
Abstract
Defective sperm function is the largest single defined cause of human infertility and one of the major reasons we are witnessing an exponential increase in the uptake of assisted conception therapy in the developed world. A major characteristic of defective human spermatozoa is the presence of large amounts of DNA damage, which is, in turn, associated with reduced fertility, increased rates of miscarriage, and an enhanced risk of disease in the offspring. This DNA damage is largely oxidative and is closely associated with defects in spermiogenesis. To explain the origins of this DNA damage, we postulate that spermiogenesis is disrupted by oxidative stress, leading to the creation of defective gametes with poorly remodeled chromatin that are particularly susceptible to free radical attack. To compound the problem, these defective cells have a tendency to undergo an unusual truncated form of apoptosis associated with high amounts of superoxide generation by the sperm mitochondria. This leads to significant oxidative DNA damage that eventually culminates in the DNA fragmentation we see in infertile patients. In light of the significance of oxidative stress in the etiology of defective sperm function, a variety of antioxidant therapies are now being assessed for their therapeutic potential. Antioxid. Redox Signal. 14, 367–381.
Introduction
Most male infertility patients produce sufficient numbers of spermatozoa to achieve a pregnancy; however, in this group of men, these cells have lost their competence for fertilization and the support of normal embryonic development. The reasons behind this loss of functionality are still not adequately resolved. As a result of this uncertainty, few therapies are available that target the actual cause of the infertility. Instead, ART is being used to overcome whatever functional deficiencies beset the spermatozoa by simply bypassing the normal physiological processes that regulate fertilization. Under these circumstances conception is achieved through the placement of defective spermatozoa in juxtaposition to the egg, or in its extreme form, by physically injecting a single spermatozoon into the oocyte in a procedure known as intracytoplasmic sperm injection (ICSI). Such assisted conception techniques pay no heed to the cause of the infertility or the risks inherent in a procedure that allows defective spermatozoa to achieve conceptions in vitro that would have been excluded from this process in vivo. Even when damaged spermatozoa are enabled to fertilize the oocyte by ICSI or, to a lesser extent, in vitro fertilization, there is no guarantee that embryonic development will be normal. DNA damage in the form of strand breaks and base adduct formation is a common feature of defective spermatozoa and the incidence of such damage is directly correlated with the normality of early embryonic development (63, 68).
Thus, the incidence of birth defects after assisted conception is double that seen in the naturally conceived population (41) and there is good evidence for an increase in imprinting disorders, notably, the Beckwith-Wiedemann and Angelman syndromes, in such children (85). Infants produced by ART are also significantly more likely to be admitted to a neonatal intensive care unit, to be hospitalized, and to stay in hospital longer than their naturally conceived counterparts (40). Large studies of Scandinavian populations using record linkage have also shown an increase in the hospitalization of ART offspring in infancy and early childhood compared with spontaneously conceived children (32, 51, 53). Further, recent independent investigations have revealed an eightfold increase in the incidence of undescended testicles in boys conceived by ICSI (62), while another study has uncovered abnormal retinal vascularization in such children (95). Notwithstanding the concerns raised in these publications, it must also be stressed that other authors have been much more reassuring about the normality of ICSI children, particularly in terms of their cognitive development and motor skills (72). Such a mixed picture for the safety of assisted conception procedures arises for a number of reasons: (i) the incidence of definable events, including overt birth defects, hospitalizations, or significant pathologies such as cancer in the offspring, is low; (ii) the study populations are generally small; (iii) the number of confounding variables influencing the health and well-being of such children is great; (iv) the duration of follow-up is short; (v) the instruments used for assessing defects in the offspring are insensitive.
If we assume that the genetic/epigenetic damage associated with ART is randomly distributed throughout the genome, then the chances that a specific gene will be directly affected is low, given that ∼95% of the genome is noncoding. It is even less likely that if a mutation did occur in a coding gene that it would both be dominant and cause a detectable phenotypic or behavioral change in the F1 generation. The incidence of even the more common spontaneous dominant mutations, such as achondroplasia, approximates to only 1 in every 10,000–30,000 births in the population at large. Thus, if phenotypic change is the sole criterion, then most ART studies are simply not adequately powered to determine whether this form of therapy has a significant effect on the normality of the offspring. Under these circumstances, the absence of evidence for paternal impacts on development cannot be taken as evidence of absence. Indeed, there are paradigms such as age or smoking where there is clear-cut support for a paternal effect on embryonic development.
In the case of paternal age, there is an exponential relationship between the age of the father and a child's susceptibility to spontaneous dominant genetic mutations such as achondroplasia, Apert syndrome (acrocephalosyndactyly), and multiple endocrine neoplasia (27). Age is also associated with an increase in the incidence of complex polygenic neurological conditions in the offspring, including epilepsy, spontaneous schizophrenia, bipolar disease, and autism, as well as an increased rate of death in the F1 generation associated with congenital malformations, injury, and poisoning (4, 100). Similarly, there is evidence for an association between heavy paternal exposure to cigarette smoke and morbidity in the offspring, including childhood cancer (48). Since both paternal age and smoking are associated with increased levels of DNA damage in spermatozoa (4, 36), the existence of such paternal effects clearly raises the possibility that children conceived using DNA-damaged spermatozoa as a result of ART may suffer from a similar range of pathologies.
The mechanisms underpinning the paternal age effects are only partly resolved. In the case of dominant genetic mutations, the conventional wisdom is that these genetic lesions are created as a result of replication error in the spermatogonial stem cell population, followed by clonal expansion of mutant germ cells as they enter a selfish pathway of proliferation in the germinal epithelium (27, 39). However, the available data do not offer unconditional support for such a model. In the case of achondroplasia, for example, the incidence of this condition in the offspring of aging males increases much more rapidly than the incidence of the major causative mutation in their gametes (45). Further accurate quantification of spontaneous mutation frequencies in the tissues of aging mice suggests that DNA proof reading and repair in the male germ line is actually extremely efficient (43). As a result, these cells have one of the lowest spontaneous mutation rates in the body; moreover, this rate does not appear to change with age (43). Thus, there may be other mechanisms contributing to the impact of paternal age on dominant genetic mutations in our species. In this context, we have suggested that a possible source of embryonic mutations would be the aberrant repair of DNA damage brought into the oocyte by the fertilizing spermatozoon. Between formation of the male pronucleus and initiation of S-phase of the first mitotic division, the oocyte engages in a rapid round of DNA repair and will not allow DNA synthesis to proceed in either the male or female pronucleus until this process has been competed. If the oocyte makes a mistake at this point, then there is the possibility of creating a mutation that, because it precedes the first mitotic division, will be represented in every cell in the body. Such a hypothesis is consistent with the increased levels of DNA damage and oxidative stress seen in the spermatozoa of aging males and the recognized importance of the oocyte as a major site of DNA repair for the male germ line (9).
A similar etiology may underpin the morbidity seen in the children of male smokers; that is, smoking induces DNA damage in the male germ line that then results in the formation of mutations in the embryo as a result of aberrant DNA repair in the oocyte. In the case of smoking there is clear evidence for oxidative DNA damage in the spermatozoa as a consequence of smoke exposure. Thus, heavy paternal smoking is thought to place the perpetrator's physiology under oxidative stress, causing a significant depletion of antioxidant vitamins such as vitamins C and E in both serum and seminal plasma (36). One of the consequences of the resultant oxidative stress is thought to be formation of the oxidized base adduct, 8-hydroxy-2′-deoxyguanosine (8OHdG), and subsequent DNA fragmentation in the spermatozoa.
The idiopathic DNA damage found in the spermatozoa of male infertility patients could also be the precursor for mutations in the embryo after aberrant genetic repair in the oocyte (Fig. 1). The DNA damage we see in infertility patients is again thought to be due to oxidative stress, given the powerful correlations that have been observed between spontaneous DNA fragmentation in these cells and 8OHdG formation (29). Determining the causes and consequences of the oxidative stress responsible for this damage will be a central element in future research designed to further our understanding of male infertility.
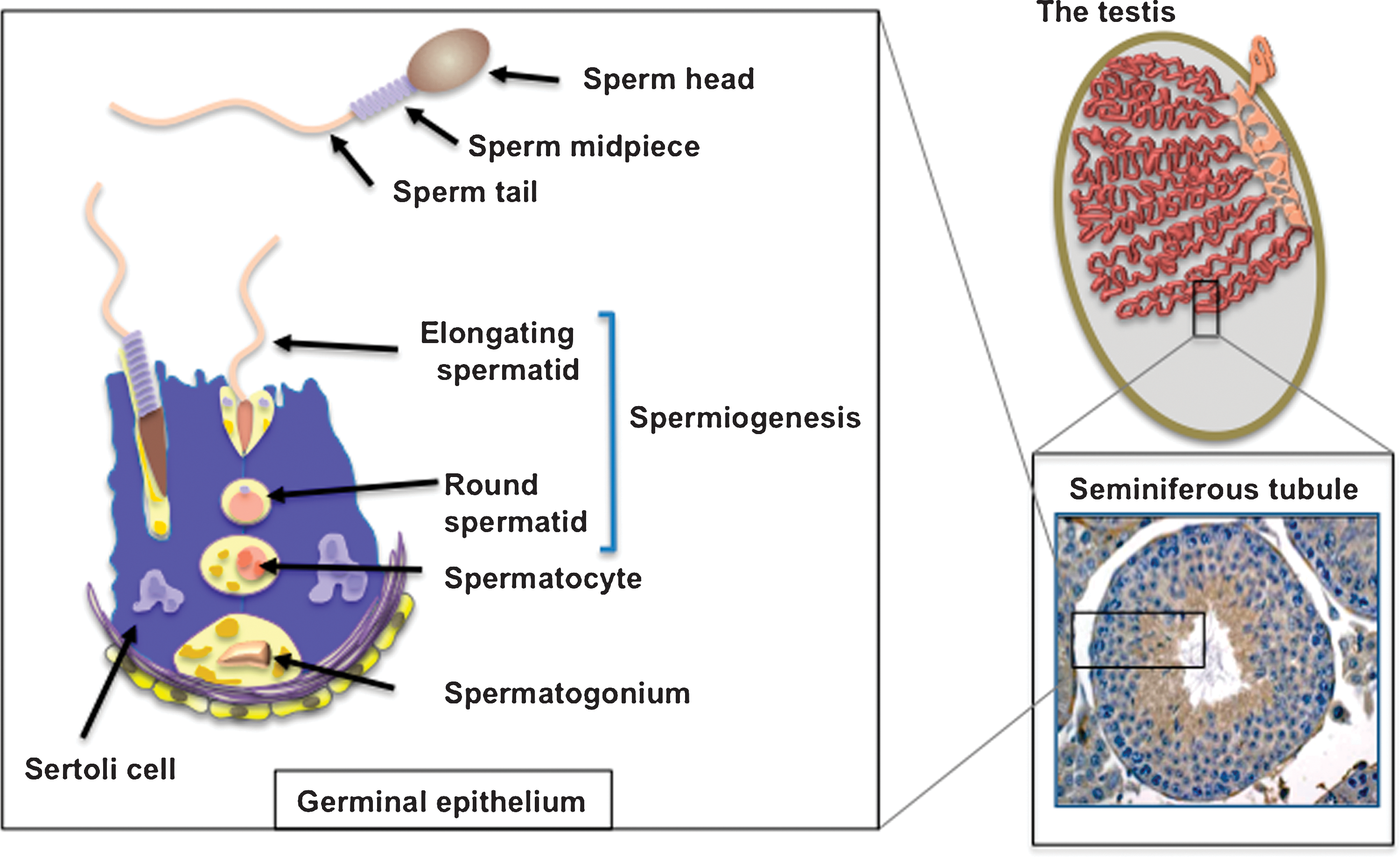
Oxidative Stress in the Testes
As germ cells are differentiating in the testes, they are supported and protected from oxidative stress by the nurse cells of the testes, the Sertoli cells. This support and protection is particularly important as these cells pass through meiosis and emerge as small haploid cells known as round spermatids (Fig. 1). By this stage of development, these cells are transcriptionally silent, and yet in the absence of any regulated gene transcription, they are able to undergo one of the most remarkable cellular transformations in biology, to become that epitome of terminally differentiated, highly specialized cells, the spermatozoon. This dramatic morphogenesis from a round spermatid to a fully differentiated spermatozoon is achieved solely on the basis of the carefully orchestrated differential translation of pre-existing mRNA species in a process known as spermiogenesis (Fig. 1). During this process these cells are exquisitely sensitive to oxidative stress. Isolated spermatids have a limited capacity for DNA repair and a limited capacity for glutathione replenishment (30). Throughout this phase of development they are highly dependent on the protection offered by the Sertoli cells, which possess high levels of superoxide dismutase (SOD) activity as well as the reductase, transferase, and peroxidase activities of the glutathione cycle (21).
Spermatozoa Are Vulnerable to Oxidative Attack
Once spermatozoa are released from the germinal epithelium, however, they are on their own and can no longer benefit from the defensive capabilities the Sertoli cell population. As isolated cells, spermatozoa are very vulnerable to oxidative attack for a number of reasons, as set out below.
Sperm are poorly endowed with antioxidant enzymes
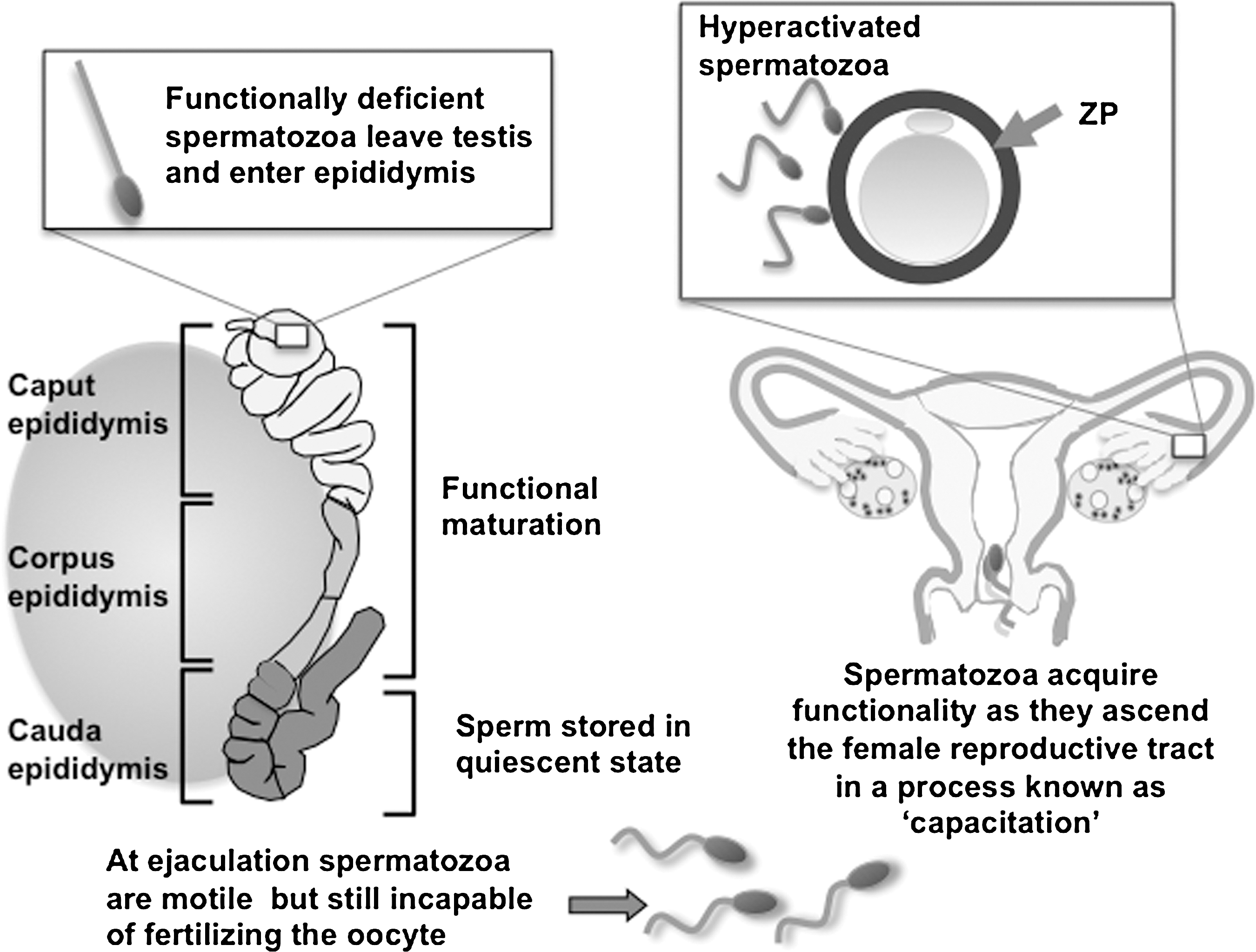
One of the reasons for the vulnerability of mammalian spermatozoa to oxidative stress is their unusual strategy of divesting themselves of most of their cytoplasm just before they are released from the germinal epithelium. Thus, the very cellular compartment that should house the antioxidant enzymes that protect most cells from oxidative stress has been discarded by the time spermatozoa are discharged into the lumen of the seminiferous tubules. What little cytoplasm there is remains confined to the midpiece of the cell in the vicinity of the mitochondria (Fig. 2). As a result, there are large areas of plasma membrane overlying the sperm head and tail that cannot be accessed by cytosolic antioxidants. Because of this relative lack of intrinsic antioxidant protection, these cells are heavily dependent on the antioxidant properties of the fluids in which they are bathed. It is for this reason that epididymal and seminal plasmas are among the most powerful antioxidant media known to man, containing several highly specialized forms of reactive oxygen species (ROS) scavenging enzymes including unique forms of glutathione peroxidase (GPx5) and extracellular SOD as well as a host of small molecular mass free radical scavengers such as uric acid, vitamin C, tyrosine, and polyphenols (1, 75, 92). Of course, this is not to say that spermatozoa are completely lacking in their own antioxidant defenses, since they do contain detectable SOD activity, an active glutathione system and, possibly, some catalase (5, 96). However, these limited defenses are easily overwhelmed, with the resultant generation of oxidative stress.
Spermatozoa are professional generators of ROS
Despite their inherent vulnerability to oxidative attack, spermatozoa are also professional generators of ROS. Indeed spermatozoa were the first cell type ever shown to generate reactive oxygen metabolites, when Tosic and Walton described the generation of hydrogen peroxide by bovine spermatozoa in a seminal article published in Nature in 1946 (89), predating the discovery of ROS generation by phagocytic leukocytes by several decades.
The purpose of this professional ROS generation appears to be the regulation of a critical biological process known as capacitation, without which fertilization is impossible. Mammalian spermatozoa are ejaculated in an immature state, completely lacking in any capacity for fertilization. However, as these cells ascend the female reproductive tract, they become physiologically transformed through the process of capacitation, so that they are ultimately competent to both recognize the oocyte and engage in the complex cascade of cellular interactions that result in fertilization (Fig. 2). This process is redox regulated. Specifically, capacitation involves a massive increase in the tyrosine phosphorylation status of the sperm tail. This is thought to facilitate a change in the movement characteristics of mammalian spermatozoa from the traditional forward, progressive type of motion (characterized by a high-frequency, low-amplitude, and symmetrical flagellar beat pattern) to a state of hyperactivation. When spermatozoa are hyperactivated, the flagella are thrown into low-frequency, high-amplitude, asymmetrical waves that are capable of generating the propulsive forces necessary to achieve two important biological goals (Fig. 3). First, during their ascent of the female reproductive tract spermatozoa become bound to the epithelium lining the isthmic region of the Fallopian tube to create a sperm store, where the gametes await a signal that ovulation has occurred and the oocyte is available for fertilization. At this point in their life history the previously quiescent spermatozoa suddenly break into a state of hyperactivation, which helps to free them for their epithelial repository and progress toward the ampulla of the Fallopian tube where fertilization will take place. Hyperactivated motility again plays an important biological role during fertilization in creating the forward thrust necessary to drive the sperm head though the zona pellucida, a tough acellular shell that surrounds the mammalian oocyte (Fig. 2). This adaptive form of movement is therefore a critical element of the biological control of fertilization. Genetically modified mice that are normal in every respect except that their spermatozoa cannot hyperactivate are sterile (49).
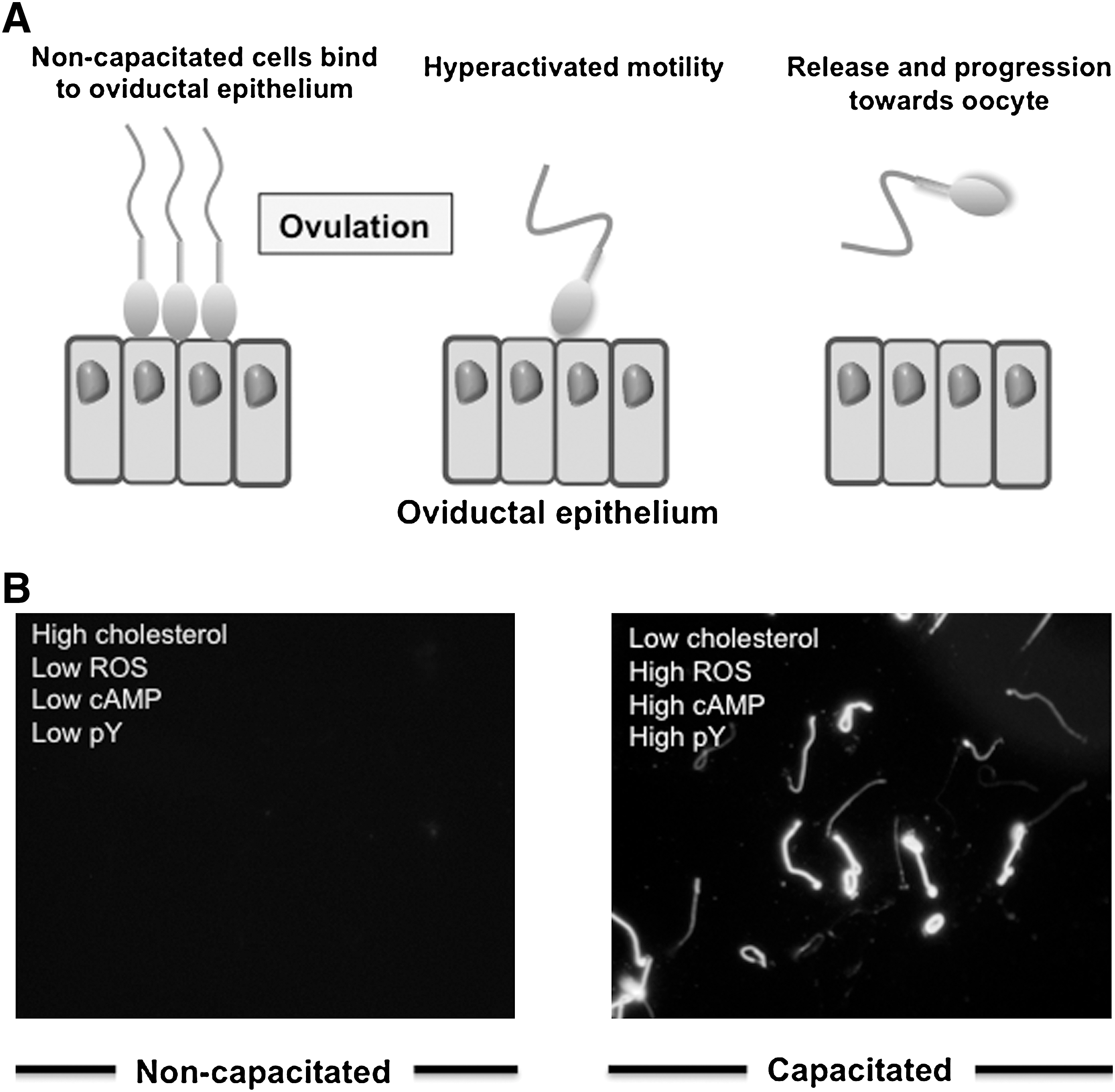
The tyrosine phosphorylation events that underpin capacitation are primarily driven by cyclic adenosine monophosphate (cAMP) generated by a soluble form of adenylyl cyclase (sAC) (94). Targeted deletion of this enzyme leads to a dramatic loss of sperm movement, including hyperactivation, via mechanisms that can be reversed by loading the cells with a membrane permeant cAMP analog (34). The cAMP increase associated with capacitation in turn activates a tyrosine kinase, Src (tyrosine kinase similar to v-src from Rous sarcoma virus), via the activation of protein kinase A. The activation of Src involves both a direct activating phosphorylation of this tyrosine kinase and an inhibitory phosphorylation of C-terminal kinase, which is a negative regulator of Src. As a consequence of these two mechanisms, Src is maximally activated and drives a global increase in phosphotyrosine expression in the sperm tail, which, in the mouse at least, is tightly associated with the induction of hyperactivated movement (15, 67). ROS facilitate this pathway via two mechanisms. First, hydrogen peroxide is a powerful negative regulator of tyrosine phosphatase activity because it induces the oxidation of a key cysteine residue at the active site of this enzyme, which must be in a reduced state for phosphatase activity to be expressed (42). Second, ROS are thought to promote the activity of sAC, stimulating the intracellular generation of cAMP and hence, via protein kinase A and Src, tyrosine phosphorlyation (8, 31, 57, 58). A summary of the available evidence (Fig. 3) would suggest that during this signal transduction cascade, superoxide anion is responsible for the stimulation of sAC, while hydrogen peroxide is responsible for the suppression of tyrosine phosphatase activity (58, 98).
The source of ROS in mammalian spermatozoa
Analysis of the source of ROS generated by capacitating populations of mammalian spermatozoa suggests that multiple redox systems are involved at the level of the plasma membrane, cytosol, and mitochondria (10). At the level of the plasma membrane several authors have claimed to detect activities consistent with the presence of an NAD(P)H oxidase (Nox) complex (6). This activity can be driven by the addition of exogenous NAD(P)H and is inhibited by the presence of diphenylene iodonium (DPI), a classical Nox inhibitor. Interpretation of these biochemical studies is complicated by a number of confounding factors. First, solutions of NADPH have a tendency to spontaneously oxidize and generate hydrogen peroxide so that it becomes difficult to determine whether the biological effects observed are due to NADPH itself or the presence of this oxidant (35). Second, we have shown that the lucigenin signals generated in the presence of exogenous NAD(P)H are the result of redox cycling of the probe itself after reduction of lucigenin to its cation radical by major intracellular oxidoreductases such as cytochrome b5 reductase (16) or cytochrome P450 reductase (17). Third, the inhibitor used in these studies, DPI, is not, as frequently assumed, a specific inhibitor of Nox activity. It selectively targets flavoproteins and, at high concentrations, can also disrupt the mitochondrial electron transport chain (59). DPI has been found to suppress ROS generation by the spermatozoa of a wide variety of species, including horse (25), bull (26), buffalo (78), mouse (32), rat (93), and human (6). Whether such results reflect ROS generation by the sperm mitochondria or the presence of an Nox is still an open question. Evidence in support of some form of oxidase in spermatozoa has been secured with the discovery of various components of the free-radical-generating Nox2 complex in mouse spermatozoa (86), the proteomic identification of dual oxidase in human spermatozoa (18), and data supporting the presence of Nox5 in equine spermatozoa (79). We have also obtained immunocytochemical evidence for the presence of Nox5 in human spermatozoa (R.J. Aitken, unpublished observations). Whether any of these Nox systems are responsible for generating the free radicals that drive sperm capacitation or create the oxidative stress associated with defective sperm function (3) is currently unknown.
More certain is the role that mitochondrial ROS play in sperm pathology. Sperm mitochondria actively leak electrons and generate ROS particularly when spermatozoa become metabolically activated after ejaculation. We have found that the mitochondria represent a major source of ROS in human spermatozoa and are responsible for creating a state of oxidative stress that correlates with a loss of sperm motility as a consequence of lipid peroxidation (54). Such results are consistent with a large volume of data indicating that oxidative stress can disrupt sperm motility (2, 11); however, they are unique in pointing out the significance of the mitochondria in this respect. Further, our evidence suggests that it is the release of ROS from complex I on the matrix side of the inner mitochondrial membrane that is the most damaging. ROS generated at this site induce high levels of lipid peroxide formation and a consequential loss of sperm function (7, 12, 54).
Spermatozoa are richly endowed with substrates for free radical attack
The vulnerability of mammalian spermatozoa to oxidative stress is not only a consequence of their inherent free-radical-generating activity and lack of endogenous antioxidant protection, but also a function of the abundant substrates that these cells present for free radical attack. For example, spermatozoa are richly endowed with high concentrations of polyunsaturated fatty acids, particularly decosahexaenoic and arachidonic acids containing six and four double bonds per molecule, respectively (55). The presence of these unsaturated fatty acids is critical for sperm function since they confer upon the plasma membrane the fluidity this structure needs to participate in the membrane fusion events associated with fertilization, including acrosomal exocytosis and union with the plasmalemma of the oocyte. The problem with such highly unsaturated fatty acids is that they are very vulnerable to free radical attack and the induction of lipid peroxidation. Polyunsaturated fatty acids are particularly vulnerable to free radical attack because the carbon–hydrogen dissociation energies are lowest at bis-allylic methylene position. As a result, the hydrogen abstraction event that initiates lipid peroxidation is promoted, generating a carbon-centered lipid radical that then combines with oxygen to generate peroxyl (ROO•) and alkoxyl (RO•) radicals that, to stabilize, abstract hydrogen atoms from adjacent carbons. These reactions create additional lipid radicals that then perpetuate the lipid peroxidation chain reaction. Lipid peroxidation is extremely harmful to spermatozoa having a dramatic effect on both sperm movement and the competence of these cells for fertilization (7, 12). As a result, the levels of lipid peroxide formation recorded after the addition of a ferrous ion promoter to human spermatozoa are highly correlated with the functional competence of these cells as revealed by a suite of inverse correlations with sperm motility, viability, their capacity for prolonged survival in vitro, and their competence for sperm–oocyte fusion (12, 38).
The other major substrate for free radical attack in human spermatozoa is the DNA in the sperm nucleus and mitochondria. While mitochondrial DNA is the more vulnerable to DNA damage and is a sensitive marker of oxidative stress in the germ line (83), this DNA is destroyed shortly after fertilization, so its integrity is of little consequence to either the functionality of the spermatozoon or the normality of the resultant embryo. It is the nuclear DNA that is critical to embryonic development.
Oxidative DNA Damage in the Germ Line
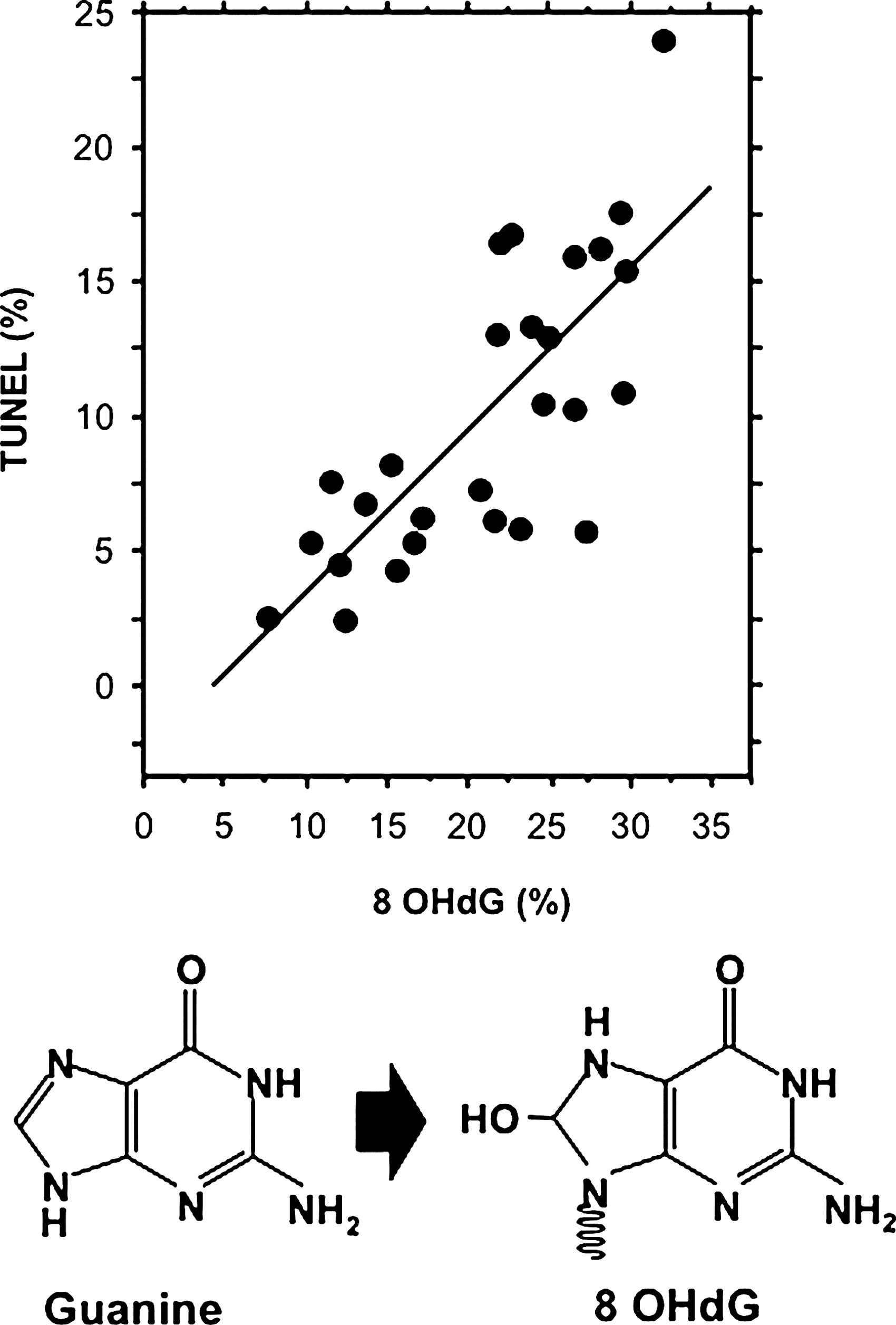
A small number of studies have examined the damaged nuclear DNA from human spermatozoa in a search for clues to its origins. These studies have revealed that the major base adduct present in human spermatozoa is 8OHdG, a marker of oxidative stress. The levels of 8OHdG expression in human spermatozoa are consistently found to be elevated in the spermatozoa of infertile men; moreover, the presence of this oxidized base adduct has been found to exhibit an extremely strong correlation with DNA damage as measured by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (29). Indeed, the correlation between TUNEL and 8OHdG formation is so strong that we (29) and others (81) have been forced to conclude that a majority of DNA damage in the male germ line is oxidatively induced (Fig. 4). Further, the presence of 8OHdG in human spermatozoa clearly interferes with the functionality of these cells since the presence of this adduct is correlated with defects in embryonic development leading to fragmentation of the blastomeres and impaired blastocyst formation (63).
Apart from 8OHdG, biochemical analyses of DNA from infertility patients have discovered two ethenonucleosides, 1,N6-ethenoadenosine and 1,N2-ethenoguanosine, which probably arise from a reaction between DNA and 4-hydroxy-2-nonenal, the main aldehyde released during lipid peroxidation (14). These findings are again consistent with oxidative stress being a major factor in the etiology of DNA damage in the male germ line.
In another study, uncharacterized bulky DNA adducts were found to be significantly more common in the spermatozoa of male factor infertility patients than in a cohort of healthy donors (44), and their presence was negatively correlated with sperm concentration and sperm motility. Polycyclic aromatic hydrocarbon–DNA adducts have also been found to be more prevalent in the spermatozoa of infertile versus fertile men (37). While details of the environmental and genetic factors responsible for creating these DNA adducts still await resolution, the one thing that does appear to be certain is that a vast majority of the spontaneous DNA damage seen in male infertility patients is oxidative in nature (Fig. 4).
This link between 8OHdG formation and DNA strand breakage could be interpreted in one of two ways. First, the link may be causative; that is, the formation of oxidative base adducts disrupts DNA integrity by labilizing the glycosyl bond that attaches the base to the ribose unit with the resultant generation of an abasic site. Abasic sites have a strong destabilizing effect on the DNA backbone and can subsequently result in strand breaks. Alternatively, the relationship may be indirect. Oxidative base damage and DNA fragmentation may simply be independent witnesses to the same fundamental underlying process—the ability of spermatozoa to undergo apoptosis. Under these circumstances the DNA strand breakage and oxidative stress may be independent facets of the same apoptotic process mediated by endonuclease activation and mitochondrial ROS generation, respectively. Such considerations have encouraged us to consider the detailed nature of apoptosis in the male germ line.
Oxidative Stress, Apoptosis, and DNA Damage in the Germ Line
The ability of germ cells to undergo apoptosis is expressed very early in life when the testes are differentiating and adjustments have to be made to achieve the optimal germ cell-to-Sertoli cell ratio for spermatogenesis to proceed normally. During this developmental process, excess premeiotic spermatogonia are removed by apoptosis. If this process is impaired by functional deletion of the proapoptotic protein, Bcl-2-associated X protein, or overexpression of antiapoptotic factors such as BclxL or Bcl-2, then a male infertility phenotype is generated associated with a perturbed germ cell to Sertoli cell ratio (77). Later in life, p53 and Fas are involved in the removal of germ cells that are damaged as a result of exposure environmental toxicants or chemotherapeutic agents (23). However, of greatest relevance to the present discussion is the concept that DNA damage in the spermatozoa of subfertile males is the result of an abortive apoptotic process. That is, that apoptosis was initiated during spermatogenesis but failed to run to completion because the extensive remodeling of germ cells after meiosis effectively removes the intracellular machinery needed to effect cell death.
Apoptosis in sperm
There is no doubt that spermatozoa have the potential to exhibit many of the features of apoptosis, including activation of caspases 1, 3, 8, and 9, annexin-V binding, mitochondrial generation of ROS, and DNA fragmentation (4, 20, 54, 70). Although many of the reagents that have been shown to induce apoptosis in somatic cells such as staurosporine, lipopolysaccharide, 3-deoxy-D-manno-octulosonic acid (Kdo), and genistein are ineffective these cells, they will default to the intrinsic apoptotic pathway in response to oxidative stress. Thus, exposure of human spermatozoa to hydrogen peroxide can readily trigger an apoptotic cascade characterized by the activation of caspase 3 and the appearance of annexin-V binding positivity (61). Further pre-exposure of human spermatozoa to the antioxidant, melatonin, will prevent this apoptotic response (33). We have also demonstrated that the activation of an apoptotic cascade after hydrogen peroxide exposure results in the stimulation of mitochondrial free radical generation and the subsequent induction of 8OHdG formation in the sperm nucleus (A. Koppers and R.J. Aitken, unpublished observations). This cascade of cause and effect involving oxidative stress, the activation of mitochondrial ROS generation, induction of oxidized DNA base adduct formation, and DNA fragmentation has been observed following (i) the direct addition of hydrogen peroxide to spermatozoa, (ii) the indirect creation of oxidative stress with radiofrequency electromagnetic radiation (28), and (iii) the activation of apoptosis using the phosphoinositide 3 kinase (PI3 kinase) inhibitor, wortmannin (A. Koppers and R.J. Aitken, unpublished observations).
We have also examined this apoptotic cascade to determine whether the induction of apoptosis is followed by the activation of endonucleases that then move into the sperm nucleus to induce DNA fragmentation. This analysis revealed that while the initial stages of the intrinsic apoptotic cascade are similar in spermatozoa and somatic cells, the former differs in one important respect from the conventional process. Thus, even though endonucleases may be released from the mitochondria (endonuclease G and apoptosis-inducing factor) or activated in the cytosol (caspase-activated DNase) during apoptosis, the physical architecture of spermatozoa prevents these nucleases from translocating to the nucleus. This limitation is due to two major factors: first, the chromatin is so densely packed that proteins cannot penetrate into its internal structure; second, the mitochondria and a majority of the cytoplasm are located in the sperm midpiece, whereas the chromatin is located in the sperm head (Figs. 1 and 5). As a result, these enzymatic mediators of apoptotic DNA cleavage cannot be directly involved in the cleavage of sperm DNA. Rather, apoptosis is associated with mitochondrial ROS and 8OHdG formation, which secondarily induces the nonenzymatic fragmentation of the DNA.
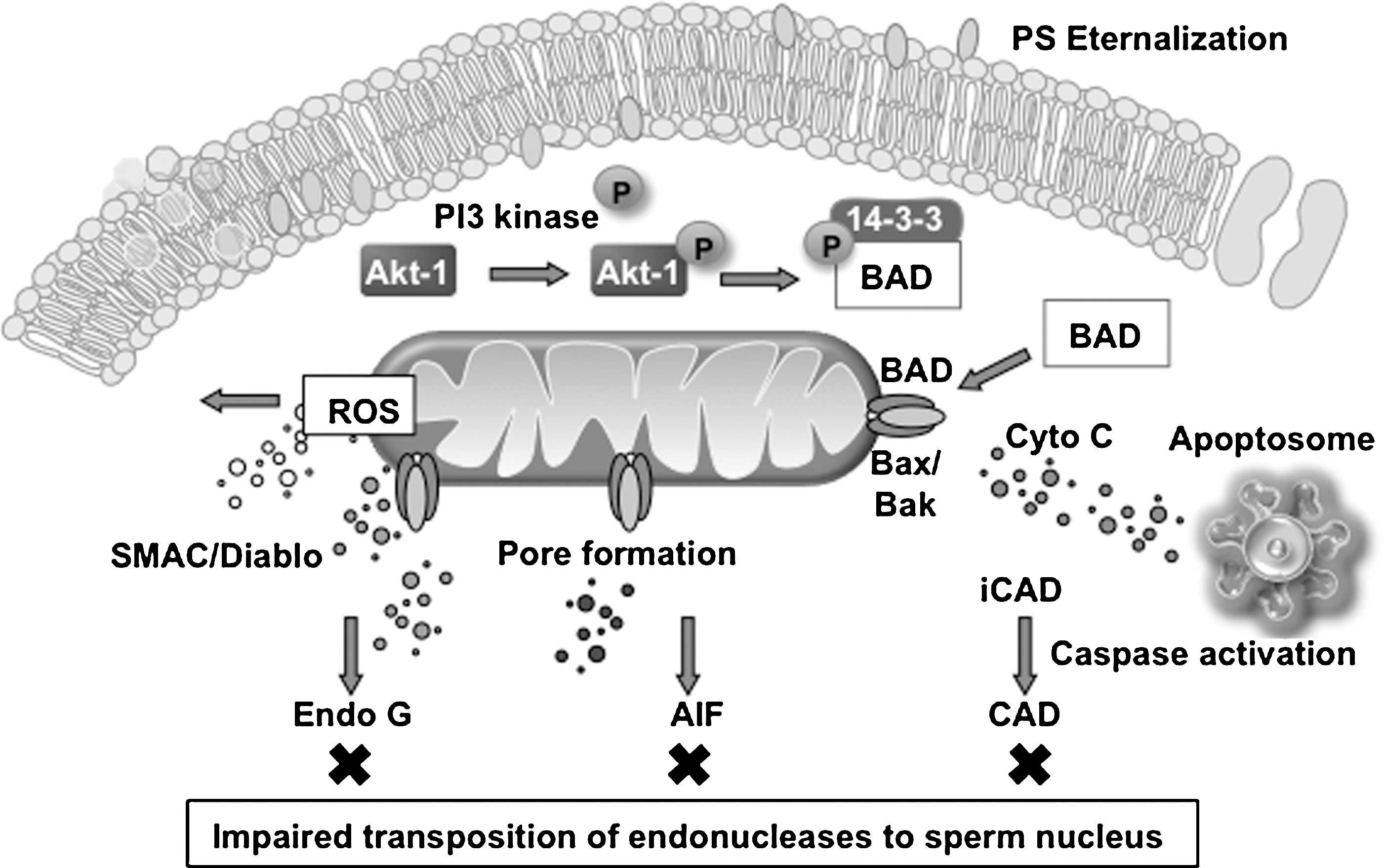
The only other possibility is that to compensate for the inability of endonucleases to move into the sperm chromatin during apoptosis, spermatozoa are endowed with an endonuclease already integrated into the body of the chromatin, which only has to be activated for DNA cleavage to commence. In this context, Sotolongo et al. (88) have described endogenous nucleases that would fit these criteria. It is possible that this endonuclease activity is involved in the final stages of apoptosis when cell viability is being lost and the plasma membrane is no longer in a condition to exclude divalent cations such as calcium (Fig. 5). This observation would be in keeping with our recent observation that a vast majority of TUNEL-positive cells in human semen samples are dead (66).
Life and Death in the Germ Line: Apoptosis versus Prosurvival
To date, no specific chemical triggers for apoptosis in human spermatozoa have been identified. Rather, entry into this pathway appears be induced by oxidative stress, frequently associated with cell senescence. One of the major reasons why spermatozoa have to undergo a regulated, programmed senescence relates to their ultimate disposal post coitum. Thus, after insemination, the female tract responds to the presence of millions of dead or moribund spermatozoa by triggering a massive leukocyte infiltration. The phagocytic activity exhibited by these cells must be silent; in other words, the spermatozoa must be efficiently phagocytosed and removed from the tract in the absence of an oxidative burst or the production of proinflammatory cytokines. This phagocytosis must be silent even though spermatozoa are patently foreign cells containing potent antigens; otherwise, every act of intercourse would be followed by an unwelcome inflammatory reaction. There are many examples of silent phagocytosis in biology and a common feature of this phenomenon is expression of apoptotic markers, such as phosphatidylserine (PS), on the surface of the target cell. This apoptotic marker is thought to instruct the phagocyte that the target cell should be engulfed in a nonphlogistic manner (56). It is therefore possible that the activation of this apoptotic cascade in oxidatively stressed spermatozoa is an adaptation that permits the efficient removal of these cells from the female tract by phagocytic leukocytes, without provoking an inflammatory response.
The second important purpose of apoptosis in the germ line is that it prevents damaged cells from participating in the process of fertilization. Thus, as soon as these cells sense that they are oxidatively damaged, they initiate an apoptotic cascade, one of the early features of which is a rapid loss of motility that prevents these cells from participating in fertilization. This preventative mechanism is not perfect; otherwise, we would not see paternally mediated defects in embryonic development or morbidity in the offspring. Further, this natural mechanism for preventing fertilization by defective human spermatozoa can, and is, circumvented by assisted conception therapy, particularly ICSI. In this light, selecting spermatozoa for ICSI on the basis that they do not express markers of apoptosis such as PS externalization might reduce the risk that DNA-damaged spermatozoa are used in assisted conceptions (80).
Although spermatozoa are supposed to engage the apoptotic process once they have entered the female reproductive tract, the DNA damage we see in the spermatozoa of male infertility patients could represent the premature entry of these cells into this pathway of programmed senescence while they are still being processed in the male reproductive tract. This hypothesis might explain why male infertility is so frequently accompanied by subclinical levels of leukocytic infiltration (76).
If entry into this truncated apoptotic cascade is the default pathway for spermatozoa, what does normally prevent them from engaging this process? An important attribute of this terminally differentiated cell is just how long it has to survive in vivo. Possessing virtually no capacity for membrane or DNA repair, this transcriptionally and translationally silent cell has to survive for up to a week in the epididymis waiting for ejaculation, followed (uniquely in our species where there is no estrus to synchronize the act of insemination with the moment of ovulation) by several more days in the female tract waiting for an egg to arrive. The maintenance of sperm viability throughout this prolonged solitary phase in its life history involves exposure to prosurvival factors generated by the female reproductive tract. The latter are thought to maintain cell viability by stimulating phosphorylation of the key regulators, PI3 kinase and Akt (protein kinase B). As long as these enzymes are phosphorylated, they can, in turn, phosphorylate a key constituent of the apoptotic cascade known as Bcl-2-associated death promoter (BAD). Phosphorylated BAD is bound to a regulatory protein called 14-3-3 and in this state cannot interact with the cell's mitochondria. However, if the cell should become senescent for any reason, then a reduction in PI3 kinase/Akt phosphorylation occurs, which leads to the dephosphorylation of BAD. The latter then escapes from the embrace of its 14-3-3 regulator and moves rapidly to the mitochondria, where it triggers pore formation, cytochrome C release, and the initiation of apoptosis (Fig. 6). The study of such prosurvival factors is still in its infancy, but one candidate to emerge from recent studies is prolactin. Spermatozoa possess several variants of the prolactin receptor (including their own unique isoform) and respond to the presence of this hormone with the stimulation of PI3 kinase/Akt phosphorylation and the prolongation of sperm survival in vitro (73).
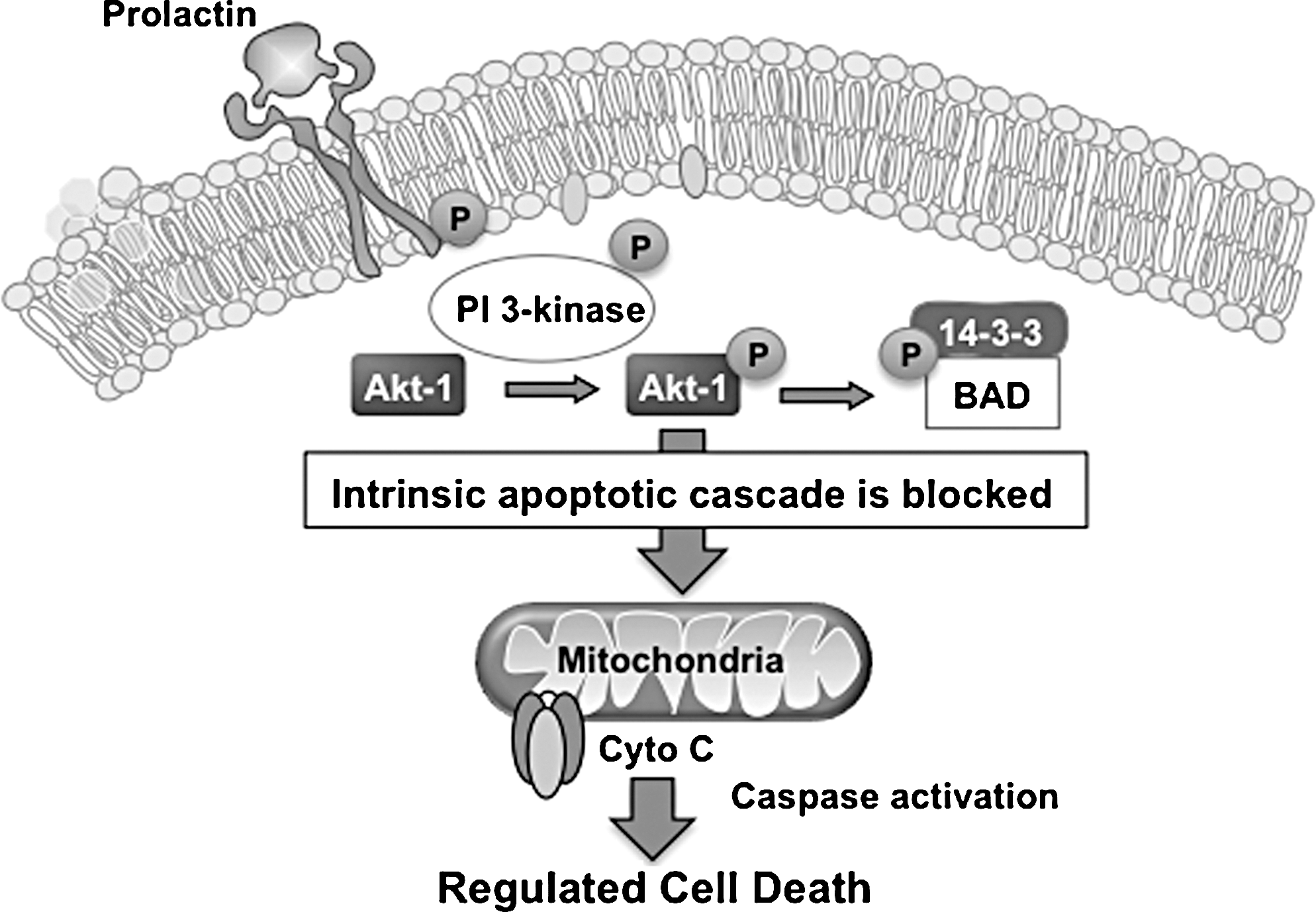
In summary, spermatozoa walk a tightrope between the opposing forces of prosurvival on the one hand and the intrinsic apoptotic cascade on the other. When the prosurvival pathway is weakened, the spermatozoa revert to an apoptotic state, which, in these cells, is a truncated affair that represents a form of programmed senescence allowing millions of redundant, moribund spermatozoa to be quietly and efficiently removed from the female tract after insemination, while reducing the risk that defective spermatozoa will participate in the process of fertilization (Fig. 5). Male infertility may involve the precocious entry of defective spermatozoa into this apoptotic pathway. The reason why these cells would be so defective that they have to prematurely enter apoptosis may have its roots in the quality of the underlying process of spermiogenesis.
Significance of Spermiogenesis in the Etiology of DNA Damage
Spermiogenesis, the process by which haploid round spermatids differentiate into spermatozoa, is a key event in the etiology of DNA damage in the male germ line (Fig. 7). During spermiogenesis the chromatin becomes extensively remodeled to compact the entire haploid genome into a 5 × 2.5 μm sperm head. In achieving this feat two important events occur, both of which are relevant to the etiology of DNA fragmentation in human spermatozoa. First, physiological DNA strand breaks are introduced by topoisomerase to relieve the torsional stresses involved in packaging the DNA during sperm differentiation. Normally, these physiological strand breaks are corrected by a complex process involving H2Ax phosphorylation, formation of poly(ADP-ribose) (PAR) by nuclear PAR polymerases and topoisomerase (65). It has been postulated by several authors that if the spermiogenesis should be disrupted for any reason, then the restoration of these cleavage sites might be impaired and the spermatozoa, lacking any capacity for DNA repair in their own right, will be released from the germinal epithelium still carrying unresolved physiological strand breaks. Experimental disruption of this repair process has clearly demonstrated the potential of such mechanisms to generate spermatozoa carrying a high frequency of DNA strand breaks. For example, knockout mice deficient in enzymes involved in PAR turnover (PAR polymerase 1 and PAR glycohydrolase) display DNA strand breaks associated with varying degrees of subfertility (65). Similarly, the transition proteins that move into the sperm nucleus during spermiogenesis between the removal of histones and the entry of protamines are thought to play a key role in maintaining DNA integrity. If these proteins are functionally deleted, then spermatozoa are generated with poor fertilizing potential, poor chromatin compaction, and high levels of DNA fragmentation (99). Such studies clearly indicate that functional disruption of chromatin repair mechanisms during spermiogenesis can result in the production of spermatozoa carrying high levels of DNA fragmentation. However, because disruption of these physiological nick-and-repair mechanisms during spermiogenesis can result in DNA-damaged spermatozoa, it does not mean that they are actually involved in the damage we see in male infertility patients.
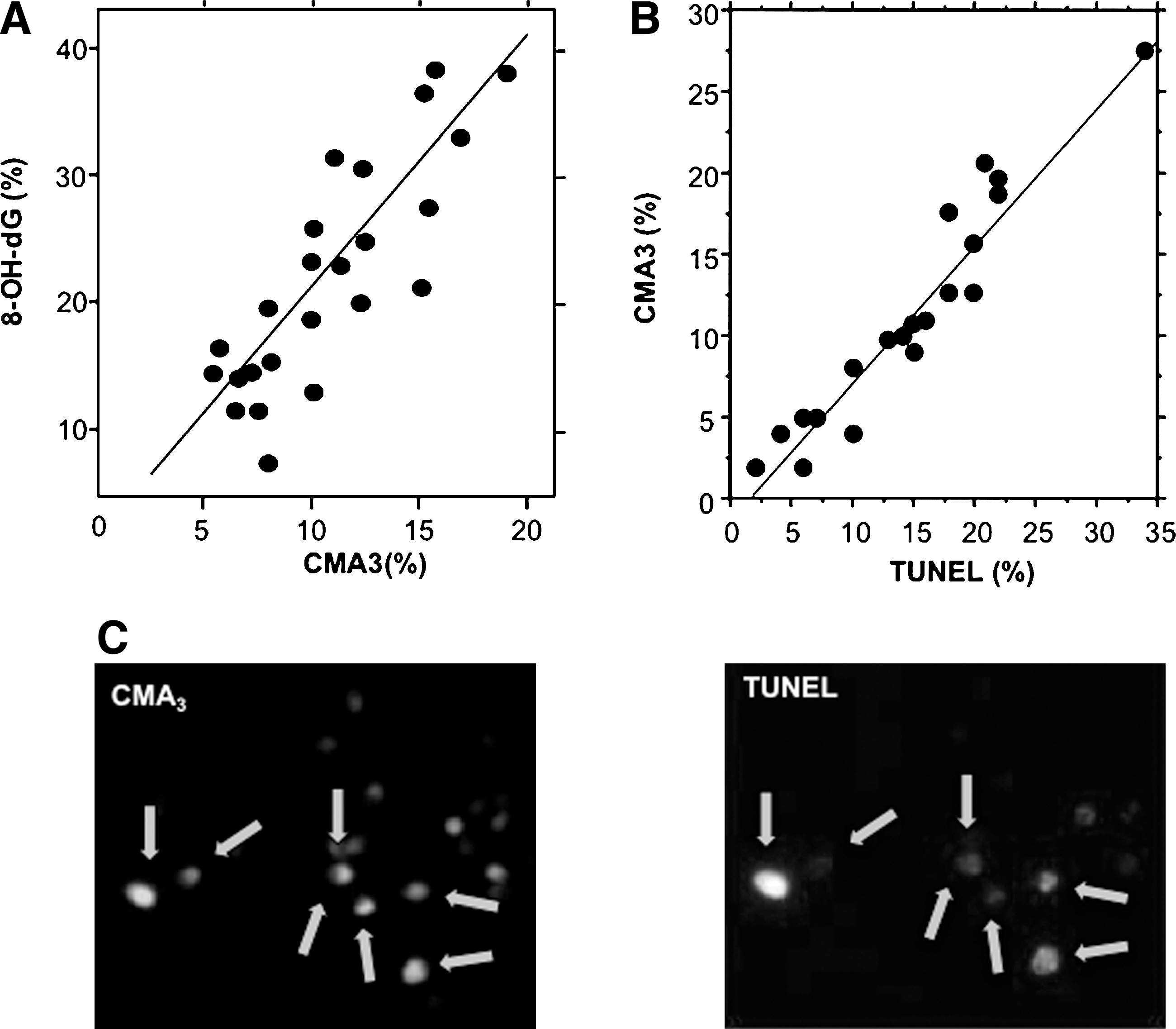
On the one hand, such a mechanism would not be consistent with the tight correlation that has been observed between DNA fragmentation and 8OHdG formation, which points to oxidative stress as the key factor driving this damage (29). On the other, there is abundant evidence linking sperm DNA damage in the patient population with the disruption of spermiogenesis, as evidenced by studies employing the fluorescent probe, chromomycin (CMA3). This probe binds to sperm DNA in the sites normally occupied by protamines (the minor groove of CG rich DNA); as a result, the amount of this dye taken up by sperm nuclei is negatively correlated with the efficiency of sperm protamination during spermiogenesis. Using this dye many independent laboratories have generated data demonstrating an excellent correlation between DNA fragmentation and poor chromatin remodeling during spermiogenesis (22, 29) (Fig. 7). This proposed link between defective spermiogenesis and DNA damage is further supported by the fact that several independent studies have also recorded correlations between DNA damage in human spermatozoa and elements of the conventional semen profile (specifically sperm count and morphology) that, in turn, reflect the efficiency of the spermatogenic process (4, 46).
Not only is the disruption of spermiogenesis correlated with DNA damage, but it is also specifically correlated with oxidative DNA damage as reflected by 8OHdG (29) (Fig. 7). We postulate that this relationship exists because the poorly remodeled chromatin detected by CMA3 is particularly vulnerable to oxidative attack by ROS originating from a number of potential sources, including infiltrating leukocytes, depleted antioxidant systems, and excessive free radical generation by the spermatozoa's own mitochondria. We further propose that of all these potential sources, the sperm mitochondria are the most important (54). Moreover, when mitochondrial ROS production is induced, it is frequently as part of an apoptotic cascade triggered by exposure to stress factors such as hydrogen peroxide, heat, or radiofrequency electromagnetic radiation (4, 28). Therefore, it seems reasonable to propose that defective spermiogenesis generates cells that are poorly protaminated and, as a result, possess regions of their chromatin that are still nucleohistone rich and vulnerable to oxidative attack. In addition, these defective cells have a tendency to undergo premature senescence characterized by the activation of an intrinsic apoptotic pathway during which mitochondrial ROS is activated and oxidative DNA damage induced (Fig. 7).
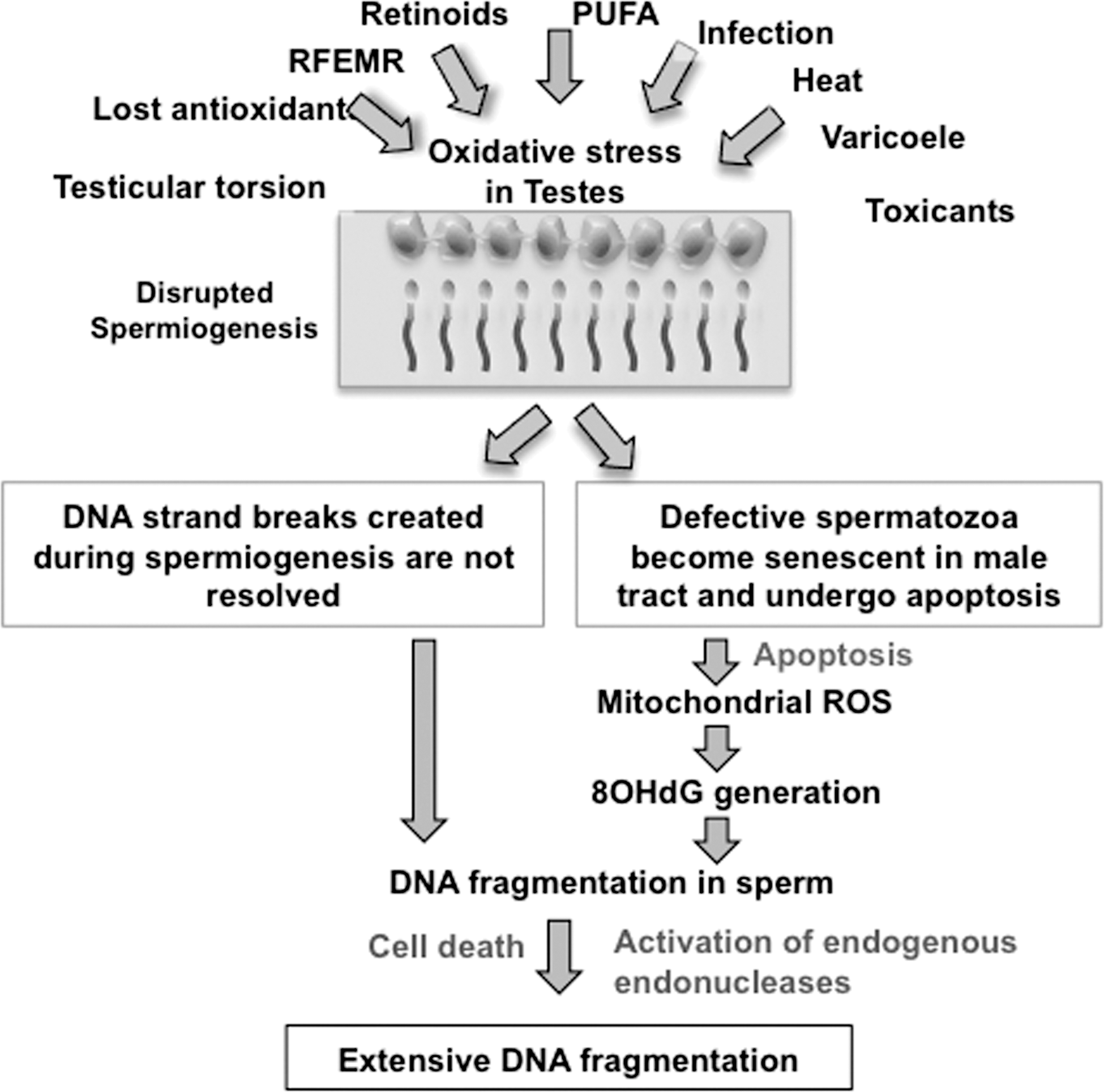
If this model for the induction of DNA damage in the germ line is correct, it suggests that the origins of this condition ultimately lie in defective spermiogenesis. Intriguingly, one of the major factors known to disturb spermiogenesis is oxidative stress. Thus, in a number of toxicological studies, spermiogenesis has been impaired in response to oxidative stress created by the systemic administration of such toxicants as cyclophosphamide (74), bacterial lipopolysaccharide (64), deltamethrin (47), streptozotocin (52), aroclor 1254 (13), methyl-parathion (71), or formaldehyde (69). Proof that oxidative stress is causally involved in the induction of defective spermiogenesis was secured by demonstrating the ameliorating impact of antioxidants, such as Satureja khuzestanica essential oil (74), lipoic acid (13), quercetin (52), and melatonin (69). In certain instances, antioxidant administration improved both testicular function and sperm DNA damage (74), reinforcing the notion that oxidative stress can underpin both the disruption of spermatogenesis and the induction of DNA damage in spermatozoa. In addition, the induction of oxidative stress with methyl parathion has been shown to specifically affect chromatin remodeling during spermiogenesis and induce DNA damage in spermatozoa (71). Such results encourage speculation that oxidative stress is a major determinant of the quality of spermiogenesis. When this process becomes disrupted, spermatozoa are produced that are vulnerable to oxidative stress, 8OHdG formation, and ultimately DNA fragmentation as a consequence of their susceptibility to apoptosis (Fig. 8). Powerful support for this hypothesis has been provided by Zubkova and Robaire (102). These authors found that the induction of systemic oxidative stress through the administration of the glutathione-depleting drug l-buthionine-[S,R]-sulfoximine both impaired spermiogenesis, as evidenced by an increase in CMA3 staining of the sperm chromatin, and increased DNA fragmentation in the spermatozoa.
As to why oxidative stress should so effectively disrupt spermiogenesis, it may be significant that this process is an object lesion in the translational control of developmental processes. By this stage of germ cell development, gene transcription has ceased with the result that spermiogenesis is driven by differential mRNA translation. Importantly, this process is highly susceptible to oxidative stress, which interferes with aminacyl-tRNA synthetase activity and reduces mRNA translation fidelity (60).
Clinical Considerations
It should be clear from this review that DNA damage in the male germ line has a profound effect on embryo quality (68), pregnancy loss (101), and the health and well-being of the offspring (5), and that most of this DNA damage is oxidatively induced (29, 81). The involvement of oxidative stress in the etiology of DNA damage in the germ line clearly has clinical relevance. For example, none of the culture media that are in current use for assisted conception therapy contain antioxidant scavengers to prevent the ROS generated by senescent apoptotic cells from impacting on the oocyte, the zygote, or the remaining spermatozoa in the inseminate. Similarly, no rigorous double-blind cross-over trials have been conducted on the possible use of oral antioxidant therapy as a prophylactic treatment to reduce the amount of DNA damage in human spermatozoa before the initiation of ART (90). The development of optimized antioxidant regimes capable of limiting DNA damage in the germ line will be a high priority task for the future.
The relationship between cell vitality and DNA damage is also highly relevant to the selection of individual spermatozoa for ICSI (66). Selection methods, such as hyaluronate binding, swim-up, or the removal of apoptotic cells, that increase motile sperm concentration are likely to significantly reduce the risk of DNA damage being present in the spermatozoon selected for injection. It should also be stressed that because of such selection procedures, the levels of DNA damage in the selected spermatozoon will not necessarily reflect the damage present in the ejaculate as a whole. Such a discrepancy may explain why the analysis of DNA damage in human sperm suspensions does not correlate with the outcome of ICSI therapy (87). However, the fact that such assays are correlated with the outcome of in vitro fertilization or intrauterine insemination therapy (87, 101) may be because the incidence of DNA damage in the ejaculate as a whole is reflective of the overall quality of the underlying spermatogenic process (24, 87). Determining whether DNA damage is present in the individual spermatozoon selected for ICSI is a challenge that might be met by simply increasing the sensitivity of the testing procedure. Thus, Simon et al. (87) were able to observe a relationship between DNA damage and pregnancy outcomes with ICSI if they increased the sensitivity of their COMET assay by the inclusion of an endonuclease (formamidopyrimidine DNA glycosylase) to cut the DNA at sites characterized by the presence of an oxidatively modified base (87).
Conclusions
High levels of DNA damage in human spermatozoa constitute a significant clinical issue because the presence of such lesions is associated with impaired fertility, an increased incidence of miscarriage, and an enhanced risk to the health of the offspring. DNA damage in spermatozoa is a particular issue for ART programs that are able to achieve conceptions in vitro that could never have occurred in vivo. Given the exponential increase in the uptake of ART in developed countries and the likelihood that DNA-damaged spermatozoa are frequently being used to achieve human conceptions within such programs, it is critical that we understand the etiology of this damage and take steps to control it.
The available evidence suggests that most of this DNA damage is oxidative and associated with impaired spermiogenesis and the enhanced generation of ROS by the spermatozoa. We hypothesize that this pathological state is frequently initiated by the creation of oxidative stress in the testes, as a result of such factors as age, testicular torsion, varicocele, cryptorchidism, and exposure to toxicants or excessive electromagnetic radiation (28, 29, 91, 100) (Fig. 8). The oxidative stress then perturbs spermiogenesis, possibly by interfering with the fidelity of protein translation (Fig. 8). This disruption of germ cell differentiation leads the generation of spermatozoa that may suffer from three kinds of defect: (i) their DNA may still retain nicks introduced by topoisomerase during the early stages of spermiogenesis to relieve the torsional stresses associated with DNA compaction, (ii) the DNA is also poorly compacted because of a lack of protamination and, for this reason, is vulnerable to oxidative attack, and (iii) these defective cells have a tendency to prematurely enter a state of programmed senescence (apoptosis), which involves the activation of mitochondrial ROS generation. The latter is self-perpetuating: lipid peroxides generated as a consequence of oxidative stress binding to mitochondrial proteins, disturbing the electron transport chain and stimulating yet more ROS generation. As a net consequence of this cascade, the spermatozoa lose their mitochondrial membrane potential, express PS on the sperm surface, activate caspases, lose motility, and exhibit oxidative DNA damage that leads to the creation of abasic sites and DNA fragmentation.
In the future, research in this area will focus on resolving the causes, consequences, and prevention of DNA damage in the germ line. In terms of causation, it will be important to determine whether the DNA damage we see in ejaculated spermatozoa originates before or after spermatozoa are released from the germinal epithelium. Similarly, we do not yet have a true perspective on the extent to which DNA damage in spermatozoa is simply an indirect reflection of impaired spermiogenesis, the direct result of environmental factors acting on the germ cells, activation of intrinsic apoptosis, or a consequence of depleted antioxidant protection in the male tract—or combinations of all these factors. We also do not know whether the tendency to exhibit sperm DNA damage is a constant phenomenon that is genetically determined and appears at puberty, or whether it responds in a more dynamic fashion to changes in the environment.
In terms of the consequences of such damage it would be interesting to know the relationship between DNA damage and telomere integrity. The telomeres are arranged around the periphery of the sperm nucleus in a position that suggests that they might be vulnerable to oxidative attack (97). The selection of spermatozoa using a swim-up procedure that selects for motile cells exhibiting low levels of DNA damage is known to isolate spermatozoa exhibiting long telomeres (82). Whether spermatozoa suffering from oxidative DNA damage possess shortened telomeres is unknown but may be very relevant to health and particularly the longevity of the progeny. Interesting relationships may also exist between neurological conditions that are frequently driven by oxidative stress in the brain and oxidative stress in the testes. For example, amyotrophic lateral sclerosis is a devastating neurodegenerative disorder characterized by a loss of motor neurons, muscle wasting, paralysis, and death. The etiology involves oxidative stress frequently associated with elevated markers of oxidative damage and mutations in the SOD1 gene (19). Intriguingly, amyotrophic lateral sclerosis males are subfertile (50). So, decades before the appearance of a neuropathology in response the oxidative stress suffered by such patients, there is a fertility readout of their condition. Whether the brain and the testes are similarly affected with other kinds of neurodegenerative disease involving oxidative stress is an intriguing possibility. A recent report of massive amyloid deposition in the testes of azoospermic patients suggests that this concept may be worthy of further investigation (84).
Footnotes
Acknowledgments
The authors gratefully acknowledge the support of the Australian Research Council, the National Health and Medical Research Council, and the NSW Department of State and Regional Planning for financial support.