
Abstract
Cellular senescence is induced in response to DNA damage, caused by genotoxic stresses, including oxidative stress, and serves as a barrier against malignant transformation. Tumor-suppressor protein p53 induces genes critical for implementing cellular senescence. However, the identities of p53 target genes and other regulators that achieve senescence under oxidative stress remain to be elucidated. Effector genes for oxidative stress–induced cellular senescence were sought, based on the fact that transcription factor Bach1 inhibits this response by impeding the transcriptional activity of p53. pRb became hypophosphorylated more rapidly in Bach1-deficient MEFs than in wild-type cells, suggesting that pRb activation was involved in their senescence. Bach1-deficient MEFs bypassed the senescence state when the expression of a subset of p53 target genes, including p21, Pai1, Noxa, and Perp, was simultaneously reduced by using RNAi. Combined knockdown of p21 and pRb resulted in vigorous re-proliferation. These results suggest that oxidative stress–induced cellular senescence is registered by multiple p53 target genes, which arrest proliferation redundantly, in part by activating pRb. Our elucidations contrast with previous reports describing monopolistic regulations of senescence by single p53 target genes. Antioxid. Redox Signal. 14, 2441—2451.
Introduction
Transcription factor Bach1 is a key inhibitor of cellular senescence induced by reactive oxygen species (ROS) (17). Bach1 forms a complex with p53 together with HDAC1 (histone deacetylase 1) and Nco-R (nuclear co-repressor), promotes histone deacetylation by directing HDAC1 to the p53 target genes, and represses their expression at the transcriptional level to inhibit cellular senescence. Bach1-defiecient MEFs enter the senescent state more rapidly than wild-type cells under 20% O2 conditions because of hyperactivation of p53 (17). In a parallel, p53-independent pathway, Bach1 represses another set of target genes, including heme oxygenase-1 (HO-1), a critical gene for protection against oxidative stress (39). The Bach1 target genes in this category possess a Maf-recognition element (MARE) to which Bach1 binds by forming a heterodimer with the small Maf oncoproteins (22, 27, 39). Thus, Bach1 plays important roles in the regulation of senescence and oxidative-stress response and may integrate these cellular responses.
To clarify the downstream networks of p53 in the process of oxidative stress–induced cellular senescence, Bach1-deficient MEFs were used in this study by exploiting their hypersensitivity to oxidative stress (17). We found that no single gene among the known senescence effectors was essential for the maintenance of senescence. A combined knockdown of four genes (p21, Pai1, Noxa, and Perp) resulted in a weak but reproducible reproliferation, suggesting that multiple p53 subpathways contribute to senescence in a redundant manner. Combinatorial knockdown of p21 and pRb resulted in vigorous reproliferation of senescent cells. Considering the necessity of p21 and pRb for senescence and more rapid reduction in pRb phosphorylation in Bach1-deficient MEFs, these observations indicate that the p53–pRb crosstalk was critical for senescence in Bach1-deficient MEFs. Our findings suggest that Bach1 inhibits the process of oxidative stress–induced cellular senescence by modulating expression of the specific downstream subpathways of p53.
Materials and Methods
Isolation and culture of mouse embryonic fibroblasts
MEFs were isolated from 14.5-day-old embryos of various genotypes, as previously described (17). MEFs from single embryos were plated into a 60-mm-diameter culture dish and cultured with DMEM (Gibco, Carlsbad, CA) containing 10% fetal bovine serum (Cell Culture Bioscience, Tokyo, Japan), 1% NEAA (Gibco), 1% penicillin/streptomycin (Gibco), and 4.2 μl 2-mercaptethanol (Wako Junyaku, Osaka, Japan) at 37°C. Cells were cultured in 20% or 3% oxygen by subculturing in a 60- or 100-mm-diameter culture dish every 2 or 3 days, and cell number was determined at each passage. Senescent cells were prepared by several passages for at least 4 weeks (about seven to nine and five to seven passages in wild-type and Bach1-deficient MEFs, respectively) under 20% oxygen until they stopped proliferation and showed flattened morphology with SA-β-gal staining. Senescence induced by H2O2 was performed by the protocol indicating that confluent fresh MEFs be treated with 550 μM H2O2 in serum-containing medium for 2 h, left to recover overnight, as described previously (25), and then used in RNA interference.
RNA interference
Stealth RNAi duplexes were designed for the target genes by using the BLOCK-iT RNAi Designer (Invitrogen Corporation, Carlsbad, CA). For the knockdown of the target genes, 2.0 × 106 cells were transfected with 6 μl of stock Stealth RNAi duplexes (20 μM) by using the basic nucleofection solution for MEFs (VPD-1004; Lonza, Koeln, Germany). Stealth RNAi Negative Control duplexes (Invitrogen Corporation) were used as negative control RNAi. The transfected cells were divided into three parts and cultured in 60-mm-diameter culture dishes. Cell number was determined at the indicated days, and data are reported in figures as relative values compared with cell numbers at day 1. Sequences of the Stealth RNAi used in this study were as follows:
p53 RNAi, 5′-UUACACAUGUACUUGUAGUGGAUGG-3′
cdkn2a RNAi 5′-UUAGCUCUGCUCUUGGGAUUGGCCG-3′
HO-1 RNAi, 5′-AUCACCAGCUUAAAGCCUUCUCUGG-3′
p21 RNAi, 5′-UUGGAGUGAUAGAAAUCUGUCAGGC-3′
Pai1 RNAi, 5′-UUGACUUUGAAUCCCAUAGCAUCUU-3′
Noxa RNAi, 5′-AUACCAGGCAUUUCCAUCAACCGGC-3′
Perp RNAi, 5′-UUAUCGUGAAGCCUGAAGGUCUGUG-3′
Notch1 RNAi, 5′-ACGCCGCUGUGAGUCAGUCAUCAAU-3′
Lrdd RNAi, 5′-AGAACUUCUCACCCUCAAACAUCUC-3′
Gpx1 RNAi, 5′-ACUUGAGGGAAUUCAGAAUCUCUUC-3′
Fat1 RNAi, 5′-AAAUGAUGUGAACAACUUUCACGGG-3′
Bcas3 RNAi, 5′-AGAACUAGUUGAUAUGACCGUGGUC-3′
Pcdh7 RNAi, 5′-UGUACAGGCUCAUCUCUGCAUUCCG-3′
Prl-3 RNAi, 5′-CUUUGAACCUCAGUCUCUGCUUAGG-3′
Rb1 RNAi, 5′-UUUAGUCGGAGAUAUGCUAGACGGU-3′
Senescence-associated β-galactosidase staining and cell-cycle analysis
MEFs were stained overnight for senescence-associated (SA) β-galactosidase, as previously reported (15). Incubated for 16 h with 10 μM BrdU with BrdU Flow Kits (BD Pharmingen, San Jose, CA), cytometric analyses of MEFs were performed on a FACSCalibur with CellQuest software (Becton Dickinson, Franklin Lakes, NJ).
Expression profiling, RNA amplification, and Q-PCR
Preparation of total RNAs from various cells was carried out by using the Total RNA Isolation minikit (Agilent Technologies, Palo Alto, CA). Agilent whole-mouse genome (4x44K, G4122F) arrays were used for this study, as described previously (17). The analysis and clustering of genes were performed by using the Genespring software package (Agilent Technologies). The quantitative PCRs were performed with a LightCycler (Roche). Primers used were as follows:
p53, (forward) 5′-GGAGGAGTCACAGTCGGAT-3′
(reverse) 5′-GCTTCACTTGGGCCTTCA-3′
p16, (forward) 5′-TGCGGGCACTGCTGGAAG-3′
(reverse) 5′-GGTAGTGGGGTCCTCGCAGTT-3′
p19ARF, (forward) 5′-GCTCTGGCTTTCGTGAACA-3′
(reverse) 5′-TCGAATCTGCACCGTAGTTG-3′
p21, (forward) 5′-CCTGGTGATGTCCGACCTGTT-3′
(reverse) 5′-GGGGAATCTTCAGGCCGCTC-3′
Pai1, (forward) 5′-TTAGTGCAACCCTGGCCGAC-3′
(reverse) 5′-TGCGGGCTGAGATGACAAA G-3′
Noxa, (forward) 5′-GTGCACCGGACATAACTG-3′
(reverse) 5′-AGCACACTCGTCCTTCAAG-3′
Perp, (forward) 5′-GCTGCAGTCTAGCAACCACA-3′
(reverse) 5′-GAAGCAGATGCACAGGATGA-3′
Prl-3, (forward) 5′-TGCTGAAGGCCAAGTTCTAC-3′
(reverse) 5′-TGAGCTGCTTGCTGTTGA-3′
HO-1, (forward) 5′-GGGTGACAGAAGAGGCTAAG-3′
(reverse) 5′-GTGTCTGGGATGAGCTAGTG-3′
Immunoblotting analysis
The immunoblotting analysis used antibodies against pRb (554136; BD Pharmingen), HO-1 (a kind gift from Prof. Shigeru Taketani, Kyoto Institute of Technology), α-tubulin (B7; Santa Cruz), ppRb (phospho-Rb Ser780) (C84F6, CST), c-myc (N262; Santa Cruz). Whole-cell extracts and SDS-PAGE were performed as described previously (17).
Statistical analysis
Data are presented as mean ± SD. All the statistical analyses were done by Student's t test or Welch's correction. A value of p < 0.05 was considered indicative of significance in all tests.
Results
p53-dependent cellular senescence of Bach1-deficient cells involves precocious pRb hypophosphorylation
Senescent wild-type MEFs are known to bypass senescence on the knockdown of p53 (16). We developed a similar assay by using Bach1-deficient MEFs to examine whether p53 was also indispensable for the maintenance of cellular senescence in Bach1-deficient MEFs. The knockdown of p53 in senescent Bach1-deficient MEFs resulted in resumption of proliferation (Fig. 1A). Similar results were obtained in wild-type cells. Thus, senescence of Bach1-deficient MEFs was dependent on continuous p53 activity. The effect of knockdown was confirmed by RT-PCR (Fig. 1B).
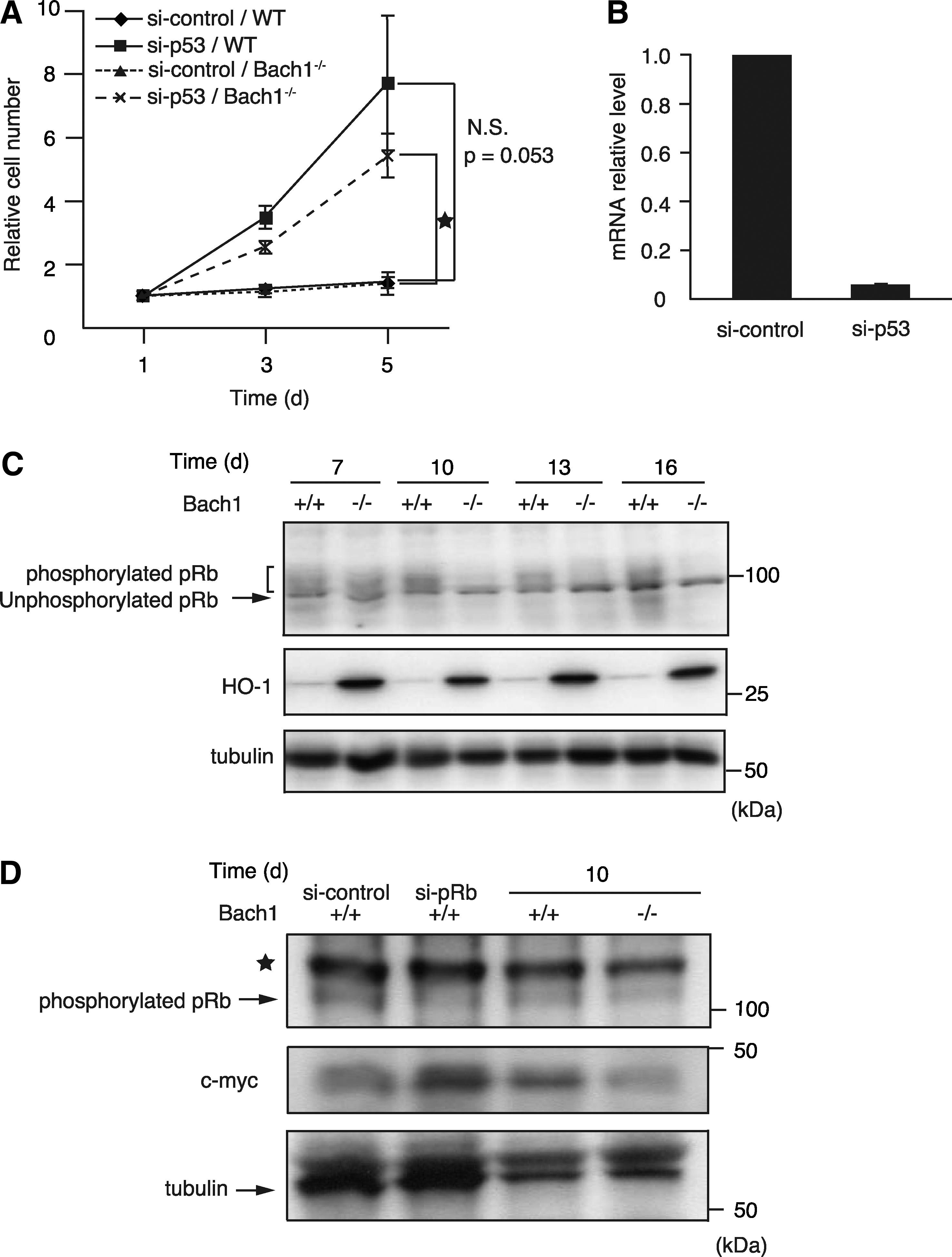
p53 activates indirectly the pRb tumor suppressor to inhibit the G1/S transition of the cell cycle, contributing to cellular senescence (8, 40). Considering the crosstalk between p53 and pRb, we examined whether pRb phosphorylation in Bach1-deficient MEFs was different from that in wild-type MEFs in the process of senescence. The levels of phosphorylated pRb and unphosphorylated pRb were compared between Bach1-deficient and wild-type MEFs during passages in vitro (Fig. 1C). Although the majority of pRb at day 7 (passage 3) was phosphorylated and migrated slower than the hypophosphorylated form in SDS-PAGE, irrespective of the genotypes, phosphorylated pRb disappeared earlier in Bach1-deficient MEFs than in wild-type MEFs on culture in vitro. To confirm these observations, antibody against pRb phosphorylated at Ser780 was used. The level of phosphorylated pRb was reduced on knockdown of pRb, verifying the specificity of the antibody. It was lower in Bach1-deficient cells than in wild-type cells at day 10 (Fig. 1D). Expression of c-Myc, one of the transcriptional targets of E2F, was increased on pRb knockdown in wild-type cells (Fig. 1D). In contrast, c-Myc expression was reduced in Bach1-deficient MEFs, indicating that the Rb pathway was more activated in Bach1-deficient MEFs than in wild-type MEFs (Fig. 1D). Taken together, these results suggest that the early accelerated pRb hypophosphorylation, presumably due to p53 activation, was involved in the rapid senescence entry of Bach1-deficient MEFs.
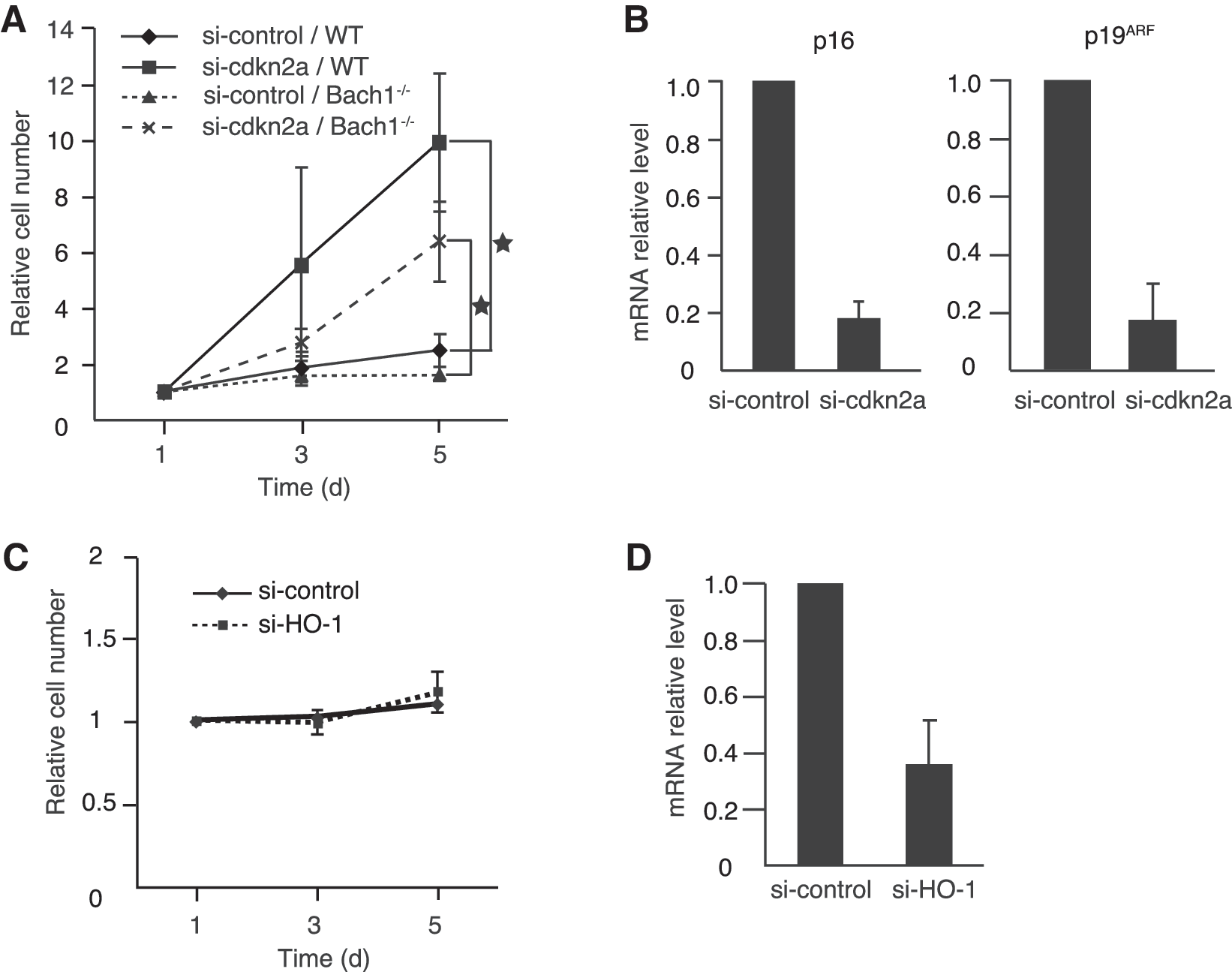
Both p16 and p19ARF are critical for the activation of pRb and p53, respectively (8). To examine whether cellular senescence in Bach1-deficient MEFs is dependent on p16/p19ARF, simultaneous knockdown of p16 and p19ARF by using siRNA targeting their gene Cdkn2a was performed in near-senescent MEFs (Fig. 2A and B). On knockdown, vigorous reproliferation was observed not only in wild-type but also in Bach1-deficient MEFs. Besides the tumor-suppressor genes mentioned earlier, HO-1 has been implicated in the regulation of cell proliferation among known Bach1 target genes. Carbon monoxide, one of the reaction products of HO-1, inhibits cell proliferation by regulating p21 and pRb (31). Consistent with the fact that HO-1 expression is repressed by Bach1 (22, 39), the expression of HO-1 was much higher in Bach1-deficient MEFs than in control cells (Fig. 1C). These previous reports and our present results suggested the possibility that the upregulation of HO-1 might be involved in the maintenance of cellular senescence in Bach1-deficient MEFs. However, knockdown of HO-1 in senescent Bach1-deficient MEFs did not affect cell proliferation (Fig. 2C). The effect of knockdown was confirmed by RT-PCR (Fig. 2D). These results suggest that HO-1 is not causally associated with the rapid cellular senescence in Bach1-deficient MEFs. Taken together, these results suggested that cellular senescence in Bach1-deficient MEFs was specifically dependent on p53, pRb, and their upstream regulators p16 and p19ARF.
Function of Bach1- and p53-co-regulated genes in cellular senescence
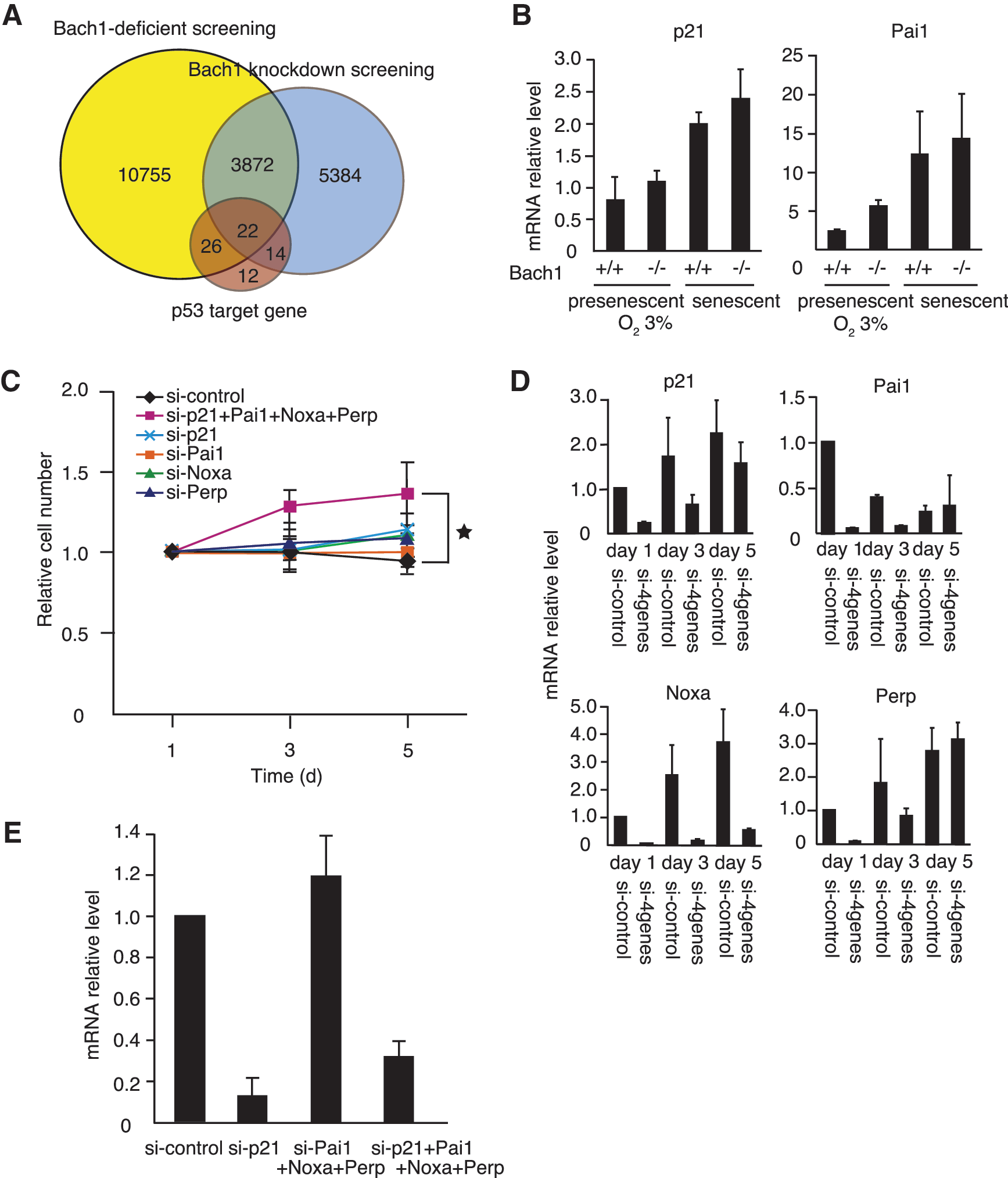
The premature senescence of Bach1-deficient MEFs is most likely due to de-repression of the p53 target genes because of the absence of Bach1. In search of the putative responsible genes, we focused on a cohort of p53 target genes that were repressed by Bach1. The messenger RNA-expression profiles of two sample sets were compared previously; wild-type and Bach1-deficient MEFs (17) and control RNAi and transient Bach1 RNAi in wild-type MEFs. Cells were cultured under conditions of 3% or 20% O2 to sort genes specifically associated with oxidative stress–induced cellular senescence (17). These profiles identified the genes whose expression was under the control of p53, repressed by Bach1, and induced more under 20% oxygen in comparison to 3% oxygen. In addition, acute knockdown of Bach1 in wild-type MEFs was performed to sort out genes that were indirectly affected by Bach1 knockout (Fig. 3A). Representative genes were selected for further studies from those that are known to regulate apoptosis, cell-cycle arrest (including pRb regulation), or ROS metabolism (Table 1). Among the candidate genes, levels of p21 and Pai1 mRNA, for example, were low in cells cultured under the 3% oxygen condition, which suppressed oxidative stress and senescence, irrespective of the genotype. Conversely, their expression increased when cells became senescent (Fig. 3B). We reported previously that expression of p21 and Pai1 mRNA was higher in Bach1-deficient, presenescent MEFs than in wild-type presenescent cells exposed to oxidative stress under 20% oxygen condition (17). Therefore, these observations also fortified the idea that cellular senescence in Bach1-deficient MEFs was rapidly induced, but its molecular mechanism seemed to be similar to that of wild-type MEFs.
A list of p53 target genes that were upregulated in Bach1-deficient MEFs in comparison with wild-type MEFs and in acute knockdown in comparison with control RNAi in wild-type MEFs under 20% O2. The same GB accession number genes are not shown.
The expression of these genes was then reduced in senescent Bach1-deficient MEFs to examine whether their increased expression in Bach1-deficient MEFs was causative of senescence. The cell proliferation, cell morphology, and SA-β-galactosidase activity staining did not change with the individual knockdown of p21, Pai1, Noxa, Perp, or the seven other genes (Fig. 3C and data not shown). To explore the possibility that several genes worked redundantly to maintain senescence, four genes were inhibited simultaneously in various combinations. A combinatorial knockdown of p21, Pai1, Noxa, and Perp showed a weak but reproducible tendency to bypass cellular senescence in senescent Bach1-deficient MEFs (Fig. 3C). RT-PCR at days 1, 3, and 5 revealed that simultaneous knockdown of these genes lasted for at least 3 days after transfection (Fig. 3D). To verify that the knockdown of one gene would not affect the expression of other genes, p21 mRNA levels were compared between combined knockdown with or without p21 (Fig. 3E). The results indicated that the combinatorial knockdown of Noxa, Pai1, and Perp did not affect p21 mRNA levels. Therefore, the effect of combinatorial knockdown could not be explained by an additional reduction of their mRNA levels by targeting others.
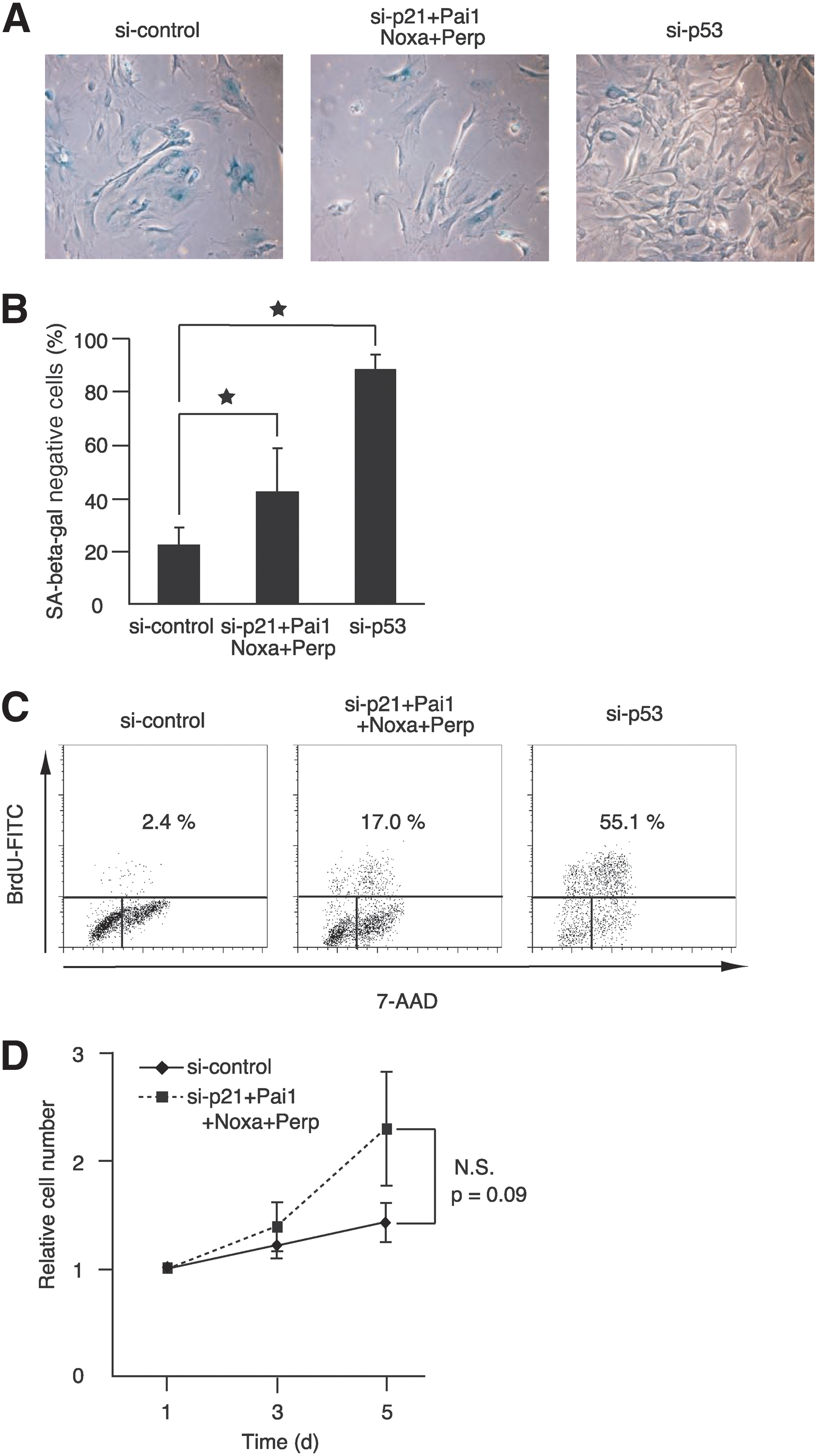
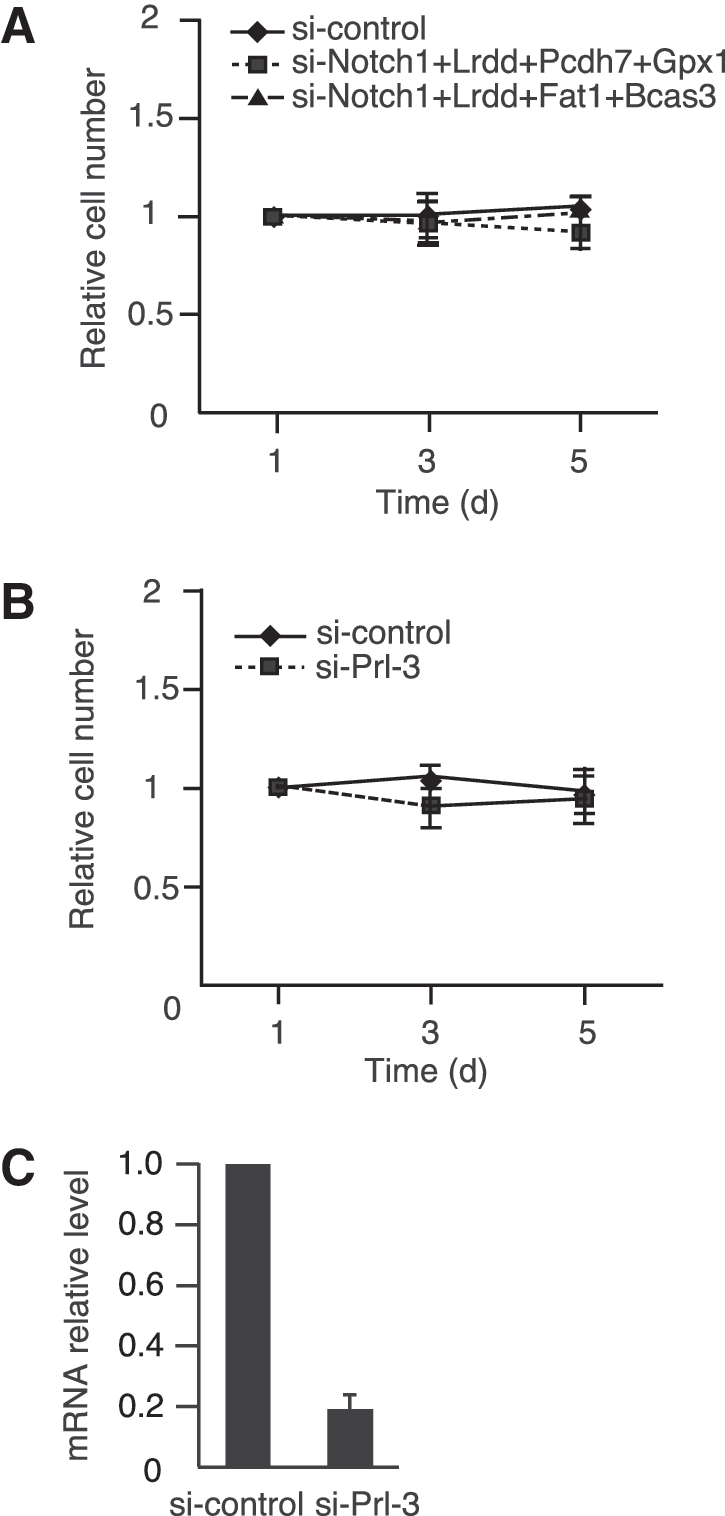
Examinations of cell morphology and SA-β-galactosidase activity confirmed that the combined knockdown of the four genes bypassed cellular senescence in senescent Bach1-deficient MEFs (Fig. 4A and B). Changes in cell morphology and expression of SA-β-galactosidase activity are independent of cell-cycle arrest but often are associated with senescence. Although senescent MEFs showed enlarged and flattened morphology, more cells showed sharper and smaller cytoplasmic regions on knockdown of the four genes, similar to p53 knockdown. Cell-cycle analysis by BrdU labeling also validated clearly that the cells resumed S-phase entry on the combined knockdown of these genes (Fig. 4C). Simultaneous knockdown of the four genes also resulted in a tendency to reproliferate in wild-type MEFs, although the effect was not statistically significant (Fig. 4D). These results suggested that the functions of the four genes were not specific to cellular senescence in Bach1-deficient MEFs. However, the effect obtained by the combined knockdown was not as profound as the knockdown of p53 in these cells. In contrast to the combination of p21, Pai1, Noxa, and Perp, other combinations did not show apparent effect on senescence, including genes such as Notch1, leucine-rich and death-domain containing (Lrdd), glutathione peroxidase1 (Gpx1), FAT tumor-suppressor homolog 1 (Fat1), protocadherin7 (Pcdh7), and breast carcinoma amplified sequence3 (Bcas3) (Fig. 5A). These genes are involved in development, proliferation, apoptosis, oxidative stress, migration, tumor suppression, and adhesion (2, 9, 10, 21, 33, 38). We also carried out knockdown of phosphatase of regenerating liver-3 (Prl-3), which has recently been reported to induce G1 arrest downstream of p53 (6). Whereas the expression of Prl-3 was higher in Bach1-deficient MEFs than in wild-type MEFs, its expression was not affected by the acute knockdown of Bach1 (data not shown). The knockdown of Prl-3 did not result in bypassing cellular senescence (Fig. 5B). The efficient knockdown of Prl-3 or other genes was confirmed by RT-PCR (Fig. 5C and data not shown). Taken together, these results suggest that a distinct subset of p53 target genes, including p21, Pai1, Noxa, and Perp, plays important roles in the execution of cellular senescence in response to oxidative stress.
Involvement of p21-pRb subpathway in cellular senescence
Because pRb hyperphosphorylation disappeared more rapidly in Bach1-deficient MEFs (Fig. 1C), we examined whether cellular senescence in Bach1-deficient MEFs was dependent on pRb. Acute knockdown of pRb in senescent Bach1-deficient MEFs resulted in weak but reproducible resumption of proliferation (Fig. 6A), consistent with a previous report of using an acute, inducible knockout of pRb in senescent wild-type MEFs (35). Efficient knockdown of pRb was confirmed by immunoblotting (Fig. 6B). The result that the bypassing effect was not so robust as p53 knockdown suggests that pRb constitutes a pathway redundant with some of the p53 subpathways for senescence. To address this assumption, we tested for a genetic interaction of pRb with the four critical p53 target genes. Combinatorial knockdown of pRb with p21, Pai1, Noxa, and Perp resulted in a more-efficient bypass of senescence than the individual knockdown of pRb or the p53 target genes subset (Fig. 6A; see Fig. 3). However, the combinatorial knockdown of p21, Pai1, Noxa, and Perp did not affect pRb phosphorylation (Fig. 6C). Because reproliferating cells were the minor fraction of the cells (see Fig. 4C), the effect on pRb, if any, may be masked at the population level. Similar effects in cell proliferation were observed in senescent wild-type MEFs (data not shown), indicating that their genetic interaction was not specific to Bach1-deficient MEFs.
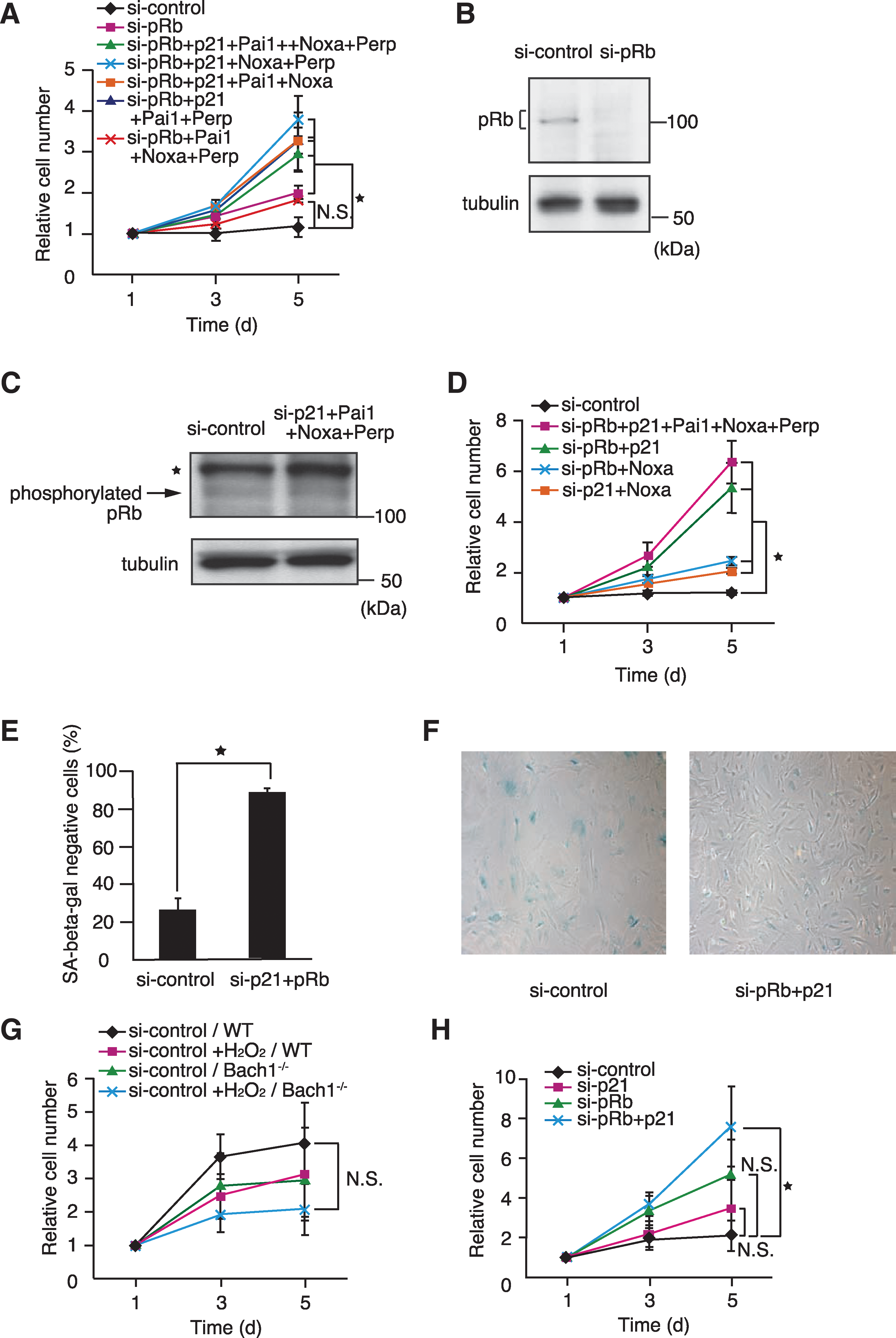
To identify critical p53 target gene(s) that collaborate with pRb, the combinatorial knockdown of pRb with three of the four genes was performed (Fig. 6A). All of the effective combinations included p21. The critical functions of p21 and pRb were confirmed by the observation that knockdown of p21 and pRb bypassed senescence as efficiently as the combination of pRb with the four p53 target genes (Fig. 6D). The cell morphology and SA-β-galactosidase activity staining confirmed that knockdown of p21 and pRb bypassed cellular senescence (Fig. 6E, F). In contrast, the knockdown of pRb and Noxa or other p53 target genes did not show any enhancement of bypassing cellular senescence (Fig. 6D and data not shown).
The involvement of p21 and pRb was further examined in the induction of senescence in response to hydrogen peroxide, another oxidative stress agent that is widely used to induce senescence acutely (25). In response to hydrogen peroxide, both presenescent wild-type and Bach1-deficient MEFs stopped proliferation (Fig. 6G). The simultaneous knockdown of p21 and pRb in presenescent Bach1-deficient MEFs resulted in restoration of vigorous proliferation in comparison to the single knockdown of either gene (Fig. 6H). Taken together, these observations indicated that both p21 and pRb were necessary simultaneously for the induction and maintenance of cellular senescence in response to oxidative stress.
Discussion
This study focused on the altered gene-expression profiles of Bach1-deficient MEFs to identify genes and their genetic interaction implementing cellular senescence in response to oxidative stress. Among the candidate genes, the knockdown experiments using senescent Bach1-deficient MEFs showed that multiple p53 target genes, including p21, Noxa, Pai1, and Perp, functioned redundantly to maintain cellular senescence. Importantly, the observed genetic interaction of p21 and pRb in cellular senescence is the first report and is reminiscent of the accelerated proliferation in p21/pRb-double deficient MEFs (11). The additive effect of p21 and pRb knockdown is explained by the canonic p21-pRb pathway (7) and implied that they constituted parallel, redundant pathways as well. This may involve other functions of p21, such as the activation of Rb family p107 and p130, which are also involved in senescence (36) or the inhibition of PCNA required for DNA synthesis (1). In contrast to p21, the other three genes (Pai1, Noxa, and Perp) can be placed within the pRb pathway because their combined knockdown did not show any additive effect. Although Pai1 is known to play an important role in pRb activation, Noxa and Perp have not been implicated in senescence thus far (3, 26), leaving their molecular functions unclear. Because combined knockdown of p21, Noxa, Pai1, and Perp was not as effective as that of p21 and pRb or p53, we surmise that an additional p53 target gene(s) is involved in the pRb-pathway activation (gene X in Fig. 7). Taken together, these observations suggest that p53 achieves cellular senescence by combined action of several, redundant genes affecting the pRb-dependent or -independent subpathways (Fig. 7). The critical role of Bach1 in regulating p53 target genes suggests that Bach1 may be involved in the process of tumorigenesis. We have found that RasV12-mediated transformation of immortalized MEFs requires Bach1 in vitro (A. Nakanome et al., unpublished data). Therefore, it will be interesting to examine possible involvement of BACH1 in human cancer.
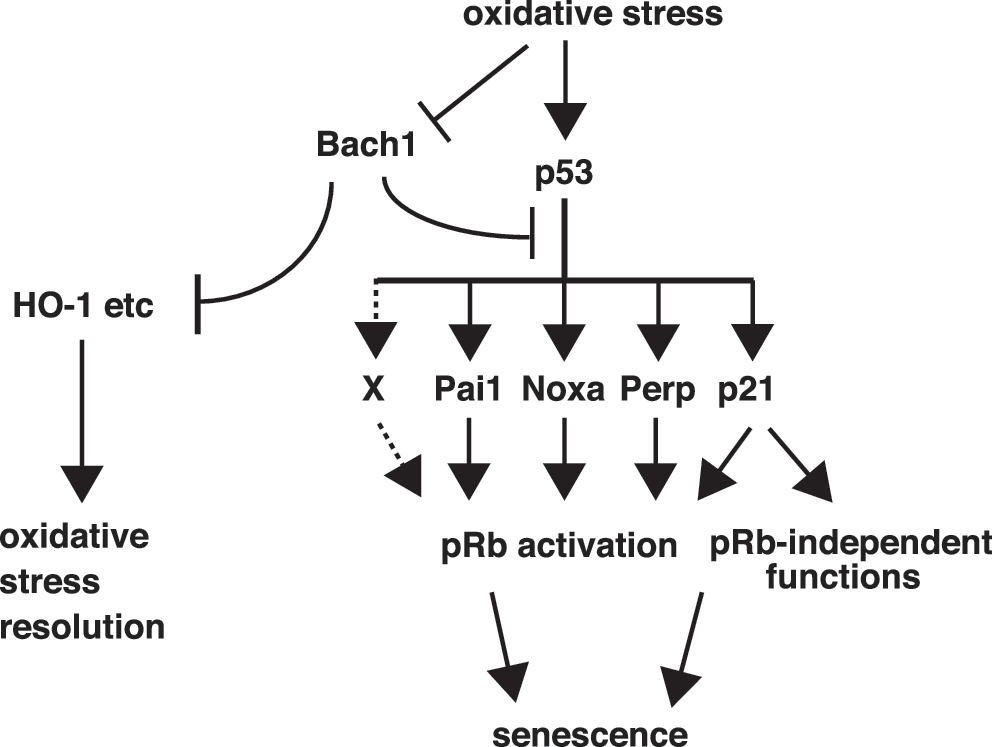
Besides the p53-dependent pathway, Bach1 acts as a repressor of the oxidative-stress response through the MARE-dependent genes including HO-1 (18, 22, 27, 31, 39). Although the knockdown of HO-1 in Bach1-deficient MEFs did not result in the reentry to the cell cycle, the possibility cannot be ruled out that HO-1 may be involved in cellular senescence. HO-1 is induced in the process of premature senescence of human fibroblasts (30) and possesses the cytoprotective function to confer a stress-resistant phenotype. It is especially noticeable that Bach1 deficiency contributes to the reduction of ROS levels with 20% oxygen conditions rather than its increase coincident with the induction of cellular senescence in MEFs (17). Therefore, when taken together with the present results, cellular senescence in Bach1-deficient cells results from higher responsibility of the p53 and pRb systems compared with wild-type cells but not from higher ROS levels. These results suggest that Bach1 plays an important role in maintaining cell homeostasis by balancing the metabolism of ROS and senescence with the distinct mechanisms (Fig. 7).
A pitfall of this study is the utilization of RNAi to assess functions of genes. Because RNAi is usually not complete, residual levels of mRNA may be sufficient to maintain cells in the senescent state. However, the observation that a transient reduction of several mRNAs simultaneously resulted in the bypassing of senescence is considered to be proof that their genetic interaction is critical in achieving senescence. The partial loss of function in the RNAi experiments may recapitulate pathologic situations in which relevant mutated genes often retain some activities. We hope that further studies on Bach1 will shed new light on the mechanisms of cellular senescence, including subsequent bypassing mechanisms toward carcinogenesis.
Footnotes
Acknowledgments
We thank Dr. Nobuyuki Tanaka, Nippon Medical School, for valuable advice and reagents, and Dr. Shigeru Taketani, Kyoto Institute of Technology, for anti-HO-1 antibody. Interpretation of data has been enriched by discussion with Prof. Keiko Nakayama (Tohoku University) and the members of our laboratory, including Tsuyoshi Ikura, Akihiko Muto, and Yasutake Katoh. This work was supported by grants-in-aid and the Network Medicine Global-COE Program from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Additional initiative supports were from the Uehara Foundation, the Takeda Foundation, and the Astellas Foundation for Research on Metabolic Disorders. This work was also supported by Biomedical Research Core, Tohoku University School of Medicine.
Author Disclosure Statement
The authors have no competing financial interest in relation to the work described.