNT2, ARNT3) and heterodimerization is essential for transcriptional regulation. Whereas the ß-subunits are constitutively expressed, the cellular concentration of the α-subunits is regulated on transcriptional and post-translational levels (347). Transcription of the HIF α subunit genes is controlled via ROS-sensitive NF-κB signaling (25). On post-translational levels, stability of the HIFα-protein is controlled in an oxygen-dependent manner via degradation by the ubiquitin–proteasome system. The immediate oxygen sensor for this degradation is a HIFα-specific prolyl hydroxylase (PHD). PHD hydroxylates two specific prolyl residues of the HIFα-subunit and requires molecular dioxygen, 2-oxoglutarate, and ascorbate as cofactor (155). Hydroxylated HIFα is then recognized by the ubiquitin E3 ligase, which ubiquitinates the HIFα protein, targeting it for proteasomal degradation (159). PHDs are nonheme iron containing proteins; three different isoforms are known. The oxygen affinity of PHD is rather low, so that the rate of HIFα hydroxylation depends on the oxygen concentration in the environment. Under hypoxic conditions, the HIFα protein is rarely hydroxylated and thus, not targeted for proteasomal degradation. It can translocate into the nucleus and heterodimerize with a HIFβ subunit to form the functional HIF complex (175). HIF-mediated transactivation is then achieved by recruitment of coactivators, such as p300/CBP (90). The intensity of HIF–p300 interaction is regulated by hydroxylation of HIF asparagine residues catalyzed by the HIF-specific asparaginyl hydroxylase FIH (factor inhibiting HIF). This asparagine hydroxylation proceeds in the nucleus and blocks the HIF/p300 interaction under normoxic conditions (205). Oxygen-dependent HIFα degradation and oxygen-dependent inhibition of HIF–p300 interaction are not the only regulatory elements in HIF signaling. In addition, oxygen independent regulatory mechanisms have been described (116). MAP kinases have been implicated in upregulation of HIF-1α by a number of nonhypoxic stimuli, which impact the cellular redox homeostasis (189, 322). Oxidative challenge may convert the active ferrous HIF prolylhydroxylase to its inactive ferric form. Consequently, HIF-1α may escape hydroxylation and subsequent proteolytic breakdown (112). Moreover, an imbalanced redox homeostasis also alters the catalytic activity of protein tyrosine phosphatases (PTPs) and protein tyrosine kinases (PTKs). Transient oxidation of thiol groups in PTPs leads to the inactivation of the enzymes via the formation of either an intramolecular S-S bridge or sulfenyl-amide bonds. Conversely, oxidation of protein tyrosine kinases PTKs leads to activation, either by direct oxidation of cysteine residues or, indirectly, by concomitant inhibition of protein tyrosine phosphatases (62). Furthermore, direct phosphorylation of HIF-1 disrupts the HIF-1/DNA complex (409). The list of HIF target genes (253, 395) includes angiogenic factors (VEGF, VEGFR-1, eNOS), genes of iron metabolism (transferrin, transferrin receptor), genes involved in brain development (EPO, Glut-1, iNOS) or genes involved in energy metabolism (Glut-3, hexokinase 1, aldolase A, and pyruvate kinase M).
During mouse embryogenesis, expression of HIF family members was observed at E4–E6 and later on between E8.5 and E9.5. Expression of HIF-2α is not restricted to the embryo but was also found in the placenta (162, 270). At various stages of intrauterine development mouse embryos experience low oxygen concentrations. At these stages, expression of HIF-1α and ARNT is upregulated specifically in the affected hypoxic regions, in particular in the developing neural tube, the heart, and the intersomitic mesenchyme at early stages of organogenesis (214, 270).
HIF has been implicated in embryo development and it exhibits its activity on various levels of the embryonic gene expression cascade (Fig. 13). Final proof for its regulatory activity was provided by in vivo gene inactivation. HIF-1α, ARNT, and HIF-2α-deficient mouse embryos die in utero between E8 and E11. HIF-1α deficiency resulted in developmental arrest and lethality at E10.5. The embryos suffer from neural tube defects, cardiovascular malformations, and pronounced cell death within the cephalic mesenchyme (156, 332). In addition, since HIF-1α plays a major role in iron homeostasis, deficient embryonic erythropoiesis was observed (442). Neuron-specific HIF-1α knockouts are viable but develop a hydrocephalus, which is accompanied by a reduction of neurons and an impairment of spatial memory. Excessive neuronal apoptosis is paralleled by vascular regression in the telencephalon but the embryos were rescued by transgenic overexpression of HIF-1α (391). ARNT-/- mice die at E10.5 due to defective angiogenesis of the yolk sac, and of the placental and branchial arches (197, 248). HIF-2α is highly expressed in the vasculature and in the organ of Zuckerkandl, which is the major source of embryonic catecholamines. Mice lacking HIF-2α die at mid-gestation as a result of defective catecholamine synthesis (389) and subsequent impaired angiogenesis, particularly in the yolk sac (306).
E. Redox effector factor-1/apurinic/apyrimidinic endonuclease-1
Redox effector factor-1 (Ref-1)/apurinic/apyrimidinic endonuclease-1 (APE-1) is multifunctional redox-sensitive protein that exhibits DNA repair activity as well as the ability to reduce oxidized cysteine residues in other proteins. Similar to other redox sensing proteins, Ref-1 contains essential cysteines (268), which are normally kept in their reduced state. Oxidation of these residues alters the DNA binding capacity and thus, the activity of transcriptional control. Oxidized APE-1 is unable to bind DNA. Ref-1 reduces cysteines in the DNA binding domain of other transcription factors, such as c-Jun and cFos and thus, restores the DNA binding capacity of AP1 (433). More general, Ref-1 functions as a regulatory protein for redox activation of various transcription factors involved in cell growth, differentiation, hematopoiesis, and stress response (384). The intracellular activity of Ref-1 can be regulated on various levels including transcription, subcellular localization, and post-translational modifications. Exposure of cells to oxidative stress rapidly promotes a transient increase in Ref-1 mRNA and protein mediated by the activity of several transcription factors, including Sp1, Egr1, CREB, and Jun/ATF4 (382). Oxidative stress induces an enhanced nuclear localization of Ref-1, which mirrors increased activity (383). Post-translational modification of Ref-1 activity is induced by phosphorylation via protein kinase C (145). The reduced activity of oxidized Ref-1 can be restored by thioredoxin, indicating the involvement of essential cysteines (141).
Ref-1 functions as normal transcription factor but also exhibits endonuclease activity. It is capable of initiating repair of apurinic/apyrymidinic sites in oxidatively damaged DNA. The relationship between the cellular redox state and DNA repair activities of Ref-1 is intriguing. The essential role of Ref-1 in mammalian development was demonstrated by genetic inactivation of the Ref-1 gene in mice (239, 434). Embryos lacking functional Ref-1 fail to develop beyond E6. Ref-1+/- animals exhibit significantly elevated markers of oxidative stress (lipid peroxide level, plasma F2 isoprostanes), but supplementation with antioxidants such as vitamins E and C restores these levels back to normal. These results are consistent with a proposed role of Ref-1 in the protection against deleterious effects of oxidative stress (256).
VII. Redox-Dependent Post-Transcriptional Control Mechanisms of Gene Expression
Once the first nucleotides of an mRNA emerge from the active RNA-polymerase, they are bound by RNA-binding proteins (RBPs) to form ribonucleoproteins (RNPs). These RNPs accompany an mRNA in ever-changing compositions all throughout its complex life cycle. This is also the case for the more recently discovered small RNA species called microRNA (miRNA). The stages of an mRNA life cycle include capping of its 5′-end, polyadenylation of its 3′-end, splicing, editing, nuclear export, translation, and finally degradation. All these processes are strictly controlled in a temporal and spatial manner, which is accomplished by a large variety of cis- and trans-regulatory elements and factors. Dysregulations may have fatal consequences, in particular when they occur at critical steps of embryogenesis. Disturbance of the finely tuned cellular redox equilibrium affects a multitude of post-transcriptional regulatory mechanisms. Similar to other biomolecules, RNA itself is susceptible to oxidative modification. In fact, in single-stranded RNA regions, the nucleotide bases are less protected from oxidation because of lacking hydrogen bond between anti-parallel double strands. Oxidative damage to RNA has often been ignored since lipids, proteins, and DNA have mainly been considered as primary targets of oxidative modification (273, 353). As in DNA, ROS can cause guanine hydroxylation in RNA, which leads to the formation of 8-oxo-7,8-dihyguanosine (179). A high degree of RNA oxidation for instance has been observed in neurodegenerative disorders such as Alzheimer's or Parkinsons's disease (283, 448).
Most of our current knowledge on post-transcriptional mechanisms during development has been gained from experiments in Xenopus and Drosophila and these data are nicely reviewed elsewhere (200, 350, 401, 427). Here we will focus on mouse embryogenesis.
A. Cytoplasmic polyadenylation and translational masking
Post-transcriptional mechanisms of gene expression regulation during the time course of embryogenesis can already be observed in the oocyte. Here large quantities of mRNA are either degraded or stored as translationally inactive RNPs until translated at later developmental stages (305, 323, 371, 373). The 3′-UTRs of several mRNA species, including cyclin B1 and the serine/threonine kinase c-mos, carry a cytoplasmic polyadenylation element (CPE) with the consensus sequence UUUUUAU (110, 379). CPE is recognized by the CPE-binding protein (CPEB), which contains a RNA-recognition domain with a zinc-finger motif and thus, is sensitive for redox alterations (125, 126, 185, 299). mRNAs bound by CPEB are deadenylated and translationally silenced (324). Protein kinase aurora-mediated phosphorylation of CPEB, which attracts the poly(A)-binding protein (PABP) to the translationally silenced mRNA, overcomes translational suppression (335). Thus, CPEB is required for translational silencing as well as activation. Translational activation of the synaptonemal complex proteins mediated by CPEB for instance is essential for murine oocyte maturation. Hence CEPB-/- mice exhibit an arrest of oogenesis at E16.5 and fail to generate diplotene oocytes, which require proper synaptonemal complex formation, and this greatly impairs fertility of CEPB-/- offspring (381).
B. Iron regulatory elements
The most extensively studied mechanism of translational regulation is the impact of iron-regulatory proteins (IRP1 and IRP2) on the expression of proteins involved in iron homeostasis. If iron is scarce, IRP1 binds to the iron-response element (IRE) in the 5′-UTR of the ferritin mRNA in a redox-dependent manner (139). The IRE-binding activity of IRP1 is diminished when specific thiol-groups are oxidized (139). In the developing embryo, IRP1 expression is at least in part regulated by HIF-1α and HIF-1α -/- mice exhibit increased IRP1 expression and abnormal erythropoiesis (442). Once the IRP1/IRE-complex is formed, it inhibits translation of ferritin mRNA. In addition, IRP1 binds to several IREs located in the 3′-UTR of the transferrin receptor (TfR) and thereby protects TfR mRNA from degradation. In abundance of iron IRP1, which contains a (4Fe–4S)2+-cluster at its catalytic center, loses its RNA affinity and functions as aconitase. Disassembly of the iron–sulfur cluster and thus induction of the RNA-binding activity is sensitive to oxidative signaling (296). Interestingly, IRP1-/- mice develop normally whereas IRP2-deficient animals suffer postnatally from progressive neurodegeneration. The first symptoms become evident at 6 months of age (209, 258). These data indicate that neither IRP1 nor IRP2 is essential for embryogenesis. For postnatal development IRP2 appears to be more important. Reduction of IRP1 expression (IRP1+/-) on a IRP2-/- background increases the extent of the neurodegeneration observed in the single knockouts (364). Complete absence of IRP expression obtained by a IRP1/IRP2 double knockout approach leads to early embryonic lethality before E6.5 (365). Blastocysts bearing IRP1/IRP2 double knockouts showed a brownish discoloration due to ferric iron sequestration and fail to develop past the implantation stage (365). These data indicate that IRP-dependent signaling is important for early embryo development and that IRP2 may compensate at least in part deficiency of IRP1.
C. Small regulatory RNA species (microRNAs)
A whole new world of expression regulation has been unveiled by the discovery of the small regulatory RNA species designated microRNAs (miRNAs). By today, 721 human and 579 murine miRNA genes have been discovered (
Because of the severe effect of targeted constitutive Dicer knockout, conditional knockout strategies have been employed to study the impact of miRNAs during development in more detail. Knockout strategies targeted to the limb mesoderm led to an absence of miRNAs and an increase of apoptotic cell death in the corresponding embryo regions (132). This induced severe developmental retardation of the limbs, highlighting the role of miRNAs in limb development. A muscle-specific knockout resulted in the reduction of muscle miRNAs and also caused strong induction of apoptosis in developing muscles (285). These embryos die perinatally. The number of studies employing conditional knockouts of the miRNA machinery or knockouts of specific miRNAs is constantly growing (369) and the vital role of miRNAs has been shown for the development of many other cells and tissues including stem cells (102, 226), the central nervous system (64, 69, 88, 183, 191, 247), the urogenital tract (115, 231, 290, 302), skin (441), liver (128), lung (237), pancreas (243), and the cardiovascular system (263, 411, 438).
A very recent study suggested the existence of an oxidative stress-response component in the expression cascade of Dicer itself (426). In fact, in several cell lines, the induction of oxidative stress caused significant inhibition of Dicer activity (426). On the other hand, apoptotic cell death induced by ROS is suppressed by the microRNA-21 (miR-21) in vascular smooth muscle cells (227). The mechanism involved expression silencing of the miR-21 target PDCD4 (programmed cell death 4) and activation of the transcription factor AP1, which in turn is a downstream component of PDCD4 (227). A similar protective effect was observed for miR-21 in cardiomyocytes (61).
In differentiating motor neurons in the developing brain, miRNAs modulate accompanying signaling events (100). Differentiation depends on hedgehog signaling, which results in the release and activation of the transmembrane protein smoothened (Smo). Smo then leads to activation of the zinc finger transcription factor family Gli (glioma-associated oncogene) and to activation of 5-lipoxygenase, which is involved in the formation of cysteinyl leukotrienes required for neurite projections (22). Almost each step of this signaling cascade is regulated by miRNAs (100).
RNPs have dual activity for miRNA-dependent expression regulation. They can stimulate the regulatory effects by stabilizing the miRNAs but they can also suppress the regulatory activity. For instance, the AU-rich element RNA-binding protein HuR/ELAV1 rescues CAT-1 (cationic amino acid transporter 1) mRNA from miRNA-122 mediated translational silencing (21). Upon stimulation, HuR leaves the nucleus, binds CAT-1 mRNA, and relocates its target from P-bodies to polysomes so that the CAT-1 mRNA becomes translationally active. However, the molecular details why HuR translocates into the cytosol and how it competes with miRNA-122 for binding to CAT-1 mRNA are not well understood. Another example of an RNP counteracting miRNA activity is represented by the dead end protein Dnd1. Truncation of Dnd1 in mice leads to an almost complete loss of primordial germ cells. This effect appears to be related to Dnd1 binding to U-rich RNA, which counteracts miRNA-mediated expression silencing (184, 444).
Taken together, these data indicate that the miRNA machinery is involved in ROS-induced intracellular signaling. However, further studies are needed to characterize its impact in more detail.
D. Alternative splicing
Alternative splicing affects the majority of genes in humans and is one of the mechanisms involved to generate phenotypic multiplicity from a limited number of genes (168, 198, 367). Therefore, it is not surprising that also genes involved in redox regulation are affected by alternative splicing. Expression of two redox-relevant selenoproteins, GPx4 and TrxR1, which have been implicated in embryogenesis, involve alternative splice mechanisms. Interestingly, both genes are driven by rather simple Sp1/Sp3-dependent, TATA-less promoters (331, 398), which limits the potential options of transcriptional regulation. The GPx4 gene generates three different isoforms with variable N-terminal sequences. These N-terminal sequences contain short targeting signals that locate the three isoenzymes to different subcellular compartments: cytosol, mitochondria, and nucleus. All three isoforms are differentially expressed during embryo development (26). For the human, TrxR (TrxR1 and TrxR2) multiple isoforms have also been described, which are generated by alternative splicing. However, the precise molecular mechanisms remain unclear and our knowledge on the specific functions of the isoforms is limited (7).
On the other hand, alternative splicing mechanisms can be regulated by oxidative stress (87). For instance, redox alterations induce elevated p53-dependent alternative mis-splicing of the pre-mRNA encoding for DNA-polymerase ß (POLB) mRNA independent of random ROS-induced DNA damage (87). For this enzyme, more than 30 splice variants have been described (363).
E. Grsf1 dependent translational control
We have recently explored translational regulation of GPx4 expression during mouse embryo development (399). GPx4 is part of the cellular antioxidant defense system since it is capable of reducing complex lipid hydroperoxides (201). GPx4 deficiency leads to abnormal development and early embryonic death (150, 439). Although transcriptional control mechanisms for GPx4 expression have been explored (135, 146, 339, 398), little is known on translational control. Identification of the RNA-binding protein guanine-rich sequence binding factor 1 (Grsf1) narrowed this gap. Grsf1 recognizes a G-rich binding motif (AGGGGA) within the 5′-UTR of the lGPx4 mRNA (Fig. 14). After Grsf1 binding, the lGPx4 messenger is recruited into translationally active polysome fractions and GPx4 protein is synthesized. Thus, Grsf1-dependent translational control of lGPx4 expression alters the cellular redox state as well as apoptotic signaling (399). Silencing Grsf1 expression (siRNA knockdown strategy) at midgestation specifically suppresses lGPx4 expression, resulting in increased membrane oxidation and apoptotic cell death leading to developmental retardation. Grsf1 knockdown embryos have an lGPx4 knockdown phenotype and can be rescued by lGPx4 overexpression. Grsf1 is not the only protein known to interact with the GPx4 messenger. The parkinsonism related protein DJ-1 associates with the 5′UTR of the GPx4 mRNA in brain (400). In contrast to Grsf1, DJ-1 binding induces translational silencing of the GPx4 messenger. Under oxidative conditions cysteine residues (Cys106 in the human DJ-1 protein) are oxidized, which causes DJ-1 to be released from the RNA/protein complex (23, 400). Translational suppression is subsequently lost, which leads to increased translation of the antioxidant protein GPx4.
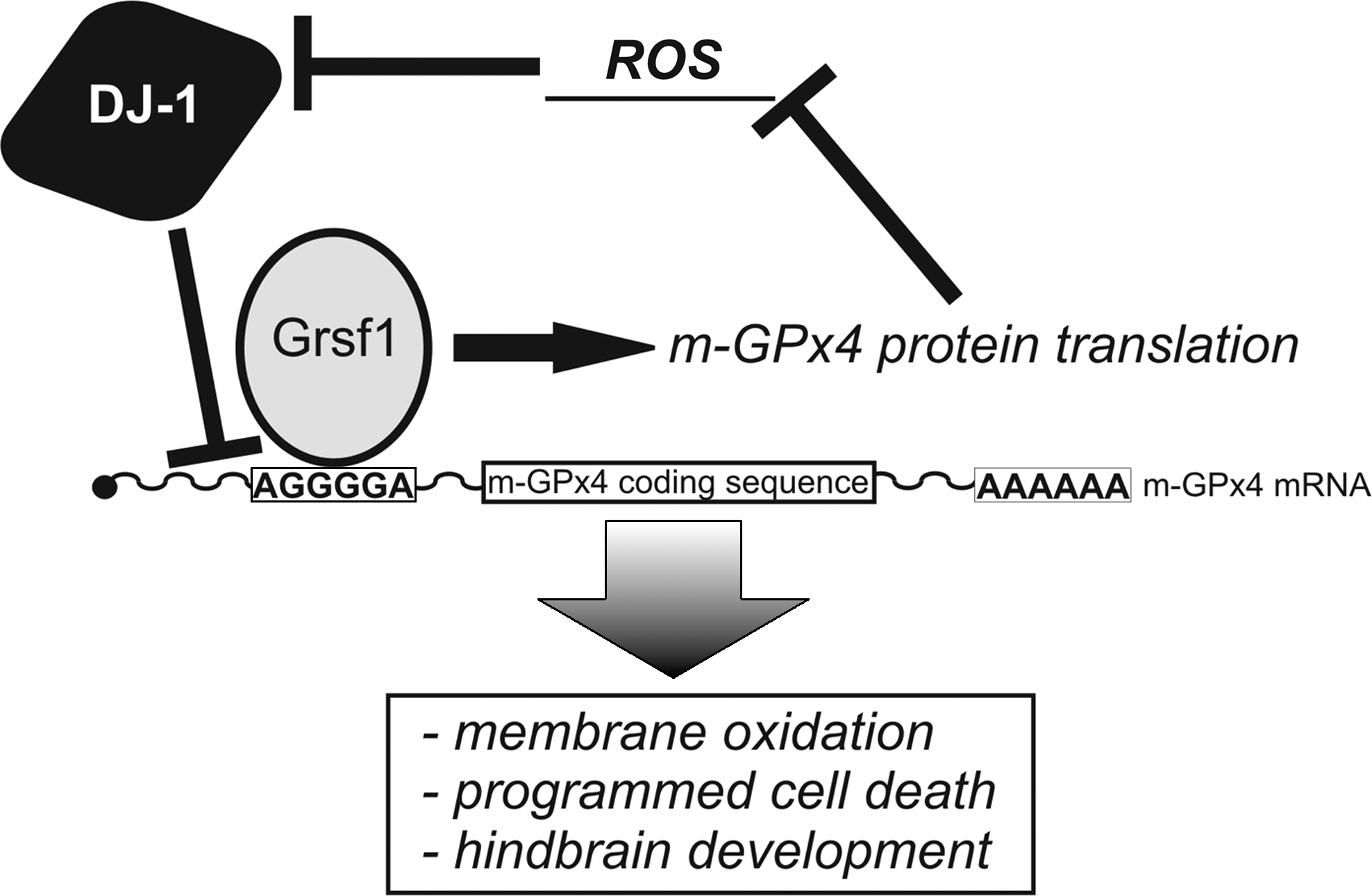
GPx4 contains the noncanonic amino acid selenocysteine (Sec) at the active site, which has been implicated in the catalytic mechanism (245, 340). Sec is coded for by the opal codon UAG, which needs to be re-coded by the complex Sec incorporation machinery. Sec incorporation requires a growing number of protein and RNA factors, including an Sec-specific elongation factor eEFsec and a Sec-specific tRNA, tRNA[Ser]Sec (30). In eukaryotes, the presence of a hairpin structure located in the 3′UTR of selenoproteins designated Selenocysteine Insertion Sequence (SECIS) is of vital importance for cotranslational Sec-incorporation. The SECIS element is bound by SECIS-binding protein 2 (SBP2), which guides co-translational incorporation of Sec into the nascent polypeptide chain. This mechanism comprises an essential component during embryo development, as revealed by knockout studies (33). For instance, knockout mice bearing a targeted deletion of the gene coding for tRNA[Ser]Sec are lethal and the embryos are resorbed shortly after implantation (28). SBP2-mediated selenoprotein synthesis is regulated by the cellular redox state. Oxidative modification of redox-sensitive cysteine residues within SBP2 protein causes it to shuttle into the nucleus, which impairs synthesis of selenoproteins (297). This mechanism very likely serves as a quick redox-sensitive switch of selenoprotein translation in general.
F. Nonsense-mediated decay
Selenoprotein expression is also dependent on the selenoprotein hierarchy, a mechanism that determines which selenoproteins are more stably expressed than others under the conditions of selenium deficiency. Although the underlying molecular principles are not completely understood, they may involve the nonsense-mediated decay (NMD), which appears to compete with co-translational Sec-incorporation (235). For instance, GPx4 is stably expressed, even under severe selenium deprivation and this may be caused by GPx4-specific masking of NMD (376, 420). The NMD pathway is a complex multistep mechanism that enables recognition of premature stop-codons upstream of exon/exon junction complexes and subsequent degradation of mutated mRNA species (274). One essential component of NMD is the helicase Rent1/Upf1, which subjects bound mRNAs to degradation in cytoplasmic processing bodies (P-bodies). These P-bodies contain the enzymes necessary for mRNA degradation (186). Rent1 is essential for early development and Rent1-/- mice die before E7.5 highlighting the importance of NMD in the developing embryo (255). NMD itself is sensitive to oxidative stress. In the event of stress such as hypoxia, stress granules are formed (4) and Rent1 targeted mRNAs are moved into these stress granules inhibiting NMD (109). The formation of these stress granules requires phosphorylation of the α-subunit of the general eukaryotic initiation factor 2 alpha (eIF2α) (109, 186). This phosphorylation appears to be a key mechanism for a cell to cope with oxidative stress (422). Interestingly, this mechanism can also lead to the activation of transcription factors such as ATF4 (activating transcription factor 4) and NF-κB (422). The ATF4 gene contains two upstream open reading frames (uORFs) that control ATF4 expression (403). Under resting conditions, both uORFs (uORF1, uORF2) are translated. Since uORF2 overlaps with the ATF4 coding region, its expression is repressed by ATF4 translation. However, when eIF2α is phosphorylated and general translation is restricted, ribosome scanning will be delayed. Consequently, uORF1 and 2 are skipped and only ATF4 is expressed (403). On the other hand, NF-κB is activated by eIF2α phosphorylation because of impaired translation of IκB (165).
G. Post-transcriptional regulation of HIF-1α expression
As outlined above, HIF-1α is a key regulator of the cellular response to stress signals and its cellular steady state concentration is mainly regulated by proteolysis (Section VI). However, HIF-1α expression is also regulated by post-transcriptional mechanisms. Several RBPs (RNA-binding proteins) have been shown to bind HIF1α mRNA (107, 124, 356). The HIF-1α 3′UTR contains AU-rich sequence elements that binds HuR (356). In addition, HuR also binds to the HIF-1α 5′UTR. Simultaneous binding of PBP (polypyrimidine tract-binding protein) at the 3′UTR and of HuR at the 5′UTR, stimulates HIFα protein synthesis in response to oxidative stress stimuli (107). Interestingly, the 3′UTR of HIF-1α mRNA is also target of CPEB, which renders HIF-1α a potential signaling component for germ cell development (124). In addition, several miRNA species against HIF-1α 3′UTR have been identified as potential regulatory elements and they are highly expressed in developing embryos. Unfortunately, their impact on HIF expression remains to be characterized (329). On the other hand, HIF-1 activation can regulate expression of miRNAs. The expression of the transcription factor Twist-1 is induced by HIF-1α, and Twist-1 promotes expression of two miRNAs (miR-214 and miR-199a) during development of the neuroepithelium and of the limb buds (213). Interestingly, the same miRNA, namely miR-199a, regulates COX-2 expression around implantation (47).
H. Post-transcriptional regulation of thioredoxin expression
The oxidoreductases thioredoxin (Trx) increase the activity of a number of transcription factors including HIF1 (7, 452) and expression of Trx itself is augmented following oxidative stress (105). Recently, the cold inducible RNA binding protein (CIRP/hnRNPA18) was identified to bind the 3′UTR of the Trx mRNA protecting it from degradation and thus, enhancing Trx synthesis (437). CIRP expression is upregulated by oxidative stress in a HIF1α independent way (423). CIRP RNA binding activity in turn is increased through phosphorylation by GSK3beta (glycogen synthase kinase 3 beta), which is an important member of both hedgehog and Wnt signaling. This relation makes Trx a downstream target of these pathways (103, 328). In embryonic stem cells, CIRP activates the ERK1/2 pathway, which plays a central role in proliferation control (8).
I. Post-transcriptional regulation of cyclooxygenase expression
Cyclooxygenase (COX)-2 is target of various post-transcriptional regulatory mechanisms. Its 3′UTR contains AU-rich elements that regulate its half-life (Fig. 15). AU-rich elements can be bound by various RNA-binding proteins such as TIA-1 (T-cell intracellular antigen-1), TIAR (TIA-1 related protein), AUF1 (AU-binding factor 1), CBF-A, RBM3, heterogeneous nuclear ribonucleoprotein (hnRNP) A1, A3, A2/B1, and HuR (71, 72, 127, 308). HuR regulates COX-2 expression post-transcriptionally by stabilizing COX-2 mRNA following cellular stress (348). Stress stimuli induce the cytoplasmic localization of the otherwise nuclear HuR where it then binds its target mRNAs. On the other hand, following oxidative challenge HuR binding at mRNA species can be impaired as indicated for certain cyclin mRNAs (1). HuR is essential for normal embryogenesis and is expressed in the placenta and in the embryo. It follows a unique spatial and temporal expression profile peaking between E10.5 and E12.5 in the embryo and slightly later between E12.5 and E13.5 in the placenta (117). Genetic ablation of HuR expression causes abnormal development of the placenta leading to embryonic lethality by midgestation (180). HuR-/- embryos are rescued from placental defects employing a conditional knockout strategy that was targeted at the epiblast only (embryo and extra-embryonic mesoderm) leaving all placenta cells unaffected (180). These conditional HuR-/- mice develop defects in skeletal ossification as well as limb and spleen development. Another protein binding COX-2 mRNA is TIAR, which induces sequestration of COX-2 mRNA into stress granules (392). Functional TIAR expression is essential for the development of primordial germ cells, since TIAR-/- mice fail to develop spermatogonia or oogonia (16). TIA-1, TIAR, and HuR, as well as the polypyrimidine tract binding protein (PTB), also bind the AU-rich elements residing in the 3′UTR of the mRNA coding for the ß1 subunit of the mitochondrial H+-ATP-synthase (ß1-F1-ATPase) (157, 321). In that case it was shown that RNPs associated with the ß1-F1-ATPase mRNA are vital for localization and translation of this mitochondrial enzyme (228). Protein binding to the ß1-F1-ATPase 3′UTR is regulated by oxidative thiol-modification in the associated RNPs during embryonic liver development (158). This allows a fast induction of ß1-F1-ATPase translation in a redox-dependent manner around birth when the liver redox equilibrium is rapidly shifted towards a more oxidized state (78). Similarly, AU-rich element-binding has been shown to be redox-sensitive for hnRNP A1 (127). It is very likely that the redox-mediated effects on AU-rich element-binding proteins are of general importance and may therefore also apply for RNP complex formation on the COX-2 mRNA. ß-Catenin, a multifunctional key player of Wnt signaling, can also function as RNA-binding protein. It binds and stabilizes COX-2 mRNA by direct interaction with AU-rich elements located in the COX-2 mRNA and by interaction with HuR. In addition, ß-catenin induces the cytoplasmic localization of its binding partner HuR (210).
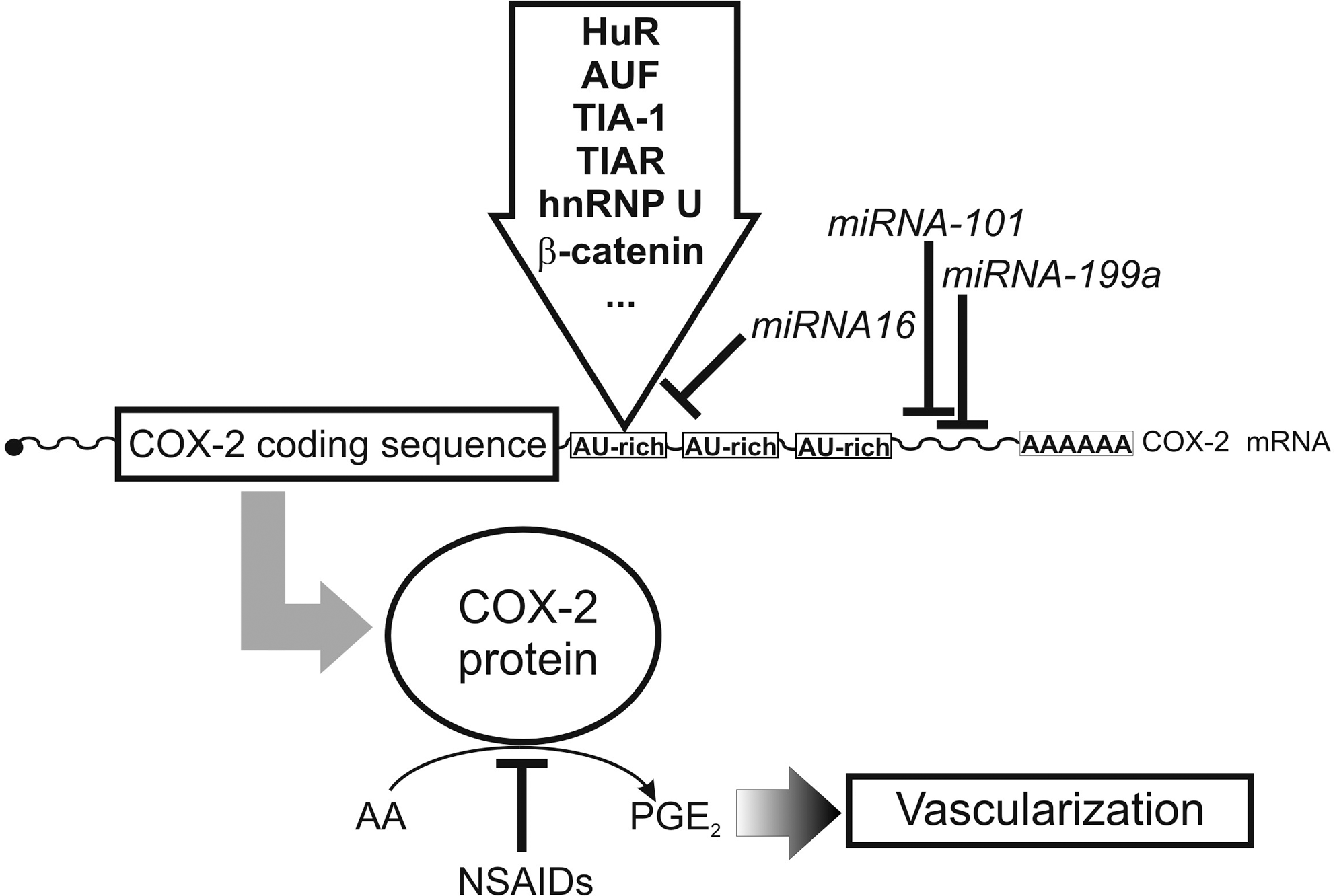
In addition, it is not surprising that miRNA species have been identified (Fig. 15) that regulate COX-2 expression by targeting the 3′UTR of the corresponding mRNA (47, 166). The microRNA miR-16 binds specifically to AU-rich elements (166). Two further miRNAs (mmu-miR-101a and mmu-miR-199a* [with miR-199 derived from the joint miRNA precursor miR-199a/a*]), have been identified, which regulate COX-2 expression in the placenta during implantation (47).
J. Post-transcriptional regulation of antioxidative enzymes
RNA-binding proteins have also been identified for other antioxidative enzymes such as Mn-SOD, GPx1, and catalase (67, 68). The selenoprotein GPx1 carries a SECIS element in its 3′UTR, which is bound by SBP2. In addition, downstream of the SECIS element a sequence has been identified that binds a regulatory protein in a redox-sensitive manner (68). This 56kDa-RBP requires free thiol-groups for binding and looses its RNA binding activity in an oxidizing environment. Interestingly, the same protein binds to the Mn-SOD mRNA and is needed for translational activation of Mn-SOD mRNA (65, 68). GPx1 and Mn-SOD mRNA binding activity of this RBP are inhibited by a yet unidentified RNA component (68). In this context, it is interesting to note that Mn-SOD mRNA has recently been identified as target for miRNA-222 that silences Mn-SOD expression (229). A similar redox-sensitive 69kDa-RBP appears to interact catalase mRNA (67).
VIII. Clinical Relevance of Redox Imbalance in Embryo and Fetal Development
In the previous sections of this review, we focused on the mechanistic aspects of how alterations of the redox homeostasis impact mouse embryogenesis. In this concluding section we will translate the lessons we learned from the mechanistic animal studies to human and will briefly summarize the clinical relevance of a disturbed redox homeostasis for human embryogenesis. For this purpose, selected disorders of human pregnancy and their impact on embryo and fetal development will be discussed.
A. Redox-dependent teratogenesis
ROS/RNS have previously been implicated in human embryotoxicity and teratogenicity (99). A variety of teratogens are capable of inducing ROS generation and a variety of pharmaceutics, pesticides, metals, and environmental contaminants may act as teratogens. In fact, approximately 2500 chemicals including phenytoin, ethanol, nicotine, and thalidomide have been identified as human teratogens (217). Many teratogens alter the maternal redox homeostasis or, after passing the placental barrier, modify the fetal redox equilibrium during key periods of embryo and fetal development (Fig. 4).
Thalidomide, a sedative-hypnotic, was prescribed in the early 1960s as tranquilizer for pregnant women who suffered from morning sickness. Unfortunately, thalidomide therapy was associated with multiple birth defects including phocomelia (380). In 1999, thalidomide was introduced as agent for the treatment of multiple myeloma because of its preventive activity in neoangiogenesis (362). Thalidomide has also been for treating symptoms of prostate cancer, glioblastoma, lymphoma, arachnoiditis, Behçet's disease, and Crohn's disease. The most sensitive organs for thalidomide intoxication are the limbs. Among mammals, a species-dependent sensitivity for thalidomide has been reported, with rabbits being most sensitive. In contrast, rats are rather resistant (130, 131). The risk of congenital malformations after exposure during the critical period of pregnancy varies between 20% and 50% (277). Thalidomide toxicity is induced via a shift in the cellular redox equilibrium resulting in the depletion of GSH, and this alteration was suggested to regulate the activity of various transcription factors (130, 131). Despite its potent teratogenic activities, thalidomide is still prescribed for treatment of severe illnesses such as erythema nodosum leprosum. In addition, patients suffering from chronic graft-versus-host disease, systemic lupus erythematosus, Behcet's syndrome, inflammatory bowel disease, prostate cancer, metastatic breast cancer, rheumatoid arthritis, uremic pruritus, severe atopic erythroderma, tuberculosis, and Kaposi's sarcoma may benefit from thalidomide administration.
Phenytoin is a widely-used anticonvulsant that can double the incidence of structural and functional birth defects when used in pregnancy (176). During human embryogenesis it induces vascular disruption, which leads to hypoperfusion and hypoxia (81). In addition, phenytoin induces oxidative DNA damage and dysmorphogenesis that can be counteracted by antioxidant treatment (429). Further evidence for the hypothesis that oxidative stress is involved in phenytoin-mediated toxicity is provided by the observation that maternal administration of catalase partially protects from phenytoin-induced teratogenicity (431). Interestingly, SOD administration was not able to do so. In fact, SOD even enhanced phenytoin teratogenicity. This data suggests that the balanced equilibrium of antioxidative enzymes is important and that excessive expression of a certain antioxidative enzymes may even be deleterious.
In humans, ethanol is a significant nutritional energy source and it is metabolized via two subsequent oxidation reactions (ethanol → acetaldehyde, acetaldehyde → acetic acid) to acetic acid. The first oxidation can proceed via three different mechanisms (Fig. 16): i) NAD-dependent ethanol dehydrogenases, ii) the NADPH+H+-dependent microsomal ethanol oxidizing system (MEOS), and iii) the peroxidase system. Among these, MEOS and the peroxidase pathway impact ROS formation (Fig. 16). Chronic alcohol ingestion compromises the maternal liver, leading to increased production of lipid peroxides and decreased expression of antioxidant enzymes. Conversely, ascorbic acid attenuates ethanol toxicity via inhibition of ROS formation and NF-κB activation (307). In addition, fetal GSH content decreased with ethanol consumption (3).
B. Maternal diabetes mellitus
Maternal diseases such as diabetes mellitus during pregnancy also impact the embryonic redox homeostasis. In fact, maternal diabetes is an important source of clinically relevant oxidative stress. Under diabetic conditions, embryonic morphogenesis is more frequently disturbed and this is at least in part related to the generation of ROS (96). In fact, antioxidative enzymes can protect diabetic embryos from glucose-induced malformations (95). Hyperglycemia-induced developmental anomalies are associated with GSH depletion (394). The teratogenic effect of diabetic serum was prevented by SOD and N-acetyl-L-cysteine in rat whole embryo culture (378). As a mechanistic element in diabetes signaling a decreased expression of the transcription factor Pax3 was suggested. Pax3 is expressed in neural crest cells and somatic mesoderm cells. Homozygous splotch (SP/SP) mouse embryos carry a loss of function Pax3 allele and develop a defective neural tube. These animals die at E16.5, owing to defective migration of neural crest cells to the heart (75). The pups of diabetic dams also show increased incidence of neural tube defects (232). Since SP/SP embryos are very similar to the phenotype seen in diabetic mice, it might well be that Pax3 dictates neural development in these animals and that changes in the glucose homeostasis might also affect Pax3 gene expression. This conclusion was supported by the prevention of these effects when antioxidants were administered or when the defects were induced in SOD overexpressing animals (50). Moreover, administration of antimycin A, a mitochondrial complex 3 inhibitor that increases superoxide production, inhibited Pax3 expression and increased neural tube defects (50).
C. Pre-eclampsia
Pre-eclampsia is a pregnancy-specific disorder and is diagnosed on the basis of hypertension associated with proteinuria in late gestation (136, 404). It is the second leading cause of maternal mortality, accounting for 12%–18% of pregnancy-related maternal deaths (19). It remains one of the most common causes of perinatal morbidity and mortality of newborns. Maternal complications include abruptio placenta (1%–4%), disseminated coagulopathy/hemolysis, elevated liver enzymes and low platelet (HELLP) syndrome (10%–20%), pulmonary edema/aspiration (2%–5%), acute renal failure (1%–5%), eclampsia (1%), and death (rare) (361). The fetal complications include preterm delivery (15%–67%), fetal growth restriction (10%–25%), hypoxia-neurologic injury (<1%), and perinatal death (1%–2%) (361). Pre-eclampsia is characterized by abnormal placental trophoblast invasion by the maternal uterine spiral arteries of the deciduas, leading to a failure to establish an adequate uteroplacental blood flow, which results in relatively hypoxic trophoblast tissue. Therefore pre-eclampsia results from a malfunctioning of the placenta (292). Classically, pre-eclampsia was described as a two-stage disorder (320). In the first stage, inadequate uteroplacental circulation leads to placental ischemia and the ischemic placenta releases signaling factor(s) into maternal circulation, which induce endothelial dysfunction (Fig. 17). In the second stage, endothelial dysfunction results in excessive inflammatory responses that cause the maternal syndrome. It is postulated that in response to ischemia and/or reperfusion within the placenta ROS can alter endothelial function. Mitochondria are more abundant in the placenta of women suffering from pre-eclampsia when compared with normal controls (172). The higher mitochondria density may lead to excessive electron leakage and thus to an increased formation of superoxide. Moreover, pre-eclamptic mitochondria show signs of increased oxidative damage (414). Another source of increased ROS formation during pre-eclampsia appears to be the xanthine oxidoreductase pathway. Under hypoxia and in response to inflammatory cytokines expression of xanthine oxidoreductase is augmented and conversion of the enzyme into its radical producing xanthine oxidase form is enhanced (385). In fact, ischemia and subsequent reperfusion induce increased xanthine oxidase activity in the pre-eclamptic placenta (249).
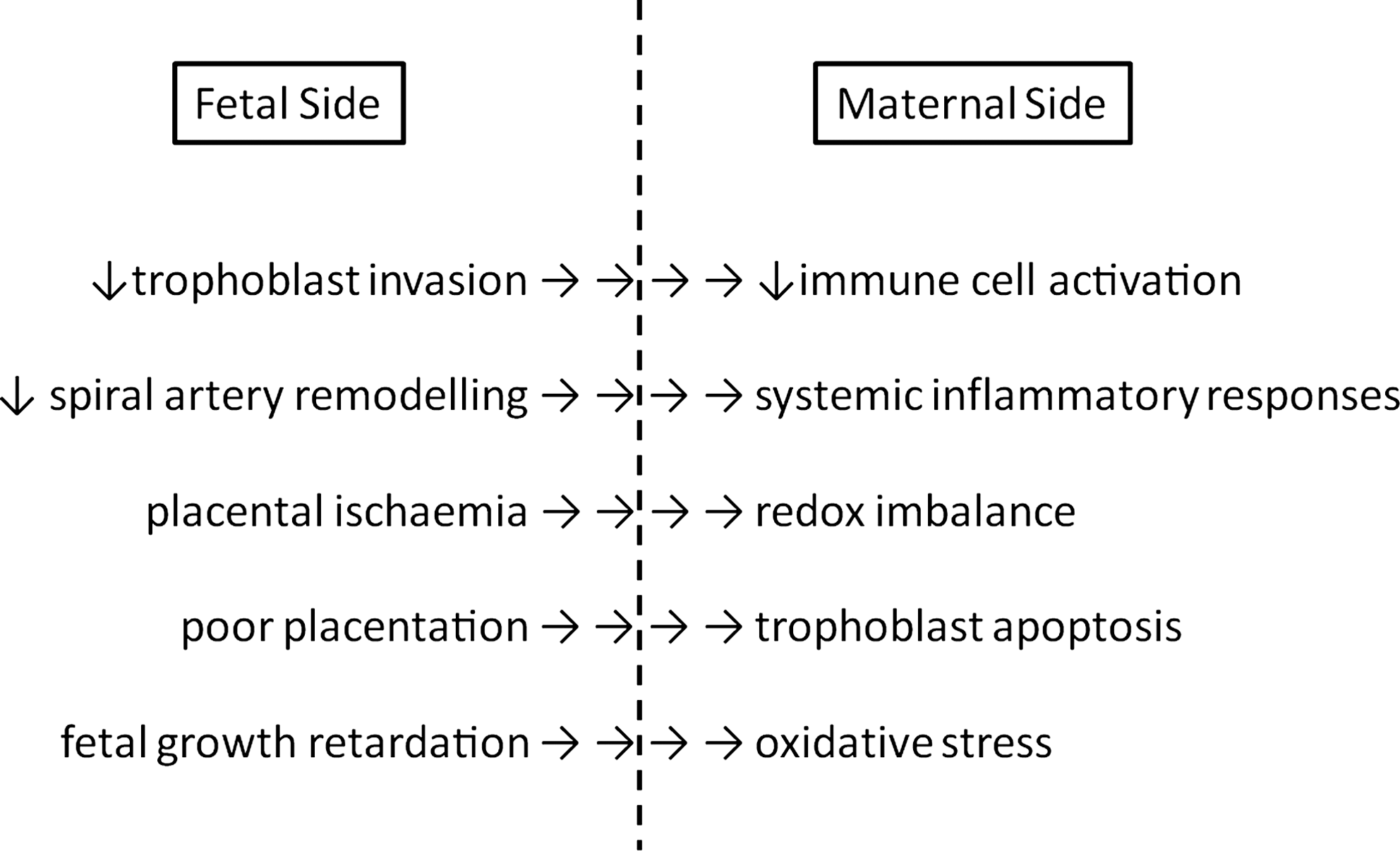
Nitric oxide syntheses (NOS) may also contribute to increased placental ROS formation in pre-eclamptic subjects. Peroxynitrite, which is formed from superoxide and nitric oxide, may attack plasma membrane proteins of the villous vascular endothelium (267) and of the invasive trophoblasts (249). In addition, stable products of lipid peroxidation such as malondialdehyde (MDA) may contribute to pre-exclampsia-associated endothelial dysfunction. In fact, MDA is increased in the plasma of pre-eclamptic women (149). Alternatively, neutrophils and monocytes passing through the intervillous space could be activated by ROS and interact with endothelial cells to generate oxidative injury. This hypothesis is supported by the observation that more neutrophils are activated in pre-eclamptic women (66). It has been shown that there is an increased deportation of syncytial fragments in pre-eclamptic placenta (194) and that the syncytiotrophoblast debris may be modified by oxidative stress (44).
D. Fetal hypoxia
Intrapartal fetal hypoxia induces metabolic acidosis and subsequently periventricular hemorrhage and impaired myocardial function (13). Prolonged or severe intrauterine asphyxia may cause permanent ischemic damage. The infant may suffer from hypoxic-ischemic encephalopathy, meconium aspiration syndrome, acidosis with decompensation, cerebral palsy, neonatal seizures, necrotizing enterocolitis, and patent ductus arteriosus (Fig. 18). Cerebral palsy is the most common birth disorder and the most severe consequence of nonfatal fetal hypoxia, affecting between 1 and 2.5 per 1000 newborns in developed countries and about 10 per 1000 live births in developing countries. The major pathological substrates are ischemic lesions in the periventricular white matter leading to spastic diplegia (pre-term) or in the basal ganglia leading to extrapyramidal or dyskinetic cerebral palsy (366). Of the survivors, up to 50% exhibit permanent neuropsychological handicaps in form of mental retardation, spastic motor deficits, learning disability, or epilepsy (39, 171). These conditions are uncommon and only reflect one extreme of the spectrum of asphyxial morbidity. Meaningful audit of obstetric practice requires parameters that can be measured objectively and which confirm clinically defined perinatal asphyxia.
A variety of clinical assessments and biochemical measurements in fetal blood have been used to assess the degree of the oxidative stress and hypoxic damage but none of these is entirely satisfactory (368). Hypoxia frequently develops during birth when uterus contraction and relaxation periods follow each other repeatedly and an increased ROS formation can be monitored at these periods. Moreover, delivery and neonatal resuscitation may also induce ROS formation. The relation between oxidative stress and fetal hypoxia has extensively been reviewed (407) before. Measurement of lipid peroxides, as footprints of fetal cellular injury from free radical activity, are used to estimate the extent of hypoxic damage in relation to intrapartum morbidity and subsequent adverse fetal outcomes.
IX. Problems and Perspectives
Embryogenesis is a long and complex process that starts with sperm–oocyte fusion and is concluded with delivery. It involves a complex four-dimensional (space and time) network of regulatory processes and redox sensitive mechansims have been implicated. In the past, redox-relevant research has been focused on early stages of embryo development, in particular on preimplantation embryos. Dysregulations in these developmental stages are often of fatal consequence and lead to abortion. These consequences are regrettable but are not of major socioeconomic impact. On the other hand, little is known on redox control mechanisms at later stages of embryo development (organogenesis in mid- and late gestation) and during delivery. If these processes are dysregulated, usually viable individuals with severe developmental defects may develop and the socioeconomic consequences of such defects are much more severe. In other words, regular development and maturation of limbs and major organs is crucial for healthy individuals and thus, more research is needed to explore the developmental consequences of dysregulations in the redox homeostasis during later stages of embryogenesis.
A major problem in the field of redox control and embryogenesis has always been distinguishing between correlation and causality. In the period of functional genomics, it has been the general strategy to silence (loss of function strategy) or activate (gain of function strategy) expression of single gene products and to search for phenotypic effects under resting conditions or after challenging. In most cases such phenotypic defects are then correlated to the genotypic manipulations without establishing a detailed causal chain connecting the manipulations with the defects. Unfortunately, this strategy has two major problems: i) Redox regulation of a certain gene product may not necessarily be connected with its developmental function and thus, connecting them causally may lead to wrong conclusions. To overcome these problems we need more information on the functional multiplicity of pro- and/or antioxidative gene products and explore their potential roles outside redox homeostasis. This is particularly important for gene products that have already been identified as moonlighting proteins (316). ii) Manipulations of a single gene in a complex, multicellular organism are likely to induce alterations in various regulatory networks and each of these are rather complex. Thus, attempts to establish linear causal chains (in fact there is no linear causal chain in a complex system like the human body) will not successfully mirror the complexity of induced regulatory alterations. For instance, manipulations of the redox equilibrium that affects protein thiol groups are likely to alter the entire pool of regulatory factors that impact each other. Thus, predictions on the overall effect are virtually impossible employing traditional deductive strategies. However, innovative algorithms of Computational Systems Biology may be considered a suitable methodology to approach this problem. However, this approach may only be successful if systems biologists strictly adhere to experimental data and also consider the experimental conditions under which the data were originally obtained. It may be strongly misleading to compare apples and oranges. It is a major task of Computational Systems Biology to develop complex but working hypotheses that can be tested experimentally.
If one summarizes the outcome of the knockout studies of the antioxidative gene products (Table 1), one comes to the conclusion that most of the knockout individuals are viable and do not exhibit major developmental defects. Only deficiencies in G6PD, GSH-synthase, Trx1/2, and GPx4 induce embryonic lethality. As reason for this disappointing outcome, it is normally discussed that deficiency of a single antioxidative gene product may be compensated for by gene products with similar functionality. However, there is little direct experimental evidence for such compensation mechanisms or even experimental data disprove such assumptions (24). One way to approach this problem would be the creation of double, triple, quadruple etc. knockout individuals, but the danger of uncontrolled side effects certainly increases which each step of manipulation.
A particular problem in redox control and embryogenesis are the conflicting results obtained with mice deficient in GSH-synthase and GSH-reductase. γGCS-HS-/- mice die at midgestation because of uncoordinated developmental apoptosis (Table 1). In contrast, Gr1a1Neu mice are viable and do not show major developmental defects. There are certainly ways to explain these unexpected observations (GSH has additional functions in embryogenesis when acting as antioxidant) but these additional functions have not been identified yet. GPx4-/- mice also die at midgestation. If the lack of its catalytic activity (GSH peroxidase) is responsible for embryonic lethality, one would expect that a lack of reduced GSH would also induce embryonic lethality. In contrast, GSH reductase-deficient mice (Gr1a1Neu) are viable and develop normally during embryogenesis. There are of course several ways to explain this discrepancy, but one of them is that GPx4 exhibits additional biological activities, which are not solely related to its catalytical function. It might be speculated that GPx4 might function as structural protein in early brain development, as it has been shown for spermatogenesis (316). Whether its catalytic activity is required for this hypothetical function can be explored by in vivo mutagenesis (knockin studies). This example emphasizes the general dilemma that certain gene products may have more functions than their name implies, and the discovery of oxygen-sensing demethylases, for instance, is a step towards coping with that problem (193, 359).
For the time being, the methodological approaches to solve problems in developmental biology are somewhat limited because of their selective character. This is in sharp contrast to the high complexity of developing biological systems such as a maturing embryo. System biology and state-of-the-art “∼omics” approaches may help to overcome these limitations. However, analyzing whole organisms rather than cultures of a single cell clone always requires more careful evaluation of the experimental settings. Finally, particular care should be taken when experimental findings obtained in murine or other vertebrate experimental setups are translated to the human situation.
Footnotes
Acknowledgments
This work was supported by a joint grant of German Academic Exchange Service (DAAD) and Hong Kong RGC (2006–2007 and 2007–2008) to HK and CCW respectively, by a CUHK Direct Research Grant 2041427 to CCW, and by a research grant of the European Commission (FP6, LSHM-CT-2004-0050333) to HK.
Abbreviations Used
Reviewing Editors: James A. Coffman, Kazuhiro Kikuchi, Robert K. Naviaux, Asher Ornoy, Maurizio Parola, Hideyo Sato, and Edward A. Schmidt