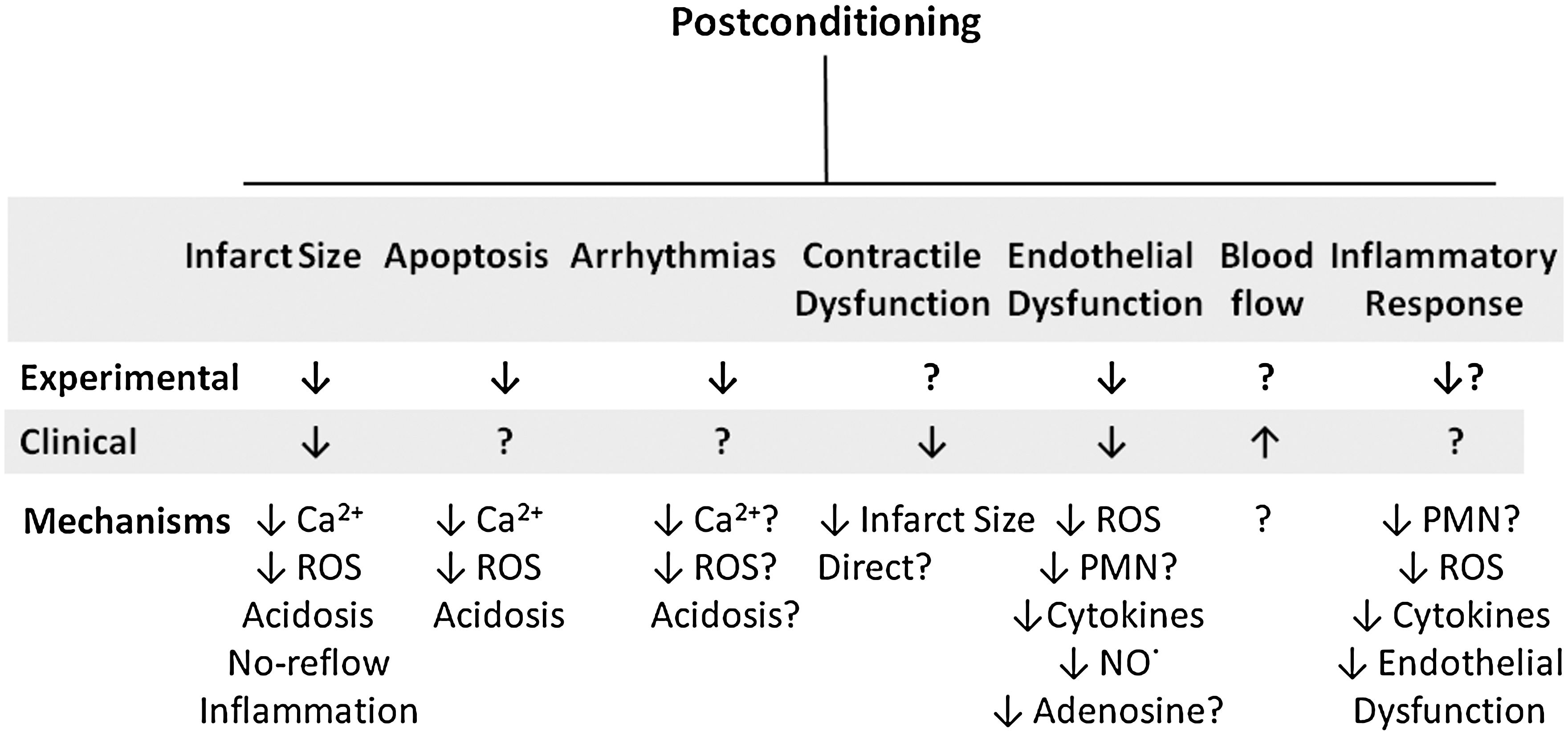
Abstract
Reperfusion is the definitive treatment to reduce infarct size and other manifestations of postischemic injury. However, reperfusion contributes to postischemic injury, and, therefore, reperfusion therapies do not achieve the optimal salvage of myocardium. Other tissues as well undergo injury after reperfusion, notably, the coronary vascular endothelium. Postconditioning has been shown to have salubrious effects on different tissue types within the heart (cardiomyocytes, endothelium) and to protect against various pathologic processes, including necrosis, apoptosis, contractile dysfunction, arrhythmias, and microvascular injury or “no-reflow.” The mechanisms by which postconditioning alters the pathophysiology of reperfusion injury is exceedingly complex and involves physiological mechanisms (e.g., delaying realkalinization of tissue pH, triggering release of autacoids, and opening and closing of various channels) and molecular mechanisms (activation of kinases) that affect cellular and subcellular targets or effectors. The physiologic responses to postconditioning are not isolated or mutually exclusive, but are interactive, with one response affecting another in an integrated manner. This integrated response on multiple targets differs from the monotherapy approach by drugs that have failed to reduce reperfusion injury on a consistent basis and may underlie the efficacy of this therapeutic approach across species and in human trials. Antioxid. Redox Signal. 14, 791–810.
Introduction
The Postconditioning Algorithm: Number of Cycles versus Duration of Occlusion-reperfusion
Postcon was defined by Zhao et al. (156) as brief periods of reperfusion alternating with brief periods of ischemia applied at the onset of reperfusion (Fig. 1). The first postcon study demonstrating a protective effect was performed in the canine model by using an algorithm of three cycles of [30 s of reperfusion + 30 s of reocclusion (ischemia)] applied immediately at the onset of reperfusion. Cardioprotection with postconditioning has subsequently been confirmed in multiple models and species by using algorithms that vary in terms of both the number of cycles applied and the duration of the reperfusion/reocclusion episodes (Table 1).
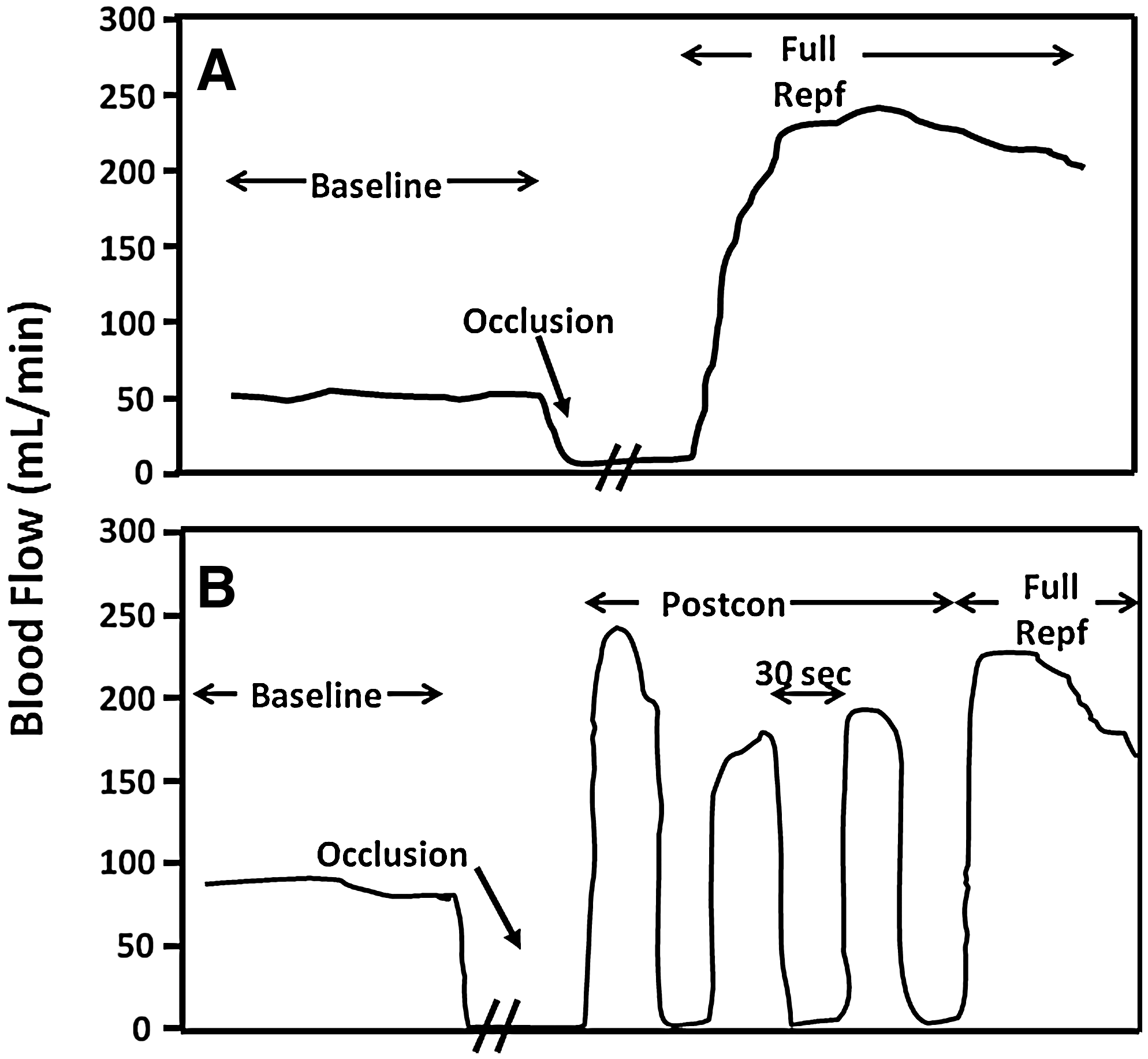
The efficacy of postcon is influenced primarily by the mechanics of the algorithm, including the number of cycles, the duration of each reperfusion/reocclusion period, and the total duration of the postcon stimulus. In addition, the importance of prompt initiation of the postcon stimulus at the onset of reperfusion has been demonstrated by studies in rats and rabbits in which the protective effect of postcon was not observed if the postcon algorithm was begun after a delay (114, 147). However, other factors, such as the duration and the severity of the index ischemia, the experimental setting (in vivo vs. in vitro), and laboratory variables (e.g., temperature, anesthetic agent) also modulate the effect of postcon. Table 2 summarizes studies in which differing effects on infarct size were reported despite the use of protocols previously shown to be effective in the hands of other investigators (24, 61). These disparities may be due to several factors, including (a) incomplete implementation of the reperfusion/ischemia sequences in the algorithm, (b) differences in delaying the postcon algorithms, and/or (c) variability encountered in protocols between laboratories (e.g., type of anesthetic, depth of anesthesia, or monitoring of blood pressure).
No single, “optimal” postcon protocol has been identified (i.e., no one protocol fits all species, which seems logical when comparing kinetics and development of infarction among species) (19, 123). It has been suggested that smaller animals require shorter durations of the reperfusion/reocclusion phases of the postcon protocol, whereas with increasing body size, algorithms with more prolonged periods of reperfusion/reocclusion are required. However, even this broad “consensus” statement has exceptions. In the pig, a protocol of 10 cycles of 15 s of reperfusion/15 s of reocclusion (88) and six cycles of 20-s reperfusion/20-s reocclusion (126) was as effective as eight cycles of 30 s reperfusion/30 s of reocclusion (121), whereas another study reported a greater infarct-size reduction by using eight cycles of 30-s reperfusion/30-s reocclusion (54). In the latter study by Iliodromitis et al. (54), increasing the number of cycles from four cycles to eight cycles of 30 s of reperfusion/30 s of reocclusion reestablished infarct-size reduction, and further demonstrated the importance of the number of cycles needed for postcon to reach the threshold for protection.
Increasing the number of cycles in the rat from three to six caused no further reduction in infarct size (66), whereas increasing the number of cycles to 60 failed to reduce infarct size (132). This demonstrates not only a lower threshold for protection but also the existence of an upper limit beyond which postcon is no longer effective.
Although it has been suggested that the optimal postcon cycle depends on maintenance of tissue acidosis or delaying the realkalinization of the previously ischemic tissue (for details, see the article by Garcia-Dorado et al. in this Forum), we still do not understand what makes one postcon protocol reach the threshold for protection while other protocols fail. Further studies exploring the physiological effects of postcon with regard to both number of cycles and the duration of the reperfusion/reocclusion phases are needed. Increased insight will be gained when the mechanisms contributing to cardioprotection by each phase of the algorithm are understood.
Infarct Size
Infarct-size reduction, the ultimate end point of myocardial I-R (92, 112), is the hallmark of the physiological effects of postcon. The infarct area may consist of myocardium that has died of necrosis or apoptosis, or by both processes. Accordingly, it is important that postcon addresses both pathways to cell death.
Since the original study by Zhao et al. (156) reporting significant reduction in infarct size by postcon (Fig. 2), myocardial salvage expressed as infarct-size reduction has been demonstrated in every species tested: mouse (65); rat (47); rabbit (147); canine (156); swine (54); monkey (145), and, most promising, in humans (127). Furthermore, the infarct-sparing effect is consistent across different experimental conditions; reduction of cell necrosis or myocardial infarct size has been demonstrated in isolated cardiomyocytes (129), isolated perfused hearts in the nonworking (Langendorff) (146) and working modes (138), and under conditions of global (10) or regional ischemia (15). In addition, even though postcon was targeted to treat patients with acute myocardial infarction (AMI), the broad-spectrum effect of postcon has been translated into other models of I/R. For example, Lauzier et al. (80) demonstrated a protective effect of postcon in a heart-transplantation model with 4 h of cold ischemia, which has been applied successfully in noncardiac organs, including the brain (151) and kidney (28).
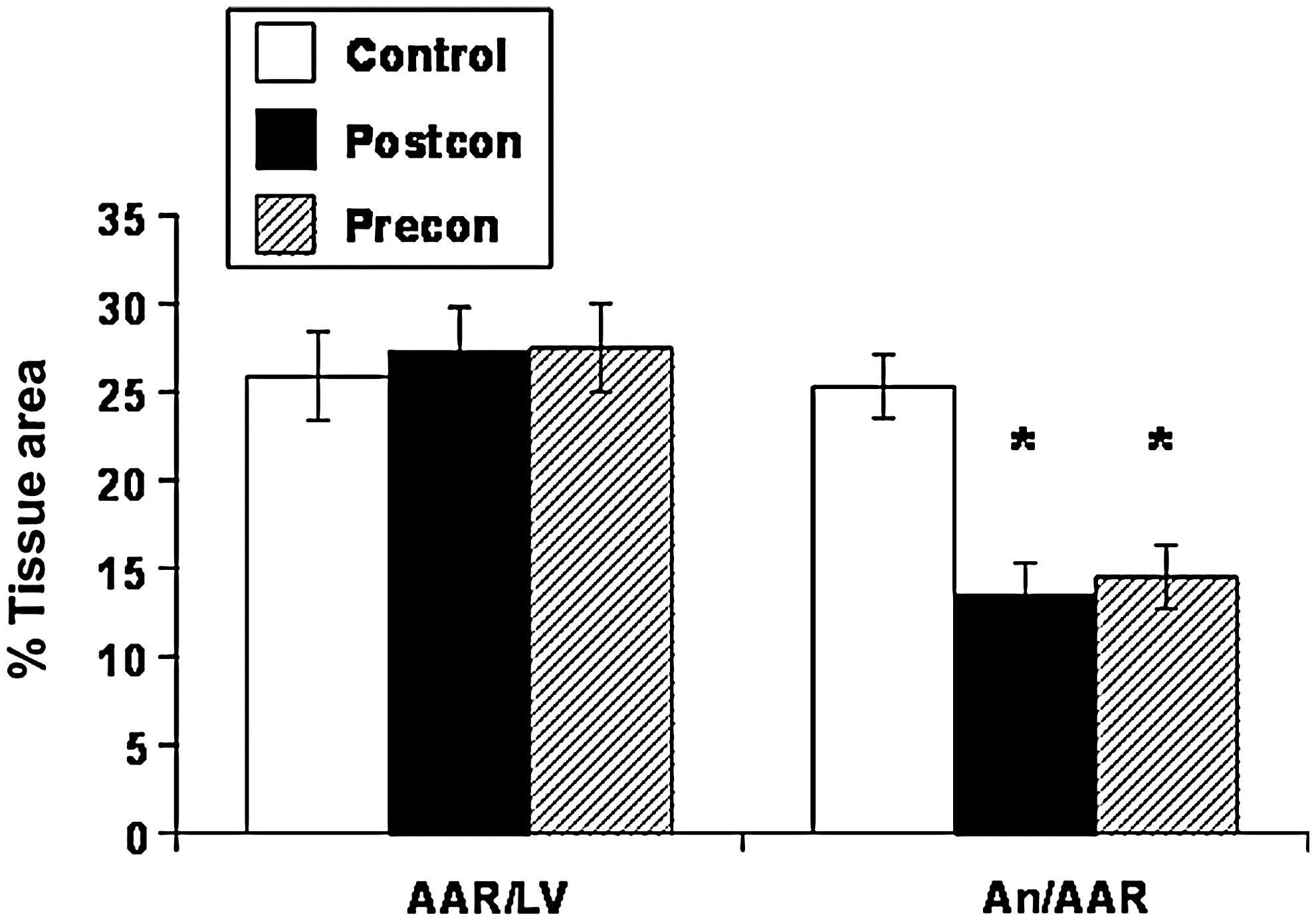
Not surprisingly, variability in the magnitude of myocardial salvage is observed among experimental studies. Some of this variability may be attributed to species differences, and the fact that not a single linear relation exists between infarct size and duration of ischemia across all species. For example, 30 min of coronary artery occlusion in rats produces infarcts occupying approximately 50% of the at-risk myocardium, whereas in dogs and pigs, this would produce relatively small necrotic areas. Postcon salvages only 25–30% of the necrotic area in rats, compared with more than 50% in the canine model (47, 156). A 30% reduction in infarct size is similar to that achieved by postcon in humans undergoing AMI (18, 94, 127, 144). Differences within the same species are more difficult to resolve and may relate to the various factors determining necrosis and infarct size. Some studies suggest that longer durations of ischemia raise the “threshold” of postcon (e.g., longer periods of the reperfusion/ischemia segments of the algorithm, or a greater number of cycles, are required to exert infarct-size reduction).
Postcon has, in some studies, failed to attenuate infarct size with longer durations of ischemia, indicative of a potential ceiling to its cardioprotective effects (84, 132). However, infarct-size reduction with postcon is preserved in other studies despite index ischemia lasting up to 3 h (154). Rodriguez-Sinovas (121) reported that postcon is protective only after 60 min, but not 48 min, of ischemia. The impact of longer periods of index ischemia on the protective effect of postcon has not been fully elucidated, and the demonstration of a threshold of protection over a time course of ischemia still must be identified.
A few studies fail to report a reduction of infarct size by postcon (24, 124) or report a paradoxic increase in infarct size with postcon when applied after shorter durations of ischemia (96, 97). For example, Manintveld et al. (97) report an increase in infarct size from 38 ± 3% to 50 ± 3% by using an in vivo rat model with 30 min as the index ischemia. Negative results may also be related to incomplete or imprecise postcon algorithms. On reperfusion, coronary arteries can go into spasm or cannot reverse the compression achieved by the ligature, thereby failing to reestablish the blood flow that constitutes the other half of the postcon algorithm. Few of these negative studies have actually examined whether successful reperfusion during the postconditioning algorithm was achieved.
It is still unclear whether postcon provides as powerful protection as the “gold standard” maneuver of preconditioning (precon). Potentially, precon has the ability to target both ischemia and reperfusion, whereas postcon targets only reperfusion (48). In this regard, notable differences in the efficacy of pre- and postcon have been reported in the rat; that is, Kin et al. (64) demonstrated a 67% reduction in infarct size with precon, whereas postcon afforded a 23% reduction in infarct size. However, other studies have compared the effects of pre- and postcon and found similar protection by both strategies.
Apoptosis
Apoptosis is genetically programmed cell death in which cells self-destruct in response to coordinated signals, as opposed to necrosis, which is an acute and explosive cell death. Some evidence suggests that apoptosis is initiated during ischemia and executed during reperfusion (35). Mitochondria figure prominently in the pathway to apoptosis, as the organelle serves as a molecular switch directing the cell to pursue an apoptotic pathway or a necrotic pathway. Apoptosis is triggered when the voltage- and Ca2+-dependent mitochondrial permeability transition pore (mPTP) opens, causing rapid collapse of the mitochondrial membrane potential and impaired ATP synthesis. In addition, opening of the mPTP permits an influx of solutes up to 1,500 kDa in size; this results in matrix swelling, rupture of the mitochondrial membrane, release of cytochrome c, and subsequent activation of downstream caspases. Opening of the mPTP occurs in the presence of membrane depolarization, high calcium levels, inorganic phosphates and reactive oxygen species (ROS), and normalized tissue pH. The role of the mPTP, ROS, and realkalyzation of the reperfused tissue is discussed in depth by other authors in this forum.
Sun et al. (129) first demonstrated that postcon reduced myocardial apoptosis (evaluated by TUNEL staining and DNA laddering) in primary cultured rat neonatal cardiomyocytes exposed to 3 h of hypoxia and 6 h of reoxygenation. The cardiomyocytes were postconditioned by 5 min of reoxygenation and 5 min of hypoxia, repeated for three cycles, applied before full reoxygenation. Postcon of hypoxic cardiomyocytes reduced the number of apoptotic cells, and also attenuated the triggers of apoptosis (i.e., intracellular Ca2+ and the generation of high levels of reactive oxygen species) relative to a control group exposed to only full reoxygenation (128, 129). Later, Wang et al. reported that postcon also reduced the generation of the oxidant peroxynitrite in hypoxic-reoxygenated adult rat cardiomyocytes (142). Treatment with the peroxynitrite scavenger uric acid reduced peroxynitrite levels. Because peroxynitrite is derived from the biradical reaction between ·O2 and nitric oxide (NO), this study gave further support for the concept that postcon reduces apoptosis by attenuating ·O2 generation. The antiapoptotic effects of postcon were also demonstrated in an in vivo rat model of regional coronary artery I-R. By using the externalization of phosphatidylserine on cell membranes during early apoptosis as a specific ligand for annexin V, Taki and colleagues (131) reported that 99mTc-annexinV uptake in the AAR myocardium was suppressed by ∼50% with postcon relative to a control group with full reperfusion. Interestingly, precon suppressed 99mTc-annexin V binding in at-risk myocardium to a similar extent.
Cytokines may activate neutrophils and vascular endothelium and may trigger the generation of ROS (see the section later on inflammatory response). Cytokines may also contribute to the pathogenesis of apoptosis after I-R. Kin et al. (64) reported in anesthetized rats subjected to 30 min of left coronary artery occlusion and 3 h of reperfusion that postcon (three cycles of 10 s reperfusion/10 s of reocclusion applied at the onset of reperfusion) reduced the translocation of nuclear factor (NF)-κB to the nucleus, decreased the release of tumor necrosis factor (TNF)-α, and attenuated the generation of ROS during reperfusion, all of which were found to be associated with a reduction in apoptosis in the at-risk myocardium. In addition, postcon reduced caspase-3 activity and the appearance of DNA fragmentation (ladders) in the AAR myocardium. In other studies, Penna et al. (110) showed in isolated perfused rat hearts subjected to 30 min of ischemia and 2 h of reperfusion that postcon decreased the proapoptotic factors cytochrome c and caspase-3, while increasing the cytosolic antiapoptotic regulator proteins Bcl-2 and Pim-1 relative to hearts with full reperfusion only. Therefore, postcon rebalanced the pro- and antiapoptotic regulating proteins favoring less apoptosis. Furthermore, mitochondrial morphology was preserved by postcon. Li and colleagues (87) further emphasized the importance of mitochondria in postcon's antiapoptotic effect in a report showing that postcon prevented the decrease in endogenous ARC (apoptosis repressor with caspase recruitment domain) protein levels observed after hypoxia/reoxygenation.
Clinically, the presence of apoptosis must be shown by surrogate plasma markers. A report by Zhao et al. (155) used soluble Fas and Fas ligand (FasL) as markers of apoptotic activity in patients with AMI undergoing percutaneous coronary intervention (PCI). This study demonstrated that Fas and FasL levels were significantly increased at baseline in patients with AMI compared with those in a control group. With the 60-s postcon algorithm, postcon decreased plasma FasL at day 7 after restoration of reperfusion, but the 30-s algorithm did not.
Incidence of Reperfusion Arrhythmias
Reperfusion of a previously occluded coronary artery not only causes de novo injury in the form of necrosis, apoptosis, and endothelial dysfunction, but can also initiate reperfusion arrhythmias (98). That reperfusion arrhythmias could be terminated by repetitive occlusions of the coronary artery was first reported in a case study in 1994 (42). Subsequently, Na et al. (102) demonstrated that a ventricular premature beat (VPB)-driven protocol reduced the incidence of ventricular fibrillation (VF), whereas a fixed “postcon” protocol of 5-s reperfusion and 35-s reocclusion of the coronary artery failed to reduce the incidence of VF. Indeed, the term postconditioning was first introduced by Na et al. (102) with respect to prevention of arrhythmias. Galagudza et al. (37) and Sasaki et al. (122), by using postcon periods of 2 and 5 min, respectively, demonstrated that postcon completely terminated reperfusion-induced ventricular arrhythmias. Halkos et al. (47) reported that postcon was associated with a lower frequency of ventricular dysrhythmias in the in vivo canine model. Kloner et al. (67) used a postcon protocol of four cycles of 20-s reperfusion/20-s reocclusion in the in vivo rat “stunning” model of 5-min ischemia and 10-min reperfusion. Postcon significantly reduced the frequency of ventricular tachycardia (VT), the duration of VT, and the incidence of VPB. Dow et al. (23) tested whether postcon reduced arrhythmias by endogenous signaling pathways relevant to cardioprotection (i.e., PI-3 kinase, Akt, and by G protein–coupled receptor agonists). However, the antiarrhythmic effect of postcon was not abolished by infusion of inhibitors of PI3-K, adenosine, or mitochondrial KATP channels, all of which are known to be involved in the antinecrotic effect of postcon.
Taken together, these studies support postcon as a powerful antiarrhythmic intervention. Our current knowledge regarding the mechanisms of this antiarrhythmic effect is limited, but may involve delayed recovery of pH, and attenuation of intracellular calcium accumulation and ROS production, all of which are known to influence arrhythmogenesis (98, 129, 156).
Global or Regional Myocardial Blood Flow, Microvascular Injury, and No-Reflow
Restoring reperfusion to the ischemic segment is the definitive strategy to reduce infarct size. Although the involved coronary artery may be opened by thrombolysis and angioplasty with or without deployment of a stent, blood flow may not be restored to all areas of the microvasculature in the previously ischemic myocardium (57), a situation that is associated with dismal outcomes (100). This lack of blood flow after transient ischemia was first observed in the heart by Krug et al. (75) and in the brain by Ames et al. (2) in 1968, who coined the term “no-reflow phenomenon.” The no-reflow phenomenon was described in detail in the heart by Kloner et al. (68). The implication of impaired postischemic blood flow is that the nutritional blood supply is inadequate to sustain contractile function, and the decrease may be severe enough to threaten viability of the involved myocardium. Reducing the no-reflow area may translate into myocardial salvage, smaller infarcts, and may translate further into less-adverse remodeling and heart failure.
Surprisingly, little has been done experimentally to characterize the effects of postcon on myocardial blood flow or to determine mechanisms of protection related to improved blood flow. Zhao et al. (156) measured myocardial blood flow in the in vivo canine hearts reperfused abruptly or by using a postcon algorithm of three cycles of 30-s reperfusion/30-s reocclusion applied immediately before full reperfusion. Despite a greater than 95% decrease in myocardial blood flow in the area at risk during left anterior descending occlusion, an approximate twofold increase in blood flow over baseline values was noted in all groups at 15 min of reperfusion, which subsequently decreased to near-baseline values. However, the investigators did not identify potential no-reflow areas within the area at risk by the various color or fluorescent dye techniques available, and therefore did not have the appropriate resolution to identify areas of severe blood-flow defects. Similarly, the study by Halkos et al. (47) did not show a defect in postischemic myocardial blood flow in the area at risk after 3 h of reperfusion. However, a significant reduction in postischemic perfusion was observed by Mykytenko et al. (101) in the subepicardium and subendocardium of the area at risk after 3 h of reperfusion, and in the subendocardium only at 24 h in the canine model, suggestive of a low-reflow state. Postcon did not alter this impaired regional blood flow.
Hale et al. (46) used a rabbit model of I-R more systematically to elucidate whether postcon reduces the area of no-reflow. Neither a postcon protocol of four cycles of 30-s reperfusion/30-s reocclusion nor a protocol of four cycles of 60-s reperfusion/60-s reocclusion reduced the area of no-reflow. However, neither protocol reduced infarct size, making it difficult to distinguish between lack of efficacy with regard to no-reflow versus an unsuccessful postcon protocol. In contrast, Zhao et al. (154), by using a mini-swine model of severe ischemia, caused by 3 h of coronary artery occlusion and 2 h of reperfusion, observed a 23% reduction in infarct size and a 33% reduction in the area of no-reflow delineated by thioflavin S. The role of postcon on postischemic blood flow remains unresolved at this point, and requires studies that focus primarily on the no-reflow phenomenon.
Clinical studies have measured coronary blood flow after postcon during PCI procedures and show a more beneficial effect of postcon on postischemic blood flow. Laskey (78) first reported postischemic blood flow in a small (n = 17) group of patients assigned to receive standard care versus an “ischemic conditioning” stimulus consisting of two cycles of 90-s inflation of the angioplasty balloon followed by 3 to 5 min of reperfusion (balloon deflation). The control cohort had a single 90-s balloon inflation before withdrawal of the catheter. This “ischemic conditioning” maneuver was associated with an increased peak blood flow velocity, diastolic/systolic velocity ratio and coronary blood flow velocity reserve (Doppler flow wire) compared with the control cohort. The author concluded that “conditioning” improved postischemic coronary blood flow. Staat et al. (127) used blush grade, which is the speed with which contrast is washed out of the myocardium of interest, as a marker of myocardial reperfusion after the initial period of reflow (116) in patients after standard PCI or postcon. They observed that, on average, the blush grade was 25% greater in patients who were postconditioned by four cycles of 60-s deflation/inflation of the angioplasty balloon in the target vessel than the control group. In agreement with this study, Ma et al. (94) found that postischemic coronary blood flow (corrected TIMI frame count) in the target vessel was greater in AMI patients randomized to the postcon group, which was associated with lower plasma creatine kinase (CK)- MB and markers of lipid peroxidation by ROS. Thus, although these data suggest that postcon has a favorable effect on recovery of microvascular perfusion after relief of ischemia, further experimental and clinical studies are needed to establish whether postcon attenuates microvascular injury and no-reflow.
Global or Regional Postischemic Contractile Function
Short periods of ischemia followed by reperfusion that result in reversibly injured myocardium with no evidence of infarction demonstrate a transient impairment of regional and global myocardial contractile function (50), or “stunning” (12). Postischemic stunning may last for days to weeks, despite full restoration of coronary blood flow (9), depending on the severity and duration of the coronary artery occlusion, and the adequacy of myocardial blood flow. The name “stunning” has been broadened to include myocardium that has endured more-prolonged periods of ischemia: for example, ≥3 h followed by reperfusion in which necrosis is observed but the adjacent myocardium is viable but dysfunctional (70). Many causes of myocardial “stunning” may be relevant to postcon, including (a) oxygen radicals, particularly hydroxyl radicals, and derivatives such as peroxynitrite generated during the first few minutes of reperfusion (8); (b) bioavailability of Ca2+ or the sensitivity of the contractile apparatus to Ca2+ or both; and (c) molecular abnormalities in the contractile proteins, such as breakdown of troponin I by proteolysis. These mechanisms are not mutually exclusive, and likely interact in ischemic/reperfused tissue. However, the generation of oxygen radicals and alterations in intracellular Ca2+ are events that occur during early reperfusion, suggesting that a component of contractile dysfunction is due to reperfusion injury. Because postcon reduces both of the major contributing mechanisms to postischemic “stunning” (i.e., superoxide generation and Ca2+ accumulation), it seems likely that postcon would improve postischemic regional contractile function.
Unlike infarct size and apoptosis, relatively little investigative work has been done on the effects of postcon on recovery of regional contractile function, either systolic or diastolic, in the ischemic/reperfused territory. In the original article introducing postcon, Zhao et al. (156) reported that postcon did not improve systolic shortening or regional segmental work in the at-risk myocardium relative to a control group reperfused abruptly (Fig. 3). Similar results were reported by Halkos et al. (47) by using the same model; this study also reported that postischemic regional contractile function was not improved by the combination of precon and postcon. In a model of stunning, Couvreur et al. (16) reported that three different protocols of postcon (four cycles of either 15-s reperfusion/15-s reocclusion, 30-s reperfusion/30-s reocclusion, or 60-s reperfusion/60-s reocclusion) following a 10-min coronary artery occlusion did not improve left ventricular free wall thickening relative to a control group that was abruptly reperfused. Moreover, a postcon algorithm of 30-s reperfusion/30-s reocclusion—an algorithm that was effective in reducing infarct size in this model—did not attenuate stunning in a rabbit model of coronary artery occlusion/reperfusion.
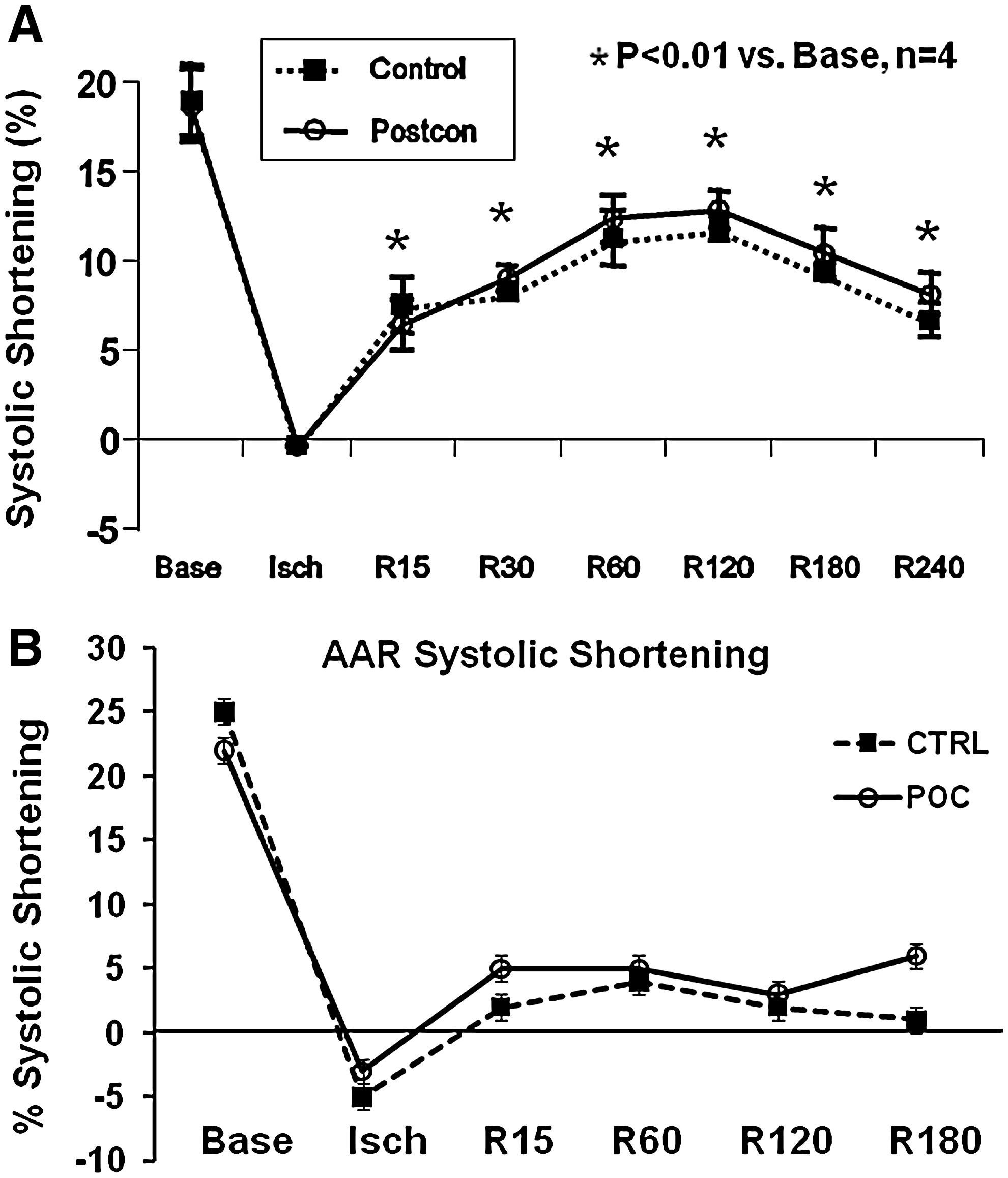
In contrast to the paucity of data obtained in models of regional ischemia, recovery of global function in isolated hearts has been extensively studied. Recently, Pinheiro et al. (115) demonstrated that postcon with three cycles of 10-s reperfusion/10-s reocclusion (but not 30-s cycles) preserved postischemic contractility in isolated rat hearts. However, Tawa et al. (133) reported that postcon using three cycles of 30-s reperfusion/30-s reocclusion after 40-min global ischemia improved postischemic contractile dysfunction, purportedly through the generation of NO. As discussed in other sections, the optimal algorithm is not yet fully defined.
The limited amount of clinical data obtained in patients with ST-segment myocardial ischemia suggest that postcon may be associated with augmented recovery of contractile function. Yang et al. (144) reported a trend toward improvement in ejection fraction at 7 days after MI (54 ± 13% vs. 44 ± 17%): in this study, patients presented within 4 to 5 h after onset of symptoms and were postconditioned with three cycles of 30-s reperfusion/balloon reinflation immediately after dilatation of the target coronary artery. In addition, Thibault et al. (135) reported that both 12-month ejection fraction and wall-motion score were improved after postcon in patients with acute ST-segment MI presenting within 6 h of symptom onset. However, it is not clear whether this improvement in global left ventricular function is derived directly from protection of the ischemic-reperfused myocardium, a favorable effect on the noninvolved myocardium, or simply a reduction in infarct size.
It is interesting to note that a direct salutary effect of postcon on contractile ability was observed in human myocardium by Sivaraman et al. (125). Right atrial trabeculae harvested from patients undergoing cardiac surgery and suspended in an organ chamber for a postcon protocol of four cycles of either 30 s or 60 s of alternating reoxygenation/hypoxia; the 60-s (but not 30-s) hypoxic-reoxygenation algorithm increased posthypoxic contractile performance of the trabeculae relative to a control segment. However, it is not known whether the force generated by these trabeculae contracting in vitro against a zero load can maintain adequate force generation against a load imposed by left ventricular pressure in vivo. Although postcon attenuates major mechanisms of contractile dysfunction, further studies are required (a) to determine what aspects of the contractile process are abnormal; (b) to define mechanisms of dysfunction that are not improved by postcon; (c) to determine contractile responses to inotropic stimulation (degree of contractile reserve) or responses to preloading; and (d) to separate systolic and diastolic components of contractile dysfunction.
Coronary Vascular Endothelial Function
The vascular endothelium is a single-cell layer covering all vascular structures. Its diminutive structure belies its complex biochemistry and prominent role in regulating vascular resistance (both vasoconstriction and vasodilation), inflammation, coagulation, and cell proliferation through the release of factors that act in an autocrine and paracrine fashion. In the normal state, the endothelium assumes a phenotype that appropriately regulates microvascular blood flow and inhibits coagulation and inflammation. Ischemia and reperfusion, however, cause endothelial dysfunction at both the functional (82) and structural levels (1) that is observed within minutes of reperfusion, changing the phenotype of the endothelium from its normal homeostatic state to one that promotes interactions between endothelial cells, neutrophils, and platelets, and one that generates oxidant species and pro-inflammatory mediators (38, 69). This dysfunction is, in part, related to an impaired release or synthesis of NO, which has vasodilatory and antineutrophil properties (81, 95). Endothelial dysfunction is thought to contribute, in part, to the pathogenesis of necrosis, apoptosis, and no-reflow areas in postischemic myocardium. In addition to the loss of the vasoregulation by the dysfunctional endothelium, a reduction of NO increases the recruitment and adherence of neutrophils to the coronary vasculature, thereby initiating the inflammatory cascade.
In the original study by Zhao et al. (156), postcon prevented postischemic endothelial dysfunction, as assessed by vasorelaxation responses to the endothelial nitric oxide synthase (eNOS) stimulator acetylcholine. Postcon attenuated the surface expression of P-selectin, suggesting a reduced state of activation in the endothelium that promotes less neutrophil recruitment and adherence. Accordingly, Zhao et al. (156) demonstrated a reduced adherence of neutrophils to postischemic coronary artery endothelium, which is physiologically linked to an impaired basal production of NO (83, 95). Zhao et al. (154) reported that, indeed, postcon increased myocardial eNOS while reducing deleterious inducible nitric oxide synthase activity. These data suggest that postcon attenuates endothelial cell dysfunction by decreasing oxidant generation, cytokine generation, and by increasing eNOS activity and NO bioavailability. The increased availability of NO may, in turn, attenuate the neutrophil-mediated inflammatory response, which may be linked to the pathogenesis of necrosis and apoptosis. In support of a postcon-induced reduction of the prooxidant phenotype induced by I-R injury, chemiluminescence data published by Mykytenko et al. (101) showed that postcon attenuated superoxide anion generation by postischemic coronary artery segments after 3 h and 24 h of reperfusion.
Clinical evidence of endothelial protection by postcon comes from several studies in which flow-mediated forearm-vessel vasodilation to transient ischemia was used as a barometer of endothelial function. Of note, this method of assessing endothelial function is based on the hypothesis that peripheral vascular reactivity reflects the physiology of vasoreactivity in the coronary artery under normal and pathophysiologic conditions (most notably, coronary artery disease). Ma et al. (94) studied AMI patients receiving PCI with or without postcon (three cycles of 30-s reperfusion/angioplasty balloon reinflation) at the time of reperfusion. They reported that postcon improved the endothelium-dependent brachial artery blood-flow responses assessed 24 h after PCI compared with a control group not receiving postcon. This study introduces a unique form of “remote postcon” (63), in that the postconditioned heart is protecting the ischemically challenged limb. Relevant to limb protection, Dragoni et al. (25) reported that postcon (three cycles of 10-s reperfusion/20-s reocclusion) failed to improve radial artery flow-mediated vasodilator responses after a 15-min index forearm ischemia. However, Loukogeorgakis et al. (93) found that, in healthy subjects, three cycles of remote postcon applied to a contralateral leg increased the flow-mediated responses to 20 min of forearm ischemia, an effect attributed in part to KATP channel activation. Therefore, remote postcon was effective in reducing endothelial injury. However, a number of questions must be answered by further research: Are the coronary arteries in patients protected by postcon after PCI? Is endothelial protection directly linked to a blunted inflammatory response? Is attenuation of neutrophil–endothelial cell interactions critical in protecting the endothelium? Is postcon linked to better physiological and clinical outcomes?
In fact, critical experimental and clinical research is needed to determine the mechanisms by which postcon attenuates inflammatory responses, and indeed how (and whether) these blunted responses are linked to any proposed reduction in postischemic injury.
Postconditioning and the Inflammatory Response to Reperfusion
I-R injury elicits an inflammatory response that is involved not only in immediate tissue damage, but also in subsequent tissue repair and healing (34). Relevant to the deleterious effects immediately after the onset of reflow, reoxygenation prompts the production of ROS both directly (within cardiomyocytes) and by triggering an inflammatory response (26). In addition, intracellular contents released from damaged cardiomyocytes undergoing necrosis interact with Toll-like receptors and thereby activate the innate immune response (5, 106). The hallmark of this response is the release of circulating soluble factors such as complement, chemokines, cytokines, and eicosanoids, and activation of neutrophils and the vascular endothelium. This leads to activation and recruitment of neutrophils into previously ischemic myocardium (33). Neutrophils have been implicated in the pathogenesis of myocardial infarction (140), although this involvement is still controversial (6, 140).
Numerous studies have suggested that cardioprotection by postcon is, in part, related to a reduction in inflammation. In the original study by Zhao et al. (156), postcon attenuated P-selectin expression, which is upregulated within minutes of the onset of reperfusion, and is involved in the recruitment and “rolling” of neutrophils (Fig. 4). In addition, Zhao et al. (156) showed that postcon attenuated neutrophil adherence to the postischemic endothelium, the production of oxygen species and the lipid peroxide byproduct malondialdehyde, and neutrophil accumulation. Mykytenko etal. (101) extended these findings to 24 h of reperfusion, whereas others demonstrated attenuated cytokine release in response to postcon both in vitro and in vivo (64, 128). Furthermore, preliminary evidence for a direct effect of postcon on neutrophil function was recently published by Granfeldt etal. (40): neutrophil function evaluated by superoxide anion production in local coronary venous blood was attenuated at both 2 and 24 h of reperfusion when compared with controls.
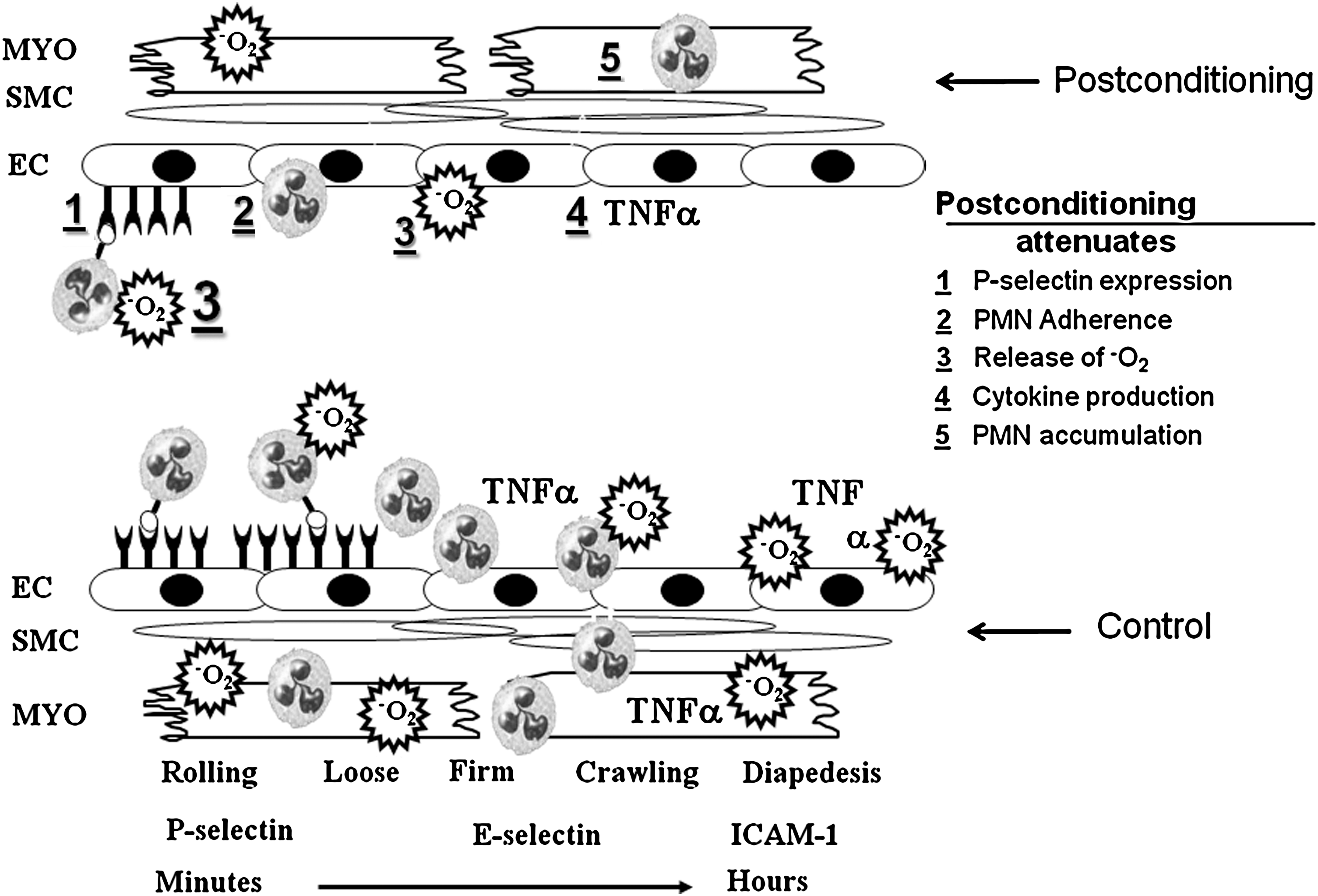
It is noteworthy that postcon is cardioprotective in buffer-perfused hearts in which circulating cells and mediators are absent, implying that the cardioprotection of postcon is not dependent on inhibition of inflammatory processes, and arguing against an inflammatory component to reperfusion injury. However, injury in cell-free systems may be caused by (a) the generation of reactive oxygen and nitrogen species by cardiomyocytes and vascular endothelial cells; (b) the depletion of endogenous antioxidants (i.e., glutathione and glutathione peroxidase); and (c) impaired delivery of perfusate secondary to microvascular injury and interstitial edema. Much of the data supporting antiinflammatory effects of postcon is indirect at this point, but studies are needed in which blood or isolated neutrophils are added to the perfusate at the onset of reperfusion to determine whether postcon attenuates cell-mediated inflammatory events in an otherwise cell-free system. Furthermore, key studies are needed to link functionally attenuated (a) neutrophil adherence to coronary endothelium, (b) neutrophil activation and accumulation, and (c) cytokine and chemokine generation to the infarct- and apoptosis-sparing actions of postcon.
Postconditioning and the “Comorbidity Conundrum”
As summarized in the preceding sections, a wealth of evidence suggests that relief of ischemia in a stuttered manner evokes a cardioprotective phenotype, as defined by the “gold standard” of infarct-size reduction. In addition, in many models, postcon-induced cardioprotection has also been associated with significant improvements in recovery of left ventricular (LV) function, attenuation in the incidence and duration of reperfusion-induced arrhythmias, improvement in endothelial function, and significant blunting of neutrophil infiltration and inflammation.
The overarching goal of all of these laboratory studies is to provide the foundation for the application of stuttered reflow in the clinical setting; indeed, initial small-scale trials conducted in patients suggest that postcon may represent a feasible and effective strategy to improve outcome in patients after MI (18, 41, 99, 127, 135, 144). It is, however, noteworthy that virtually all of our current insights into the pathophysiology and cellular mechanisms of postcon-induced cardioprotection are based on data obtained from healthy, adult animal models devoid of clinically relevant comorbidities. This issue has obvious implications for the clinical application of postcon: comorbidities including diabetes, hypertension, hyperlipidemia, and increased age are all risk factors for cardiovascular disease (31, 91), and patients exhibiting one or more of these risk factors are the specific populations in which the incidence of AMI is greatest and thus cardioprotection is most relevant. Moreover, extrapolation of data obtained in healthy adult models may be confounded by reported age- and disease-associated changes in the expression and activity of archetypal cardioprotective signaling proteins (including ERK 1/2, PI3 kinase-Akt, STAT3, p38 MAPK, and isoforms of PKC), both under baseline conditions and in response to I-R and other stressors (27, 44, 52, 58, 72, 73, 77, 103, 130, 134) (Fig. 5). These concepts raise the pivotal question: is the efficacy of postcon, as documented in healthy adult animal preparations, maintained in the setting of aging, diabetes, hypertension, and other disease models?

Efficacy of Postconditioning in Comorbid Models
Of the approximately 250 articles published to date on the topic of postcon, <10% of these studies have been conducted in models of aging, diabetes, hyperlipidemia, hypertension, and/or ventricular hypertrophy. However, despite the paucity of data, emerging evidence suggests that the efficacy of postcon is compromised in at least some of these comorbid models (Table 3).
N/a, not applicable; n/d, not determined; ?, inconclusive.
Reduction of Infarct Size
A consensus exists among currently published studies that, in rodent models of metabolic syndrome and diabetes, postcon fails to evoke an infarct-sparing effect (11, 118, 141) (Table 3). In the db/db mouse, a genetic model of type-2 diabetes, postcon exacerbated (rather than attenuated) myocardial necrosis (118). Similar outcomes have been obtained in models of acute hyperglycemia (achieved by short-term intravenous infusion of glucose before and during ischemia) by using the anesthetic agents sevoflurane and isoflurane: in hyperglycemic rats and rabbits, anesthetic-induced postcon was ineffective in reducing infarct size (51, 119). For all of these studies, the failure to initiate a protective phenotype was attributed to failed activation or phosphorylation of one or more of the cellular mediators implicated to play a role in postcon, including components of the ERK signaling pathway (11, 118, 141), PI3-kinase-Ak (11, 119, 141), and eNOS (119).
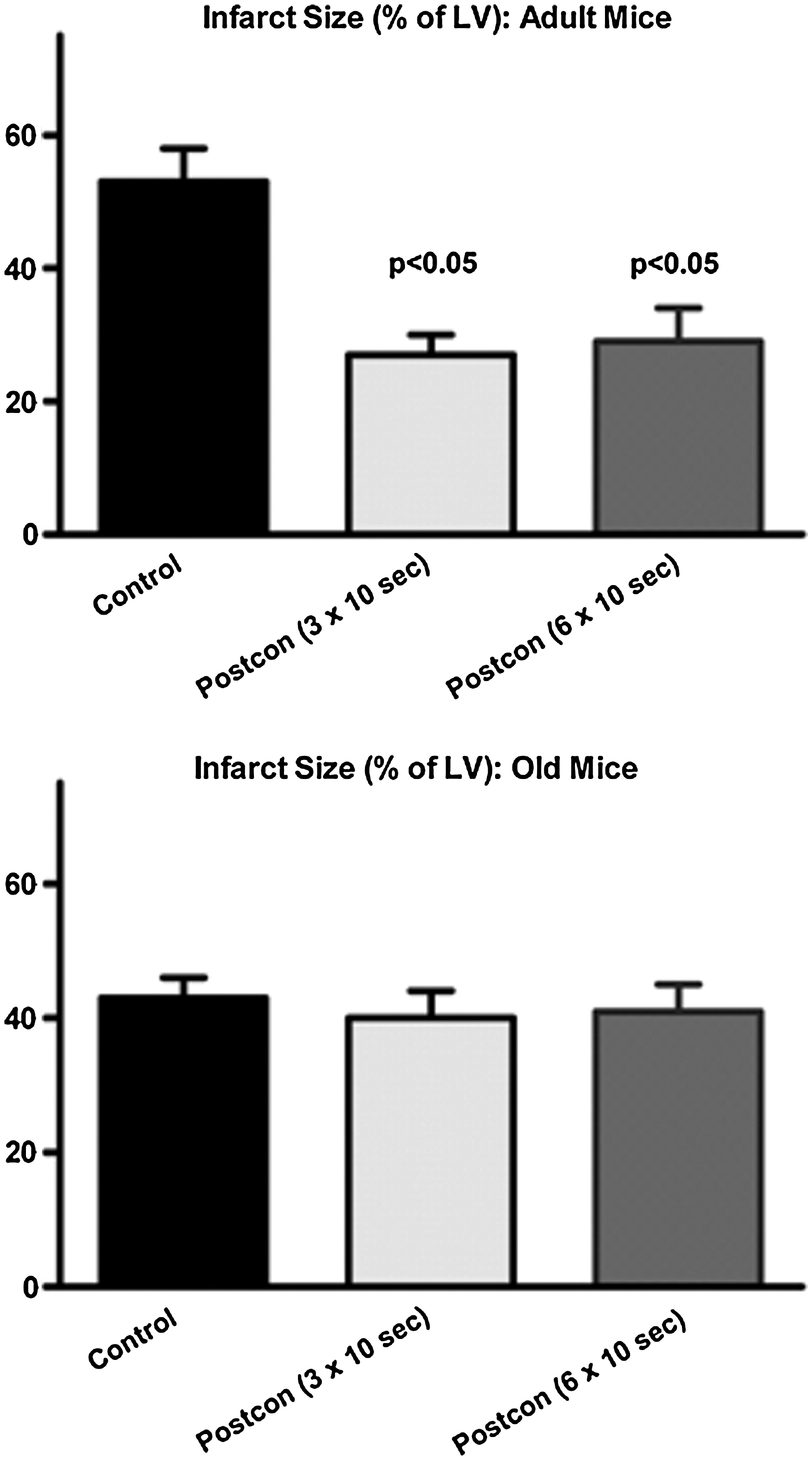
In contrast, in models of aging (7, 117, 148) and hyperlipidemia (20, 53, 55, 76, 152, 154), a range of results have been obtained: some studies have reported a loss of postcon-induced cardioprotection, whereas others observed a sustained benefit of stuttered reflow (Table 3). For the hyperlipidemic models, the dissimilar outcomes may reflect differences among protocols in the duration and composition of the high-fat diet (i.e., in rabbits, the infarct-sparing effect of postcon was lost after 6 weeks of feeding with a diet containing 0.2% cholesterol (2 g/kg food) + 6% corn oil (53, 55), whereas, in rabbits fed 1% cholesterol for 4 weeks, infarct-size reduction with stuttered reflow was maintained (20). A similar rationale can be applied to the aging models; aging is a continuum, and, as a result, disparate outcomes may reflect varying definitions of what constitutes an “old” or “aging” cohort. For example, mice older than 13 months displayed an attenuation in the efficacy of postcon: no benefit was seen with three cycles of 10 s of reperfusion/10 s of reocclusion, but significant reduction of infarct size was achieved when the stimulus was modified to five cycles of 5 s of reperfusion/5 s of reocclusion (7). However, in hearts from 2-year-old mice (a model that displays definitive morphometric and molecular hallmarks of cardiovascular aging, including hypertrophy, cardiac fibrosis, and increased myocardial expression of the senescence marker p16INK4a), no benefit was seen in any of the multiple postcon algorithms that were evaluated (117) (Fig. 6). Although mechanistic insights into the loss of postcon-induced cardioprotection in these models remain limited, alterations in NO production and nitrosative stress have been implicated to play a role in the setting of hyperlipidemia (53, 76), whereas, in aging myocardium, attention has focused on defects in ERK and STAT3 signaling (7, 117).
Interestingly, in models of hypertension or hypertrophy or both, initial reports suggest that the salutary effects of postcon are largely maintained (30, 86, 113) (Table 3). Of the two published studies in which infarct size was assessed, one demonstrated robust, postcon-induced cardioprotection in a mouse model of transverse aortic constriction (86), whereas the second observed a nonsignificant trend toward sustained reduction of infarct size with postcon in spontaneously hypertensive rats with attendant hypertrophy (113). Whether infarct-size reduction with postcon is, indeed, preserved in hypertension or hypertrophic myocardium or both remains to be substantiated in future studies.
Beyond the “Gold Standard”: Other End Points of Postconditioning-Induced Cardioprotection
Among the handful of studies that have interrogated the efficacy of postcon in comorbid models, the overwhelming focus has been on infarct size. As underscored in Table 3, only a small proportion have broadened the scope of the investigations to include an assessment of recovery of left ventricular function, incidence of reperfusion-induced arrhythmias, endothelial/vascular reactivity, or other facets of postcon-induced protection (20, 22, 30, 79, 86, 113, 148, 154). The only end point and model for which currently data exist is measurement of LV function in the setting of hypertension or hypertrophy or both: in three published studies, conducted in isolated buffer-perfused hearts, acute recovery of LV developed pressure was augmented in hypertensive/hypertrophic postconditioned groups versus matched controls (30, 86, 113). These findings are consistent with the concept that, for as-yet-unknown reasons, postcon-induced cardioprotection does not appear to be undermined in the presence of hypertension/hypertrophy. However, as with the infarct-size data, further studies are required to confirm these nascent findings. With regard to arrhythmias, endothelial function, and neutrophil infiltration, the dearth of information precludes any meaningful speculation on whether these ancillary benefits are maintained or lost in one or more of the comorbid models.
Comorbid Models versus Clinical Outcomes: Reconciling the Data
One might predict, based on the waning or total loss in efficacy of postcon in diabetic, aging, and hyperlipidemic models, that stuttered reflow would be of limited benefit in the clinical setting. Patients enrolled in the small postcon trials conducted to date displayed the typical profile of comorbidities (91), with the incidence of diabetes, dyslipidemia, and/or age older than 65 years ranging from 10% to 25%, 50% to 80%, and 20% to 35%, respectively (18, 127, 135, 144). Nonetheless, postcon applied in patients undergoing PCI for the treatment of AMI has consistently been shown to reduce indices of infarct size by 27% to 47% versus controls (18, 127, 135, 144). These data prompt the question: how can this incongruity between experimental models and clinical outcome be reconciled?
Limitations of the Models
An issue that undoubtedly contributes to the apparent discrepancy is the fact that comorbid models do not fully mimic the complexity of the patient population. Specifically, the comorbidities are relatively acute (on the order of days to weeks, rather than years), and the models are simplistic (do not display the multiple comorbid conditions seen in a substantial proportion of patients). In addition, the models typically do not incorporate the pharmacologic agents given as the standard of care in the setting of AMI and PCI, and, furthermore, do not consider the effect of the therapies administered to patients to manage their comorbid diseases (hypoglycemics and sulfonylureas, antihypertensive agents, lipid-lowering drugs, etc.). The potential relevance of this latter point is highlighted by recent data obtained in murine diabetic models: although mice with uncontrolled diabetes (displaying blood glucose values of 480 to 550 mg/dl) were refractory to postcon-induced cardioprotection, treatment to restore normoglycemia reestablished the infarct-sparing effect of stuttered reflow (118). Thus, in terms of clinical relevance of the models, the germane question may not be whether diabetes, hyperlipidemia, and so on alter the efficacy of postcon but, rather, whether stuttered reflow can evoke a protective phenotype in comorbid models managed by using standard pharmacologic therapies.
The Need for Larger Clinical Trials
Despite the consistent and compelling cardioprotection seen with postcon in the initial clinical trials (18, 127, 135, 144), it remains possible that, in one or more subpopulations, the benefits of postcon may be blunted. A logical, first approach to test this concept would be to perform posthoc analyses on the subset(s) of interest. In the three studies in which patients were prospectively assigned to receive stuttered versus abrupt reflow, meaningful subset analysis is precluded by small n values [i.e., total enrollments of 30, 41, and 38 patients (127, 135, 144)]. However, in the fourth trial, a retrospective analysis of outcome in patients after angioplasty (18), the larger enrollment of 115 patients makes posthoc analysis more feasible. When analysis was limited to the “aging” subpopulation of this retrospective study, defined empirically as patients aged 65 years or older (37 of the 115 patients enrolled), a provocative outcome was obtained: no significant infarct-sparing effect of postcon was observed [p = 0.70 (ns); Fig. 6]. This is in marked contrast to the robust, 30% reduction of infarct size seen when the full study cohort was included in the analysis (p < 0.05; Fig. 7) (18).
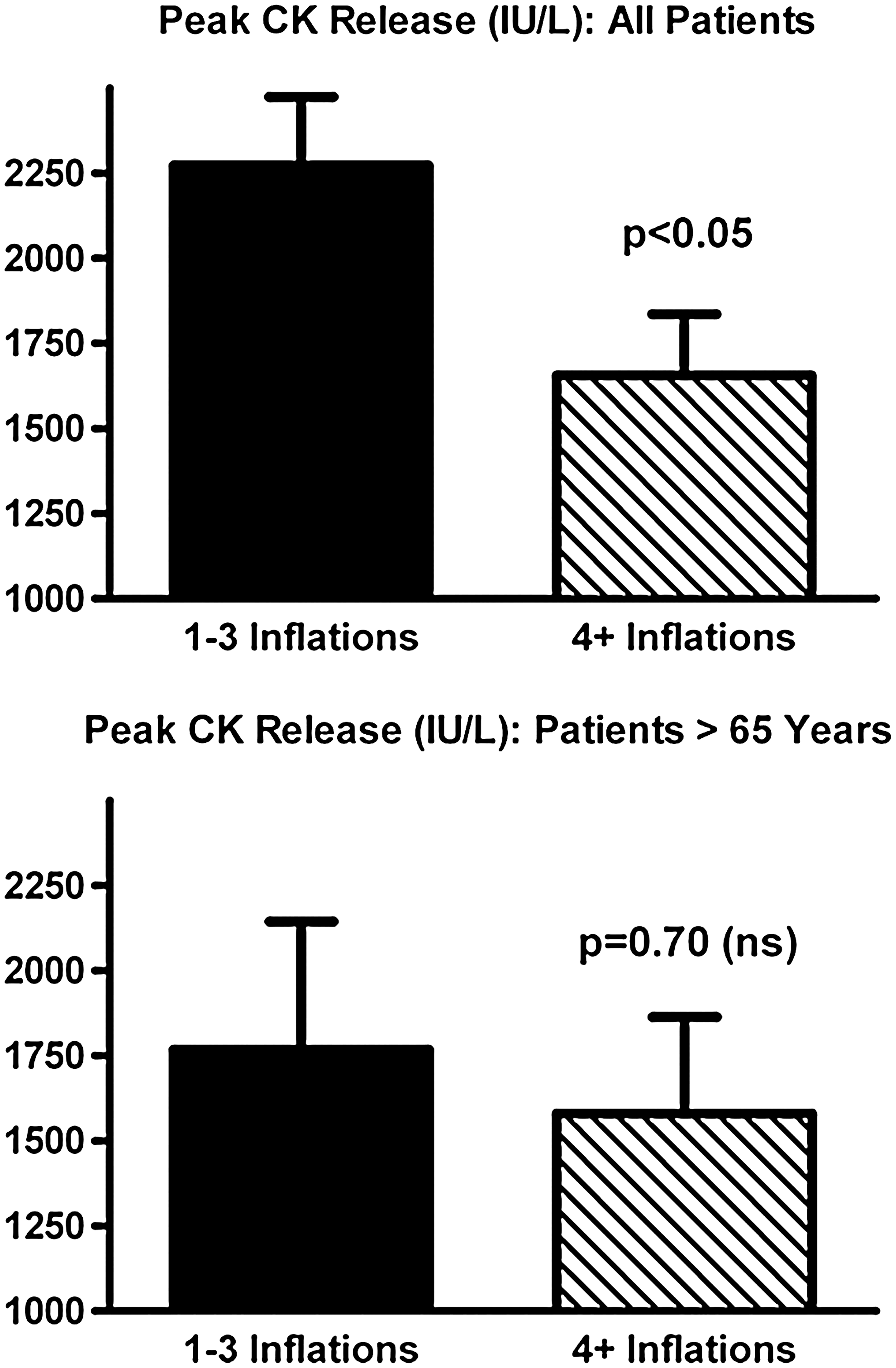
These data do not constitute proof of a loss in efficacy of postcon in aging patients, and do not undermine the compelling evidence that, as in experimental models, relief of ischemia achieved in a stuttered manner provides profound benefit against myocardial I-R injury in the clinical setting. However, the outcome of this posthoc analysis underscores the fact that larger, prospective trials with appropriate statistical power are required to discern whether postcon-induced cardioprotection is maintained or compromised in patients with diabetes, dyslipidemia, increased age, and so on, and, thus, resolve this “comorbidity conundrum.” Such studies should be large enough to permit stratification according to isolated comorbidities, multiple comorbidities, and medications used in the treatment of acute or chronic myocardial ischemia.
Concluding Remarks
Cardioprotection by postconditioning has been observed in all species tested, including humans undergoing not only AMI but also cardiac surgery. Postconditioning has broad-spectrum protective potential, which is derived from its effects on multiple cell types, including the coronary vascular endothelium, cardiomyocytes, and inflammatory cells in the heart (summarized in Fig. 8). Although much knowledge has been gained in these 7 years since its introduction, much more remains to be learned about postconditioning for us to understand its mechanisms and to be able to exploit its protective potential to our maximal advantage. Further studies are required to determine what each cycle of the algorithm does to endogenous cardioprotective mechanisms and pathways. Insights from these studies may provide a logical basis for optimization of the algorithm. In addition, how to alter postconditioning to capture the “holy grail” of improving cardiac function and staving off heart failure must still be determined. Investigation of postconditioning will likely lead to the identification of new pathways of endogenous protection in much the same way as did preconditioning. Finally, postconditioning in its conventional mechanical algorithm or a pharmacologic mimetic form is yet to be adopted by the cardiology, surgery, and other healthcare communities for use in the treatment of ischemia/reperfusion diseases. This adoption will require large-scale clinical trials that determine which patient populations respond to the protocol, and which do not.