
Abstract
The mitochondrion plays vital roles in various aspects of cellular metabolism, ranging from energy transduction and apoptosis to the synthesis of important molecules such as heme. Mitochondria are also centrally involved in iron metabolism, as exemplified by disruptions in mitochondrial proteins that lead to perturbations in whole-cell iron processing. Recent investigations have identified a host of mitochondrial proteins (e.g., mitochondrial ferritin; mitoferrins 1 and 2; ABCBs 6, 7, and 10; and frataxin) that may play roles in the homeostasis of mitochondrial iron. These mitochondrial proteins appear to participate in one or more processes of iron storage, iron uptake, and heme and iron–sulfur cluster synthesis. In this review, we present and critically discuss the evidence suggesting that the mitochondrion may contribute to the regulation of whole-cell iron metabolism. Further, human diseases that arise from a dysregulation of these mitochondrial molecules reveal the ability of the mitochondrion to communicate with cytosolic iron metabolism to coordinate whole-cell iron processing and to fulfill the high demands of this organelle for iron. This review highlights new advances in understanding iron metabolism in terms of novel molecular players and diseases associated with its dysregulation. Antioxid. Redox Signal. 15, 3003–3019.
Introduction
Cellular Iron Metabolism and Regulation
Iron is necessary for a variety of important cellular processes, such as oxygen transport, cellular respiration, and DNA synthesis, and there is also a tight link between the metabolism of iron and key metabolic processes such as cell growth, inflammatory pathways, and cell death (121). Before addressing recent findings in the metabolism of mitochondrial iron, we will first briefly describe the relatively well-characterized molecular pathways of cellular iron metabolism and their regulation (44, 46).
Cellular Iron Uptake
In the blood, the majority of iron is bound to the glycoprotein, transferrin (Tf), which contains two high affinity iron(III) binding sites (94, 121). The uptake of iron into the cell is facilitated by the binding of diferric Tf to transferrin receptor 1 (TfR1) on the cell surface, resulting in receptor-mediated endocytosis of the Tf-TfR1 complex (94, 121) (Fig. 1). In addition to TfR1, recent studies have identified two additional mediators of the Tf-mediated iron uptake pathway, namely, the myotonic dystrophy kinase-related Cdc42-binding kinase alpha (MRCKα) (30) and the mammalian exocyst complex Sec15l1 (87). MRCKα is regulated by intracellular iron levels in the same manner as TfR1 (29, 30), and facilitates the organization of the actin cytoskeleton that is necessary for the internalization of the Tf-containing endosome (30). In fact, attenuation of MRCKα expression significantly reduces Tf-mediated iron uptake (30). Sec15l1, on the other hand, may regulate Tf-mediated iron uptake via its postulated role in exocytosis, and it is notable that a mutation in Sec15l1 is responsible for the anemia found in hemoglobin-deficit mice (87, 154).
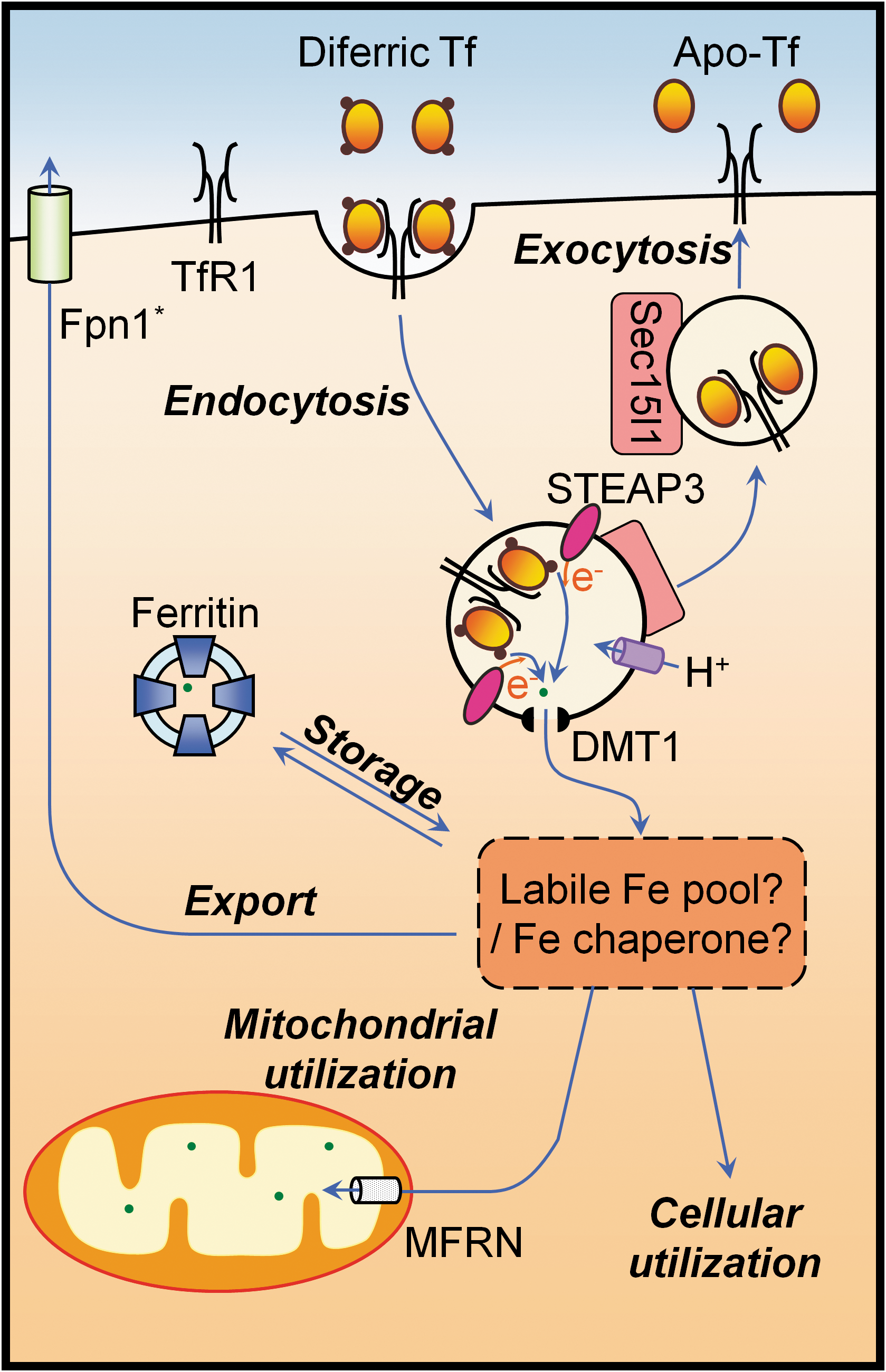
Once internalized, the acidic environment generated by a proton pump within the endosome reduces the affinity of Tf for iron, thereby releasing iron from Tf (44). In erythroid cells, iron(III) that is liberated from Tf is thought to be reduced to iron(II) by six-transmembrane epithelial antigen of the prostate 3 (STEAP3), a ferrireductase within the endosomal membrane (105) and is subsequently exported into the cytosol by divalent metal transporter-1 (DMT1) (47, 48). To date, there are several hypotheses regarding the fate of iron that has been exported from the endosome. The prevailing view is that the iron that has been released from the endosome first enters a low-molecular-weight labile or chelatable iron pool within the cytosol, before being processed in downstream metabolic pathways (67). Alternate hypotheses suggest that iron is either directly trafficked from the endosomal compartment to other membrane-bound compartments, such as the mitochondrion (155), or is sequestered by cytosolic iron–chaperone proteins (146). Unlike the prevailing view, these alternate hypotheses are consistent with studies on iron trafficking in developing erythroid cells, namely, reticulocytes (122, 135, 155).
In addition to the utilization of cytosolic iron for various cellular processes and its subsequent transport into organelles such as the mitochondrion, cytosolic iron can be stored in the cytosolic iron storage protein, ferritin. Alternatively, cytosolic iron can be exported from the cell by the only known iron exporter, ferroportin 1 (Fpn1) (41, 42).
Cellular Iron Regulation
Iron that is not tightly sequestered within a redox-inactive state has the potential to generate ROS, which can be disastrous in the environment of the cell. To minimize these reactions, cellular mechanisms exist to tightly regulate the metabolism of iron depending on intracellular iron levels and metabolic requirements. Within the cell, the regulation of iron metabolism is largely controlled by the iron regulatory proteins (IRP1 and 2), which regulate cellular iron uptake, utilization, export, and storage in a tightly controlled manner. However, at the systemic level, iron homeostasis is primarily regulated by the “hormone of iron metabolism,” hepcidin, which acts through the modulation of Fpn1 expression (52).
IRP/iron-responsive element: cellular iron regulatory system
The IRPs are RNA-binding proteins that bind to iron-responsive elements (IREs), which are specific sequences within the 3′- or 5′- untranslated regions (UTRs) of mRNAs, to modulate the expression of the encoded proteins (96) (Fig. 2). The binding of IRPs to mRNAs containing a 3′ IRE increases the stability of the mRNA [e.g., TfR1 and MRCKα (24, 29)], whereas the binding of IRPs to mRNAs containing a 5′ IRE prevents the translation of the mRNA [e.g., ferritin subunits (60, 62) and Fpn1 (89)]. This has the effect of increasing or decreasing the expression of the encoded proteins, respectively. In addition to the ferritin subunits, TfR1 and MRCKα, IREs have been identified in UTR region of mRNAs encoding proteins that are involved in iron import (DMT1) (56), iron export (Fpn1) (89), erythroid heme synthesis (δ-aminoleuvulinate synthase 2, ALAS2) (36, 39), and the TCA cycle (i.e., mitochondrial aconitase) (156) (Fig. 2).
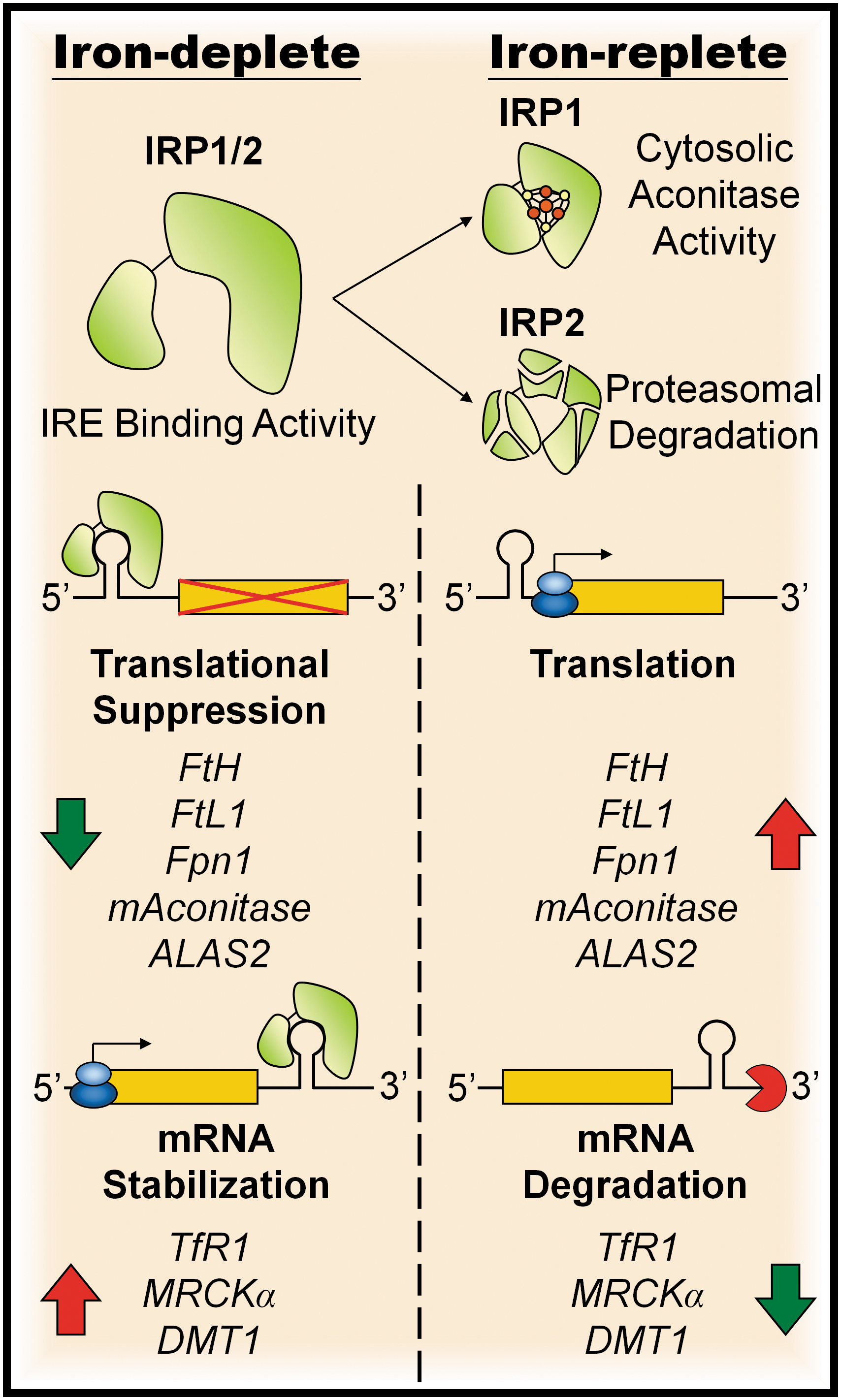
The IRE-binding activity of IRPs is regulated by cellular iron levels, such that iron-deficient conditions promote the binding of IRPs to IREs, and thus the regulation of the corresponding mRNA. In contrast, iron-replete conditions diminish the level of IRE binding by IRPs, thereby reversing the effect on the target mRNA. Although IRP1 and 2 have similar IRE-binding abilities, the mechanisms by which their IRE-binding activity is inactivated at high iron levels are categorically different. Under conditions of high cytosolic iron, IRP1 assembles an iron–sulfur cluster (ISC) to become a cytosolic aconitase (61) and thereby loses its IRE binding ability, whereas IRP2, which lacks aconitase activity, is degraded by an iron-dependent ubiquitin-proteasome system (129, 144).
Although IRP1 and 2 share high sequence similarity, studies that compared the effects of selective disruption of IRP1 and 2 have identified IRP2 as the dominating regulatory protein in homeostasis of cellular iron (91, 148). Studies in mice have demonstrated that the IRE-binding activity of IRP2 is more sensitive to varying iron levels than that of IRP1, and that under normal conditions most IRP1 exists in the aconitase form rather than the IRE-binding form (91). Additionally, deletion of IRP2 in mice causes progressive neurodegeneration (79) and mild microcytic anemia (33, 51), with disruption to the regulation of iron metabolism in several major tissue types including the liver, kidney, brown fat, and brain (91). In contrast, the deletion of IRP1 only causes misregulation of targeted proteins in tissues where endogenous IRP1 expression exceeds that of IRP2 (91). Further, mice with homozygous deletion of IRP2 and heterozygous deletion of IRP1 demonstrated more severe neuropathy than IRP2 deletion alone (139), indicating that IRP1 also contributes to iron homeostasis, but IRP2 remains the dominant regulator.
Hepcidin and cellular iron metabolism
The discovery of hepcidin, an antimicrobial peptide predominantly expressed in the liver (111), has enlightened the field of iron metabolism research. Hepcidin possesses the ability to modulate systemic iron levels presumably by binding to and inducing the internalization and degradation of Fpn1 when plasma-iron levels are high (102). The degradation of Fpn1 reduces iron release from macrophages, hepatocytes, and enterocytes (70, 71, 102, 123). The expression of hepcidin can be regulated by a range of stimuli. Although the molecular details remain unclear, hepcidin may be induced by high plasma-iron levels through signaling by TfR2 (101) and proteins that are mutated in hemochromatosis (HFE) (4, 18) and the membrane-associated form of HFE2 (also known as hemojuvelin) (64). Hepcidin may also be induced by inflammatory cytokines such as interleukin-6 (80, 100). Conversely, hepcidin expression is suppressed under conditions of anemia, hypoxia, and erythropoiesis (82, 103, 110, 145). Recently, a study has identified α2-macroglobulin as the protein that sequesters and transports hepcidin in the blood (114) and potentially that also facilitates the regulatory activity of circulating hepcidin within the body.
Mitochondrial Iron Metabolism
The mitochondrion plays an important role in the metabolism of iron. It is the only site for the synthesis of heme, a vital iron-containing prosthetic group that is required for the functioning of hemoproteins, including hemoglobin. Further, the mitochondrion is a major generator of ISCs that are essential for the electron transport ability of various ISC proteins that contain them. Consequently, a dysregulation of mitochondrial iron processing can have dire consequences for cellular iron homeostasis, as vital proteins such as IRP1 require an ISC for their proper function. Notably, a number of neurodegenerative diseases are associated with misregulated mitochondrial iron metabolism (63), some of which are discussed below.
Iron Transport into the Mitochondrion
Currently, there are three hypotheses for iron delivery into the mitochondrion. First, on the basis of studies performed with isolated mitochondria, Lange et al. (76) proposed that iron may be taken up directly from cytosol by mitochondria as iron(II), a process that appears to be driven by the membrane potential but does not require ATP (76). The second hypothesis states that iron is transported to the mitochondria in a non-labile, chelator-inaccessible form to prevent the occurrence of damaging redox-cycling reactions in the cytoplasm (138). The third hypothesis, called “kiss and run”, proposes that iron is imported into mitochondria via the transient contact of Tf-laden endosomes with the outer mitochondrial membrane (135), a process that potentially involves Sec15l1 (154) (Fig. 3).
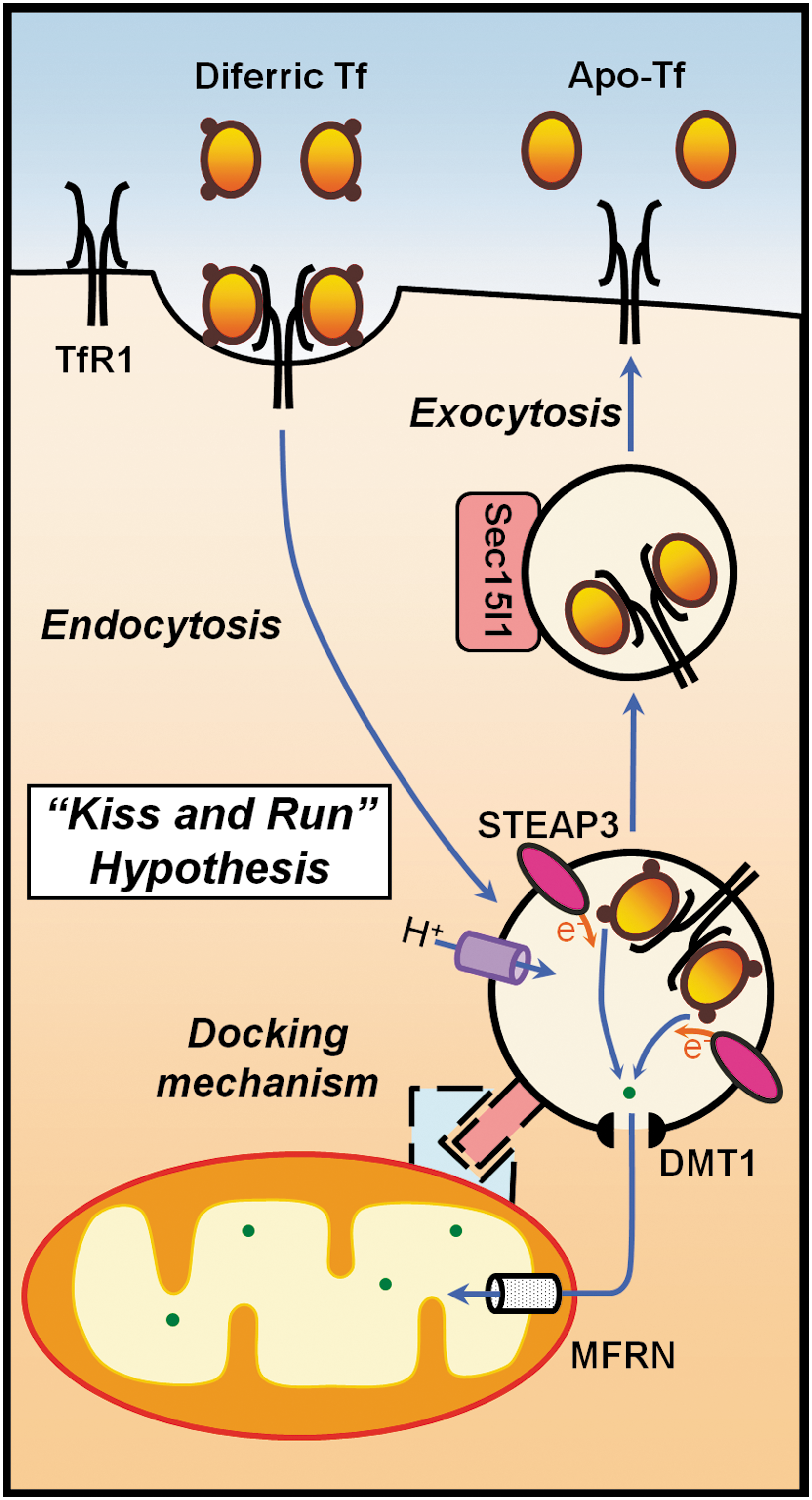
As indicated above, the exact mechanism of iron transport from endosomes to target metalloproteins is still unknown. According to one hypothesis, after endosomal iron release, iron enters the cytosol and is temporarily stored in a low-molecular-weight “labile iron pool” (67, 68). An alternate hypothesis postulates the existence of an iron chaperone and protein-to-protein contact iron transfer. For the latter hypothesis, there is supporting experimental evidence from studies on the maturation of erythroid cells, in which the majority of iron that is released from endosomes must cross both the outer and inner mitochondrial membranes to reach ferrochelatase (Fech). Remarkably, iron continually flows into erythroid cells when the synthesis of protoporphyrin IX (PPIX) is inhibited by isonicotinic acid hydrazide or succinylacetone (17, 95, 117). Considering that Tf-bound iron is efficiently used for hemoglobin synthesis (122) and that virtually no labile iron pool intermediate has been found in reticulocytes, the “kiss and run” iron transfer hypothesis proposes that, at least in erythroid cells, a direct protein-to-protein contact transfer of iron from endosomes to mitochondria occurs that bypasses the cytosol (116, 122, 135, 155) (Fig. 3). In support of this hypothesis, it was shown that 59Fe efflux from endosomes can be intercepted by the membrane-permeant chelators, dipyridyl and salicylaldehyde isonicotinoyl hydrazone, only in metabolically active cells (155). Additionally, myosin light chain kinase inhibitors caused inhibition of 59Fe incorporation from 59Fe-Tf labeled endosomes into heme, suggesting that the intracellular movement of endosomes by myosin is important for iron delivery to mitochondria (155).
A zebrafish mutant with profound anemia, frascati, brought about the discovery of mitoferrin (Mfrn) (133), an ortholog of the previously discovered yeast mitochondrial iron transporters, Mrs3 and Mrs4 (49, 97). The Mfrns are the only transport proteins known to import iron across the inner mitochondrial membrane. In mammals, there are two paralogous genes that encode two closely related proteins, Mfrn1 and Mfrn2, with ∼65% sequence identity. Mfrn1 is localized on the inner mitochondrial membrane and functions as an essential iron importer for mitochondrial heme and ISC synthesis in erythroblasts and is necessary for erythropoiesis (133). Mfrn1 is highly expressed in erythroid cells and at low levels in other tissues, whereas Mfrn2 is ubiquitously expressed, although only at low levels in erythroid cells (133). Recently, ABCB10, an inner mitochondrial membrane ATP-binding cassette (ABC) transporter, was found to physically interact with Mfrn1 and thereby enhance the stability of the protein and increase mitochondrial iron import (28). It is still unclear how iron is transported across the outer mitochondrial membrane, and whether Mfrns are responsible for all iron import into mitochondria.
Three Major Mitochondrial Iron-Processing Pathways
There are three major iron metabolic pathways within the mitochondrion: (i) heme synthesis, (ii) ISC biogenesis, and (iii) mitochondrial iron storage (Fig. 4).
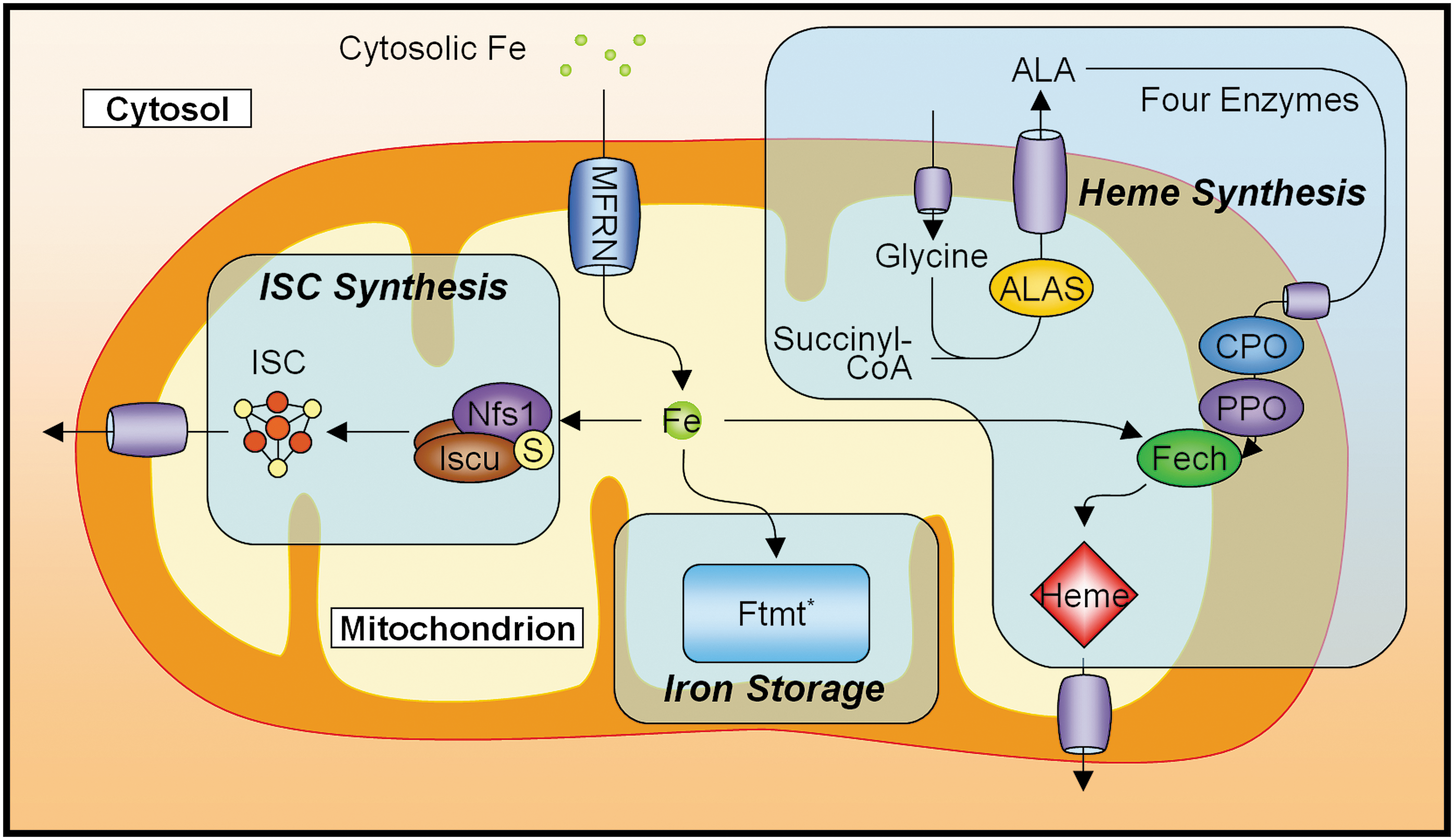
The mitochondrion is a major site of ISC biogenesis, providing ISCs for mitochondrial proteins/enzymes, and either ISCs or ISC precursors for cytosolic and nuclear proteins/enzymes (53, 85). In humans, the assembly of ISCs is facilitated by the cysteine desulferase, Nfs1, which provides sulfur (75, 141), and the splice variants Iscu1 and Iscu2, which may be differentially targeted to the cytosol and mitochondria, respectively (142), and bind iron(II) to function as scaffolds for de novo synthesis of ISCs (50). These clusters are required as cofactors by a number of proteins to perform the functions vital to the working of the cell (e.g., certain enzymes in the respiratory chain and the citric acid cycle). In addition, the production of ISCs can directly affect the regulation of cellular iron metabolism via the IRP1. Therefore, it is not surprising that defects in ISC synthesis, or in the proteins that require them, can have severe implications for health (126, 134, 151). Despite the finding that in mammals the ISC synthetic machinery also exists in the cytosol (141, 143), a defect in ABCB7, a mitochondrial transporter speculated to be involved in the exportation of a cytosolic ISC precursor (85), leads to the disruption of cytosolic ISC protein maturation (14). This defect also leads to a human disease, X-linked sideroblastic anemia with ataxia (XLSA/A) (136), which highlights the importance of mitochondrial ISC synthesis in overall cellular function.
Heme synthesis involves the sequential catalytic ability of eight enzymes and is strictly dependent on the mitochondrion, although four of the intermediate enzymatic steps occur within the cytosol [for an in-depth review, see Ponka (116)]. The biosynthetic pathway spans all compartments of the mitochondrion, as well as the cytoplasm. The first enzyme in the pathway is ALAS, which catalyzes the condensation of glycine and succinyl-CoA to form δ-aminolevulinic acid (116). Two variants of ALAS exist: ALAS2 is expressed specifically in erythroid cells, whereas ALAS1 is expressed in other cell types. Unlike ALAS1, ALAS2 possesses an IRE in its 5′ UTR, and is thus subject to regulation via the IRP system. It is due to this difference that in erythroid cells expressing ALAS2, the rate of heme synthesis may be governed by the availability of cytosolic iron (116). The final enzyme in the heme synthesis pathway is Fech, which inserts iron(II) into the heme precursor PPIX to form heme (116). Interestingly, as mammalian Fech is an ISC-containing enzyme (38), it is therefore conceivable that a disruption to ISC synthesis could also adversely affect the rate of heme synthesis. Indeed, investigations in the past have shown that defects in the ISC synthetic pathways can result in disruptions to heme synthesis (see sections below).
The final pathway that processes iron in the mitochondrion in at least some cell types is the storage of mitochondrial iron in mitochondrial ferritin (Ftmt). Ftmt has ferroxidase activity and forms shells that sequester mineralized iron, similar to cytosolic ferritin (34). However, unlike its cytsolic counterpart, Ftmt lacks an IRE and does not appear to be transcriptionally regulated by iron levels (43). Ftmt is an unique protein that is encoded by an intronless gene, and is normally only abundant in testes. However, Ftmt appears to be expressed at low levels in a number of tissue types (e.g., brain, kidney, thymus, heart, and smooth muscle), where its expression level appears to correlate with the degree of demand for oxidative metabolic activity and mitochondrial density (130). Additionally, the protein appears to be completely absent from tissues that have a known iron storage function (e.g., liver, spleen) (130). Further, previous studies utilizing a cell line that hyper-expresses Ftmt observed pronounced alterations in iron metabolism both in the cytosol and mitochondria (104). This finding indicates the importance of Ftmt in iron metabolism, and further highlights the ability of mitochondrial iron dysregulation to manifest itself as a disruption to whole-cell iron homeostasis. This suggests that the disruption of iron homeostasis within an organelle such as the mitochondrion can lead to changes in cellular and possibly systemic iron processing, which can cause devastating effects at the molecular, cellular, and tissue levels that result in disease.
As mentioned earlier, due to the mitochondrion being a highly redox-active site, the trafficking of iron within the mitochondrion must be tightly regulated to prevent the formation of cytotoxic ROS.
Regulation of Mitochondrial Iron Processing: Possible Roles for Frataxin
A mitochondrial iron regulatory system, analogous to the cytosolic IRP-IRE system, has not yet been identified. Although the details are far from clear, accumulating evidence suggests that the nuclear-encoded, mitochondrial-targeted protein, frataxin, may contribute to the regulation of one or more of the major mitochondrial iron processing pathways, including ISC synthesis, heme synthesis, and iron storage (13, 120).
Frataxin
Since its identification, frataxin has been associated with iron metabolism, particularly in reference to its apparent regulatory role in mitochondrial iron metabolism (120). Notably, frataxin deficiency is the primary cause of FA, a devastating genetic disorder characterized by severe neuro- and cardiodegeneration, as well as extensive accumulation of mitochondrial iron from perturbed cellular iron homeostasis (66). This accumulated iron ultimately leads to the production of toxic ROS (12) (vide infra). Frataxin is a 210 amino acid protein that is associated with the inner mitochondrial membrane and crest (22). However, as it appears to lack any canonical structural motif that would provide a membrane anchor, its association with the membrane may be mediated by its putative ability to form complexes with mitochondrial membrane proteins such as Fech (152). Frataxin has also been found to possess iron-binding capacity in vitro (3, 31), although contradicting findings were reported in another study (98). Studies based on FA patients, mice and other cellular models for frataxin deficiency have suggested a role of frataxin as an iron chaperone (152, 153), an iron scavenger/storage protein (25), and as an iron sensor and regulator (2) within the mitochondrion [for a review, see Richardson et al. (120)].
Frataxin as an iron chaperone
Frataxin was first discovered in yeasts that were deficient in the frataxin ortholog, Yfh1, and consequently demonstrated defects in the synthesis of ISC proteins (11). A similar association between frataxin deficiency and phenotype was subsequently observed in humans and a mouse model of FA (119, 125). Indeed, an emerging body of evidence suggests that frataxin directly participates in ISC biosynthesis (147, 153), as well as the maintenance of ISC proteins (19). Further, other studies have suggested the involvement of frataxin in heme synthesis (81, 152) (see below). It was observed that frataxin can interact directly with Iscu scaffold proteins (153) and Fech (15, 152) as an iron donor, thereby respectively facilitating the synthesis of ISC and the Fech-catalyzed insertion of iron(II) into PPIX to form heme. These results suggest that frataxin may function as a mitochondrial iron–chaperone that transports iron to these crucial pathways.
The exact role of frataxin in heme synthesis is controversial. Although experimental frataxin deficiency leads to defective heme production and alterations in expression enzymes involved in heme synthesis in vitro (132) and in vivo (65), FA patients do not appear to present with anemia. At the very least, this suggests that the level of frataxin deficiency in FA is not sufficient to impair heme synthesis in hematopoietic tissues. Moreover, it has been suggested that the role of frataxin in heme synthesis may only be indirect, resulting from impairment in ISC production, given that mammalian Fech is an ISC-containing protein. However, the similar observation that the depletion of yeast frataxin, Yfh1, disrupts heme synthesis (81), even though yeast Fech is not an ISC-containing protein, instead supports the notion that frataxin can contribute directly to heme synthesis. This hypothesis is further supported by the numerous observations of a direct and functional interaction of frataxin with Fech (81, 113, 152), namely: (i) that the Fech-binding surface on the frataxin molecule has recently been mapped (15); (ii) that human holo-frataxin binds to Fech with nanomolar binding affinity and markedly stimulates Fech activity in a cell-free system (152); and (iii) that both hemoglobinization and PPIX trigger a marked downregulation of frataxin expression in Friend cells (13) (see below). These latter observations provide support for the notion that frataxin plays a direct role in regulating Fech activity and heme synthesis.
Frataxin as an iron storage protein
Frataxin has also been proposed to function as a mitochondrial iron storage protein through the formation of frataxin oligomers that possess ferroxidase activity (1, 113). It was observed that in the presence of excess iron(II), the yeast frataxin ortholog, Yfh1 is capable of undergoing a two-step reaction to form a 48-subunit multimer that can sequester >2000 atoms of iron (112, 113). In addition, the bound iron(II) can be mobilized by physiologic chelators such as citrate for use in heme synthesis (113). If not mobilized, this bound iron becomes mineralized into the redox-inactive iron(III) state that prevents cytotoxicity (108, 113). Similarly, recombinant human frataxin overexpressed in yeast and Escherichia coli is able to assemble into multimers, although the process is spontaneous and is not induced by iron (25). Further, recombinant human frataxin has a lower iron-binding capacity, even under conditions of iron loading, and has been estimated to bind 5–10 iron atoms per molecule (∼50 iron atoms per molecule of Yfh1) (1, 25). The insensitivity of the human frataxin multimer to iron loading, and the discovery of Ftmt in human tissues (43, 83), makes the proposed iron storage function of frataxin seemingly unlikely, or at least redundant. However, Campanella et al. have demonstrated that overexpression of Ftmt in frataxin-deficient yeasts was able to rescue several phenotypes caused by the lack of frataxin (21). This finding suggests that mitochondrial iron binding and detoxifying ability may be an integral function of yeast frataxin (21).
Although the assembly of human frataxin multimers has only been observed in heterologous overexpression systems, this assembly behavior does not appear to be the product of random protein aggregation (25). Indeed, the assembly of multimeric human frataxin involves unique stable subunit interactions at a non-conserved amino-terminal region (107), and appears to serve specific functions, such as the detoxification of redox-active iron (108). The discrepancy in behavior between the function of yeast frataxin and its orthologs in higher organisms may be because yeast do not express ferritins (10). Therefore, perhaps the iron storage capacity observed for yeast frataxin is an acquired function exclusive to yeast and prokaryotes. In support of this, a study has demonstrated that an oligomerization-deficient mutant of Yfh1 can still participate in ISC and heme synthesis (6), suggesting that oligomerization is not necessary for these processes, but may still perform a secondary function.
Frataxin as a mitochondrial iron regulator
The regulation of mitochondrial iron metabolism is another emerging role for frataxin. It was first observed that in erythroid cells, frataxin may act as a metabolic switch to facilitate the distribution of iron between the major mitochondrial iron pathways (13). During erythroid differentiation, frataxin expression is downregulated, potentially to allow for higher rates of heme synthesis at the expense of decreased ISC synthesis (13). This hypothesis is consistent with the observation that an increased level of PPIX, indicating a requirement for heme synthesis, leads to decreased frataxin expression and a diversion of iron from other mitochondrial pathways, such as ISC synthesis, to heme synthesis (13). Further, in support of this hypothesis, Yoon and Cowan (152) showed that rate of heme synthesis is dependent on the molar ratio of frataxin to Fech, whereby frataxin levels higher than 1 frataxin to 1 Fech lead to decreased rates of heme synthesis. Thus, these findings suggest that the regulatory mechanism of frataxin is dependent on its expression level relative to that of its binding partners (e.g., Fech).
More recently, another regulatory mechanism of frataxin has emerged. Studies conducted with the bacterial ortholog of frataxin, CyaY, have indicated that it may act as an “iron sensor” to fine-tune ISC synthesis (2). This hypothesis proposes that CyaY negatively regulates ISC synthesis when iron is in surplus relative to available ISC apo-acceptor proteins (2). ISCs are labile species that can degrade when not rapidly bound to an acceptor protein. In the absence of a negative regulation mechanism, such as that proposed for CyaY, an increase in iron supply coupled with low ISC apo-acceptor availability will lead to the production of an excess of unbound ISCs. The free iron resulting from the degradation of these ISCs would likely result in the production of noxious ROS that could cause oxidative damage to cellular components. Therefore, one can appreciate the significance of this mechanism, whereby iron surplus leads to the binding of CyaY to IscS (bacterial homolog of Nfs1) that inhibits ISC formation (2). However, this proposed regulatory model remains to be thoroughly tested as this study did not assess the regulatory effect of CyaY under the condition of a surplus of both iron and ISC acceptors. The validity of the proposed regulatory model is also dependent on the ability of CyaY, or an unknown protein, to sense the availability of the ISC acceptor relative to the available iron. Further, the study proposed that in the case of frataxin deficiency, such as FA, the precipitation of insoluble iron is due to a surplus of ISCs relative to ISC apo-proteins from uncontrolled ISC synthesis (2). Under this scenario, FA pathology should exhibit functional ISC-containing proteins as ISCs are in excess. Instead, a deficiency of ISC proteins is observed in FA patients and mice models (66, 119, 125), thereby arguing against the extension of this model to higher organisms.
Although the precise function of frataxin is still uncertain, its involvement in mitochondrial iron metabolism is irrefutable.
Regulation of Mitochondrial Iron Transport: The ABCBs
Another group of molecules that are vital to the homeostatic regulation of mitochondrial iron are the mitochondrial transporters that either directly or indirectly facilitate the metabolism of mitochondrial iron via the import or export of substrates related to iron pathways. Disruptions in the transport of these substrates can severely impact the homeostasis of mitochondrial iron, and an accumulation of mitochondrial iron is a typical result. Recent evidence suggests that a number of these transporters physically interact with other molecules in the mitochondrial iron-processing machinery under conditions in which there is increased requirement for mitochondrial iron (27, 28, 140) (Fig. 5), potentially to facilitate the efficient utilization of iron within the mitochondria.
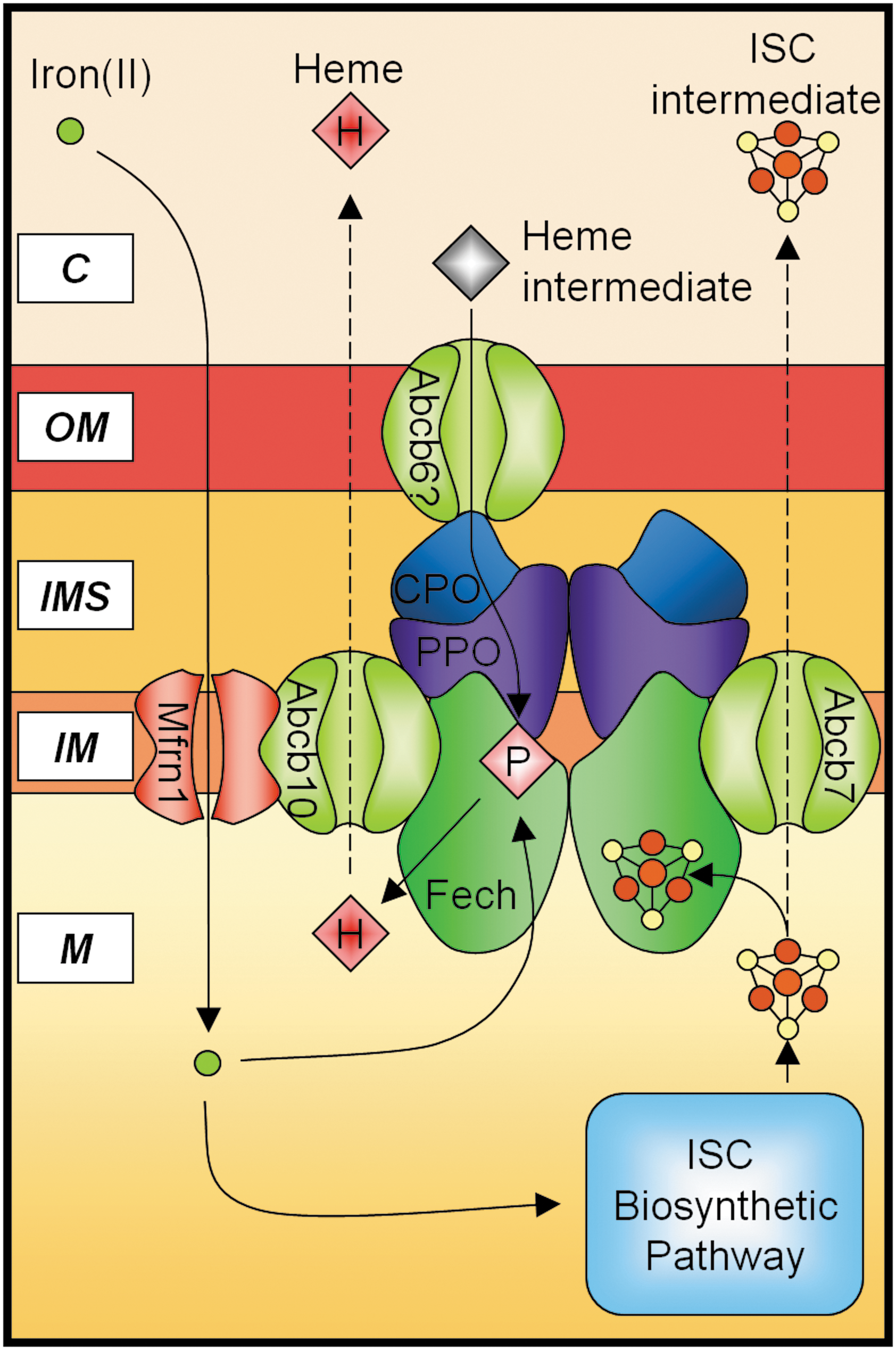
ABC Subfamily B (MDR/TAP), Member 7
The ABC transporter ABCB7 is a mitochondrial half-transporter located in the inner mitochondrial membrane, and has a proposed role in the exportation of a mitochondrial ISC intermediate (14, 115). ABCB7 is an essential protein as its targeted knockout in various tissues is lethal (115). Partial loss-of-function mutations in this gene cause XLSA/A in humans, which is characterized by mitochondrial iron accumulation in erythroblasts and the formation of ring sideroblasts (8, 109). It was first identified in yeast that the deletion of the ABCB7 homolog, ABC transporter in the mitochondrion 1 (ATM1), causes a 30-fold increase in mitochondrial iron and reduced activity of cytosolic, but not mitochondrial, ISC-containing enzymes (69). Subsequent studies have shown that ATM1 is required for maturation of cytosolic ISC proteins, and expression of normal ABCB7 in ATM1-deficient cells fully complemented the defects caused by deletion of ATM1 (14, 37). Thus, ABCB7 was proposed to be the mitochondrial transporter that is responsible for the efflux of a mitochondrial ISC intermediate for cytosolic use (14). Interestingly, ABCB7 appears to play a role in heme synthesis through an interaction with Fech (140), as patients with XLSA/A exhibit mild anemia with elevated free PPIX (8, 109).
ABC Subfamily B (MDR/TAP), Member 10
ABCB10, also known as ABC-mitochondrial erythroid protein (ABC-me), is another ABC mitochondrial transporter that is expressed in the inner mitochondrial membrane (54, 137). Similar to ABCB7, which is proposed to export mitochondrial ISC, ABCB10 has been hypothesized to participate in heme synthesis (137) by exporting an unknown substrate from the mitochondrion to the cytosol. Shirihai et al. (137) first identified ABCB10 in mouse erythroid cells under the control of the transcriptional factor GATA-1, which allows it to be highly induced during erythroid differentiation. The mature form of ABCB10 is also proteolytically processed during erythroid differentiation (28). Overexpression of ABCB10 enhances hemoglobin synthesis. Conversely, ABCB10 transcript expression is significantly downregulated by the addition of exogenous heme, whereas it is unaffected by varying iron levels (137). Further, the addition of the δ-aminolevulinate dehydratase (ALAD) inhibitor succinyl acetone alleviates the reduction in expression elicited by exogenous heme (137), suggesting that expression of ABCB10 is subjected to regulation by intracellular heme levels.
Structurally, ABCB10 resembles a half-transporter that functions as a homo- or heterodimer, which probably exports an as-yet-unknown heme biosynthetic intermediate from the mitochondrial matrix to the intermembrane space (54, 137). Recent studies in murine erythroid cells have found that ABCB10 interacts with Mfrn1, the erythroid-specific mitochondrial iron importer, to stabilize the expression of Mfrn1 and thereby increase mitochondrial iron uptake during erythroid differentiation (28), presumably to enhance heme synthesis by providing iron to Fech for PPIX metalation (9). Indeed, a subsequent study found that Fech immunoprecipitates with both Mfrn1 and ABCB10, suggesting the formation of an Mfrn1-ABCB10-Fech hetero-oligomeric complex that directly couples mitochondrial iron uptake to heme synthesis during erythroid maturation (27). Native gel separation has also identified the formation of a higher-order multimeric complex with Mfrn1-ABCB10-Fech (27, 28). This multimeric complex could potentially also consist of the terminal heme biosynthetic enzymes as shown previously (72, 90, 118), thereby tightly regulating the synthesis of heme with mitochondrial iron uptake in the developing erythron, to avoid the build-up of toxic intermediates (Fig. 5). The strong association between ABCB10 and the heme synthetic pathway suggests that ABCB10 may be a mitochondrial heme exporter. However, the occurrence of similar interactions in the human homolog remains to be tested.
ABC Subfamily B (MDR/TAP), Member 6
ABCB6 was first identified as mtABC3 in humans, and exhibits 31.1% identity to yeast ATM1 and 34.1% identity to human ABCB7 (92). The expression of ABCB6 in ATM1-deficient cells rescues the phenotype of mitochondrial iron accumulation and mitochondrial DNA damage, similar to ABCB7 (92). The protein is highly expressed in heart and skeletal muscles (92). Further, the lethal neonatal metabolic syndrome, a disorder of mitochondrial function associated with iron metabolism, may be caused by defects in ABCB6 (92). In addition, ABCB6 has been speculated to be the coproporphyrin importer (74).
Diseases Related to Dysregulation of Mitochondrial Iron Metabolism
Many molecular players and mechanisms involved in iron metabolism have been discovered and elucidated by the analysis of diseases or mutant animals. The regulation of mitochondrial iron is no exception. As the function of the mitochondrion is fundamental to the viability of the cell, mitochondrial damage can have severe pathological consequences (84, 131). As a consequence of its high and dynamic redox activity, a dysregulation of mitochondrial iron processing predisposes the mitochondrion to iron accumulation, and the subsequent formation of noxious ROS that this accumulation engenders. Accordingly, neuronal and cardiac cells are often the most susceptible to mitochondrial dysfunction, due to their high mitochondrial content, and disorders stemming from mitochondrial dysfunction often manifest as neurological and cardiological symptoms. It is therefore important to identify molecules in mitochondrial iron metabolism. Genetic disorders that exhibit mitochondrial iron loading are “tell-tale” signs of dysregulation of mitochondrial iron and are therefore essential to the understanding of mitochondrial iron metabolism. The following section describes some of these disorders that have provided researchers with a glimpse into the growing field of mitochondrial iron metabolism.
Friedreich's Ataxia
FA (Online Mendelian Inheritance in Man, OMIM #229300) is an autosomal recessive spino-cerebellar disorder that affects ∼1 in 50,000 Caucasians (23). It is the most prevalent inherited ataxia, and is characterized by progressive neuro- and cardiodegeneration and mitochondrial iron accumulation (7, 23). A GAA repeat expansion hinders the transcription of the FRDA gene (128), causing a marked reduction in expression of frataxin (22). This genetic defect accounts for ∼98% of FA cases, whereas the remainder is due to point mutations (23).
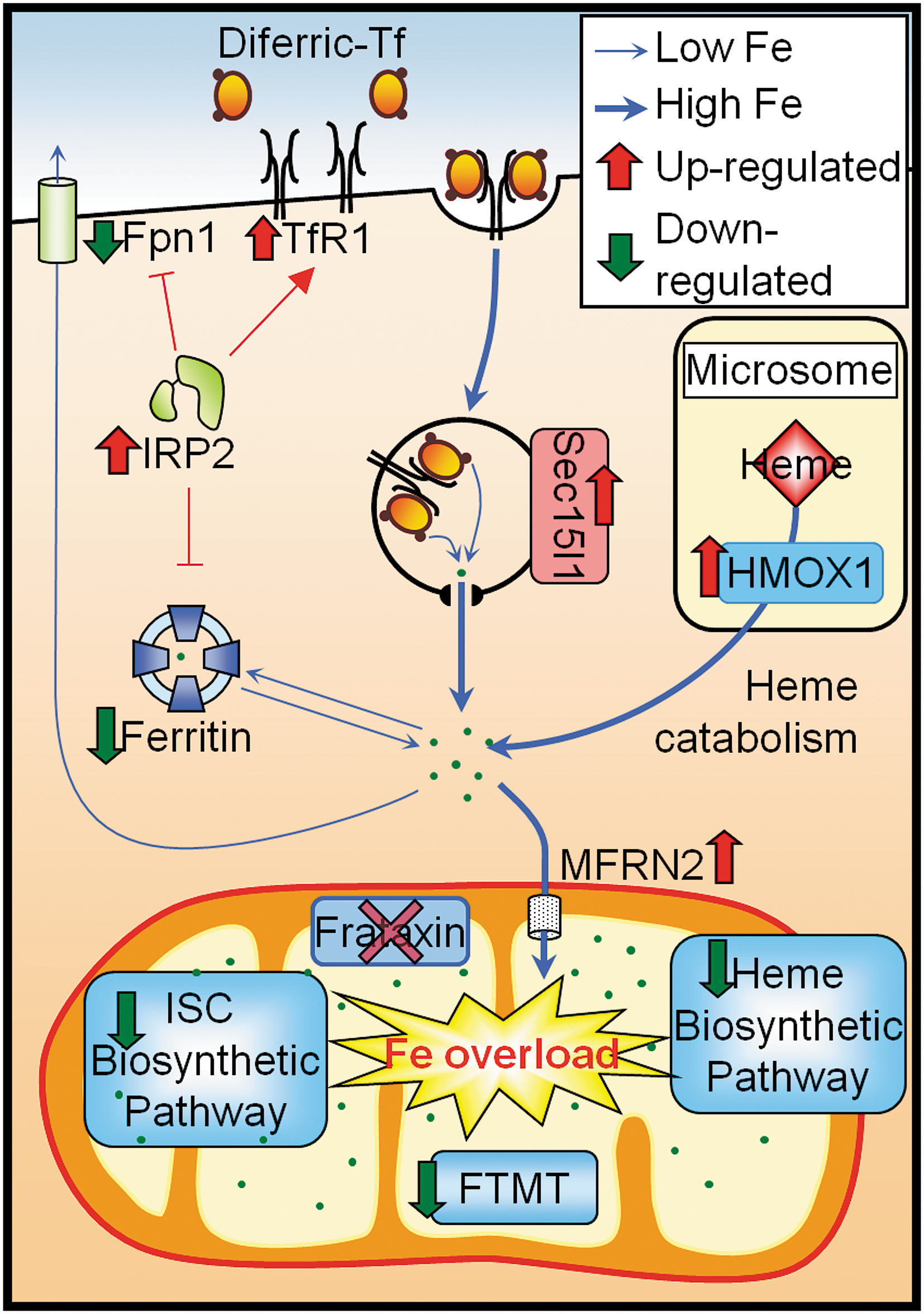
Patients suffering FA exhibit progressive neurological disability and cardiomyopathy with a tendency for diabetes mellitus (45). The pathogenesis of FA is associated with mitochondrial iron accumulation that results in ROS induced toxicity (66, 149). As such, iron chelation therapy has been shown to be beneficial in reducing both neurologic and cardiologic FA pathology, presumably by preventing oxidant-mediated cell death (12, 16, 32, 149). In addition to mitochondrial iron accumulation and oxidative damage, FA patients also exhibit a deficit of ISC enzymes, leading to decreased energy metabolism, as well as perturbed heme synthesis (66). Recent studies utilizing a conditional frataxin knockout mice model of FA have demonstrated that mitochondrial iron dysregulation can result in changes in whole-cell iron regulation, whereby increased iron uptake is coupled with targeted trafficking of iron to the mitochondria, where frataxin is deficient (66, 149). Further, frataxin deficiency causes reduced ISC and heme synthesis, which is characterized by the decreased expression of enzymes that are involved in mitochondrial iron processing, diminished levels of heme and ISC products and depressed mitochondrial iron storage (66) (Fig. 6). Results from this study also demonstrated the mechanism of mitochondrial iron loading, which has confirmed frataxin's role as a central regulator in mitochondrial iron metabolism (66).
X-Linked Sideroblastic Anemia
XLSA (OMIM #301300) is another genetic disorder in which mitochondrial iron accumulation occurs (Fig. 7A). XLSA is caused by a dysfunction in heme synthesis, specifically due to mutation in the erythroid-specific ALAS2 gene, located at Xp11.21 (35). A deficiency in ALAS2 results in impaired heme-synthesis, hemoglobin production, and hyperferremia; and death from hemochromatosis frequently occurs. The formation of ring sideroblasts in XLSA is characteristic of iron-loaded mitochondria clustered around the nucleus of nucleated erythroid cells. The accumulation of mitochondrial iron in XLSA suggests that iron continues to enter the mitochondrion despite dysfunctional heme synthesis. Together with the in vitro observation that the inhibition of heme synthesis in reticulocytes leads to mitochondrial iron loading (117, 122), these observations suggest that the uptake of mitochondrial iron for heme synthesis may be regulated by a heme product (116). Further, as the upregulation of Ftmt is significantly correlated with the XLSA phenotype, the accumulation of mitochondrial iron may be partially facilitated by Ftmt (26) (Fig. 7A).
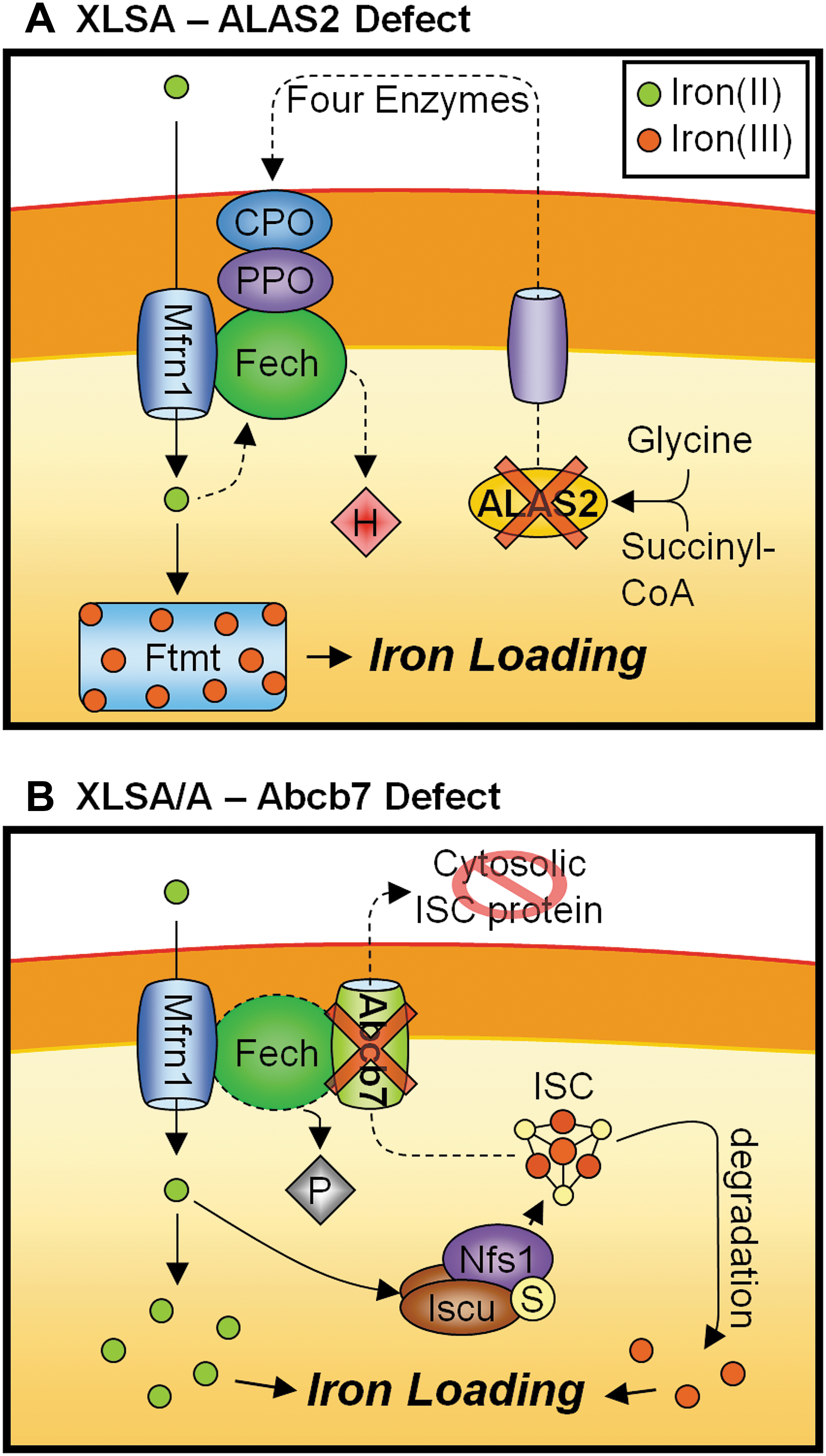
Importantly, XLSA differs from the phenotypically related disorder, XLSA/A, which is also caused by mitochondrial dysfunction (see section below), in that it lacks the neuropathy typical of mitochondrial dysfunctions. This discrepancy is probably due to the fact that ALAS2 expression is confined to erythroid cells, whereas the ubiquitously expressed ALAS1 is responsible for heme synthesis in other tissues including neural tissues (116).
It is worth noting that a new form of human sideroblastic anemia has been recently identified to be due to the mutation in an erythroid specific mitochondrial transport protein, SLC25A38 (55). This mutation causes an autosomal recessive disorder, but is also characteristic of a non-syndromic form of severe microcytic anemia with systemic iron overload, similar to that of XLSA. Based on sequence and functional analysis, it is hypothesized that SLC25A38 facilitates heme synthesis by importing glycine, one of the initial substrates of heme synthesis, into the mitochondrion (55).
X-Linked Sideroblastic Anemia with Cerebellar Ataxia
XLSA/A (OMIM #301310) is caused by mutation in the human gene ABCB7, mapped to chromosomal location Xq13.1 (14, 136) (Fig. 7B). This mutation leads to a recessive disorder characterized by a mild sideroblastic anemia with an early onset, non-progressive cerebellar ataxia. Although the symptoms of mitochondrial iron loading in erythroblasts and formation of ring sideroblasts is similar to that of XLSA, the XLSA/A disorder also features elevated free erythrocyte PPIX levels, and lacks excessive parenchymal iron loading (109). This difference is because ABCB7, which is deficient in XLSA/A, does not have a direct role in heme synthesis. Previous studies have shown that ABCB7 and its yeast homolog ATM1 are involved in the exportation of a mitochondrial ISC intermediate, and facilitates the maturation of cytosolic ISC proteins [see above ABC Subfamily B (MDR/TAP), Member 7 section] (5, 37, 115, 136). However, the mild hypochromic microcytic anemia that occurs in XLSA/A patients indicates defective heme synthesis in the developing erythroid cells. Further, the elevated PPIX levels (109) and the mitochondrial iron loading (109) suggest that the defect lies in the final step of heme synthesis: the incorporation of ferrous iron into PPIX to form heme by Fech (Fig. 7B).
Although it is conceivable that disruptions in ISC synthesis can impede Fech's function, as Fech is an ISC-containing protein (38, 77), exactly how a disruption in ISC export can have the same effect is unclear. The activity of Fech in XLSA/A patients has not been reported, but normal Fech expression was observed in a yeast ATM1 mutant (77). However, unlike the mammalian homologs, yeast Fech does not contain a [2Fe-2S] cluster at its carboxyl-terminus (40). Therefore, mutations in ABCB7 and its orthologs may have different effects in yeast and human cells.
Interestingly, a study utilizing mouse erythroleukemia cells showed direct involvement of ABCB7 in heme synthesis via its interaction with Fech (140). In particular, this study demonstrated a parallel increase in ABCB7 and Fech mRNA during erythroid differentiation. Further, in vitro pull-down assays using an anti-Fech antibody revealed that the ABCB7 protein interacts with the ISC-containing carboxyl-terminus region of Fech. Moreover, the transfection of anti-sense oligonucleotides directed against ABCB7 in differentiated mouse erythroleukemia cells showed a strong decrease in intracellular heme content. Conversely, the overexpression of ABCB7 leads to marked increases in both heme synthesis and Fech protein expression (140). These results indicate that ABCB7 positively regulates Fech's expression and heme synthetic activity during erythroid cell differentiation (140). Thus, in XLSA/A, in which ABCB7 is deficient, the anemia observed could be due to the dysregulation of Fech activity resulting from a disruption of the interaction between ABCB7 and Fech. However, this result is yet to be confirmed in humans.
Sideroblastic Anemia Associated with Glutaredoxin 5 Deficiency
Another disorder that highlights the close association between heme and ISC synthesis is the sideroblastic anemia caused by glutaredoxin 5 (Glrx5) mutations (20, 150, 151) (Fig. 8A). Glrx5 is an important mitochondrial protein for the synthesis and assembly of ISC centers, with deficiency of Glrx5 resulting in marked mitochondrial iron accumulation and decreased activity of mitochondrial and cytosolic ISC proteins (124, 151). Consequently, the activity of IRP1 was strongly affected by Glrx5 deficiency, whereby the IRE binding activity was activated in the absence of an ISC center (150), leading to dysregulated cellular iron homeostasis. Thus, the microcytic anemia observed in human and other vertebrates with reduced Glrx5 was primarily mediated by the decrease in erythroid-specific ALAS2 as a result of an increase in IRP1 RNA-binding activity (20, 150) (Fig. 8A). Consistent with this result, it was found that Glrx5 may have an important role in erythropoiesis, as Glrx5 is highly expressed in erythroblasts and was strongly induced during erythroid differentiation (151). Further, Fech expression was greatly reduced in Glrx5-deficient erythropoietic cells, possibly due to the inability to assemble a [2Fe-2S] cluster (151). Reduced Fech expression further hampers the synthesis of heme and is thought to contribute to the pronounced hypochromic anemia observed in the human patient (151). Thus, these studies showed that the disruption to heme synthesis in Glrx5-deficient erythroid cells is secondary to reduced ISC synthesis, and further highlights the tight linkage between mitochondrial iron-processing pathways (Fig. 8A).
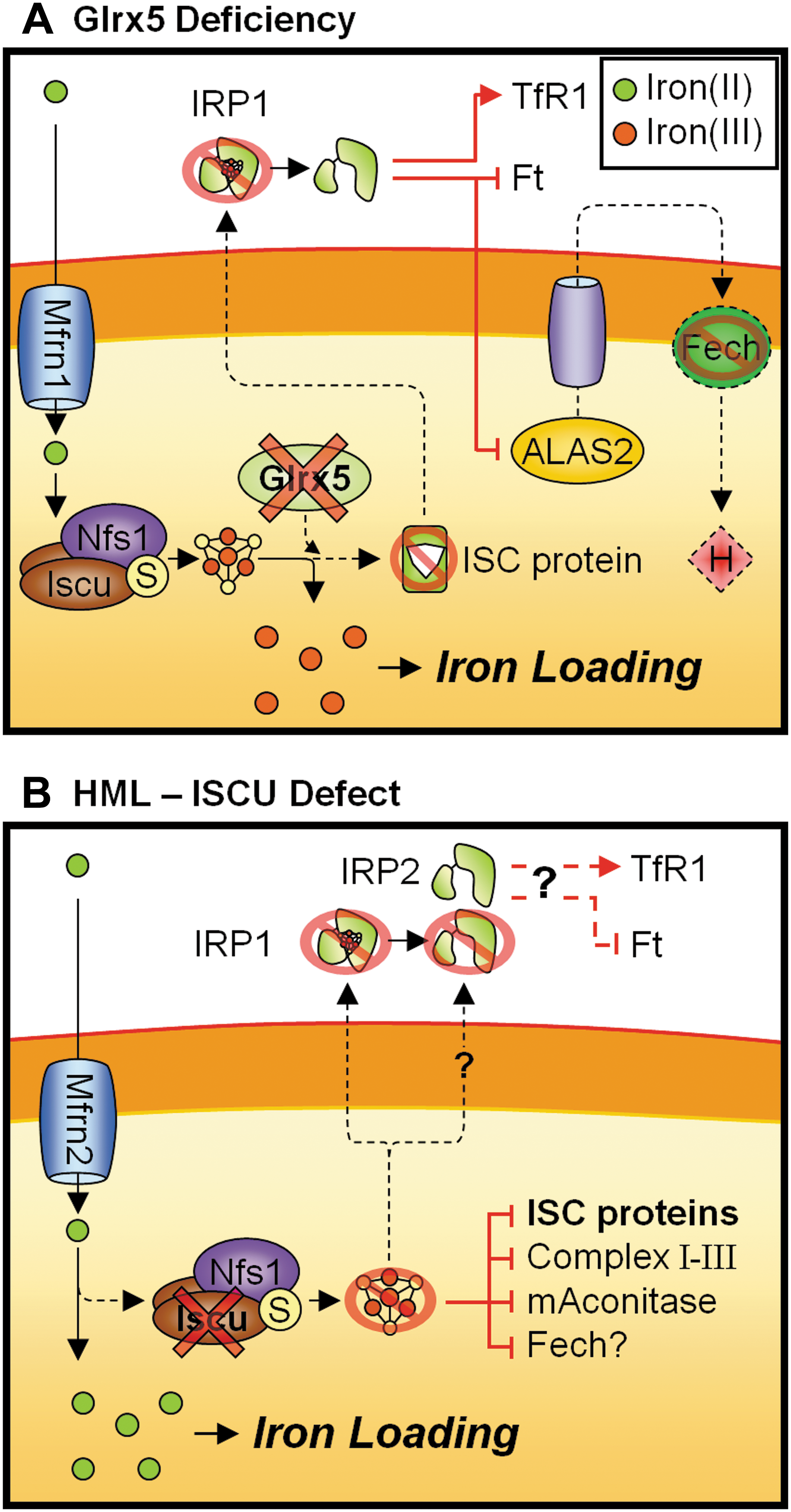
Hereditary Myopathy with Lactic Acidosis
Hereditary myopathy with lactic acidosis (HML; OMIM #255125) is caused by an intronic mutation in the human gene for the ISC synthesis scaffold protein, Iscu, affecting both cytosolic (Iscu1) and mitochondrial (Iscu2) splice variants (93, 106) (Fig. 8B). A detailed report of this disorder was first published in 1964 that describes patients of northern Swedish origin affected by moderate to severe myoglobinuria, acidosis, and exercise intolerance (78). Subsequent investigations have identified mitochondrial iron deposition and abnormalities in ISC-containing proteins, including subunits of complexes I–III of the respiratory chain and mitochondrial aconitase in skeletal muscle of HML patients (58, 59, 88). These findings suggest that the cause of this disorder is due to dysfunction in the biogenesis, processing, or assembly of ISCs (58). Consistent with this notion, two recent reports concurrently identified an intronic G>C mutation within chromosome region 12q23.3–24.11 that results in a splicing defect in the Iscu gene, leading to the generation of a novel Iscu transcript containing an additional pseudoexon that results in a truncated Iscu protein (93, 106). The resultant splice alteration leads to a decrease in the total Iscu mRNA levels and drastically reduced protein levels in patient muscles (93).
Consistent with reduced ISC synthesis, decreased activity of cytosolic (IRP1) and mitochondrial aconitases were observed in HML patient muscles (93). IRP1 protein level and the amount of activatable IRP1 available for IRE-binding was also found to be reduced in patients (93). Despite abnormal IRP1 IRE-binding activity, accumulation of mitochondrial iron was observed in the patients' skeletal muscle (59, 73, 93). This is potentially mediated by IRP2 due to the higher muscle-specific expression (57), although expression and activity of IRP2 have not been assessed in HML patients (Fig. 8B).
Unexpectedly, the pathological phenotype and reduction of Iscu protein appear to be confined to skeletal muscles as fibroblasts harvested from affected patients appear to be asymptomatic (73, 93). Moreover, dysfunctional ISC-proteins and iron accumulation were not uniformly distributed within skeletal muscle fibers (73, 93). The tissue- and cell-specific Iscu deficiency is hypothesized to be due to the balance of naturally occurring pseudoexon-carrying transcript and the normal transcript (73, 93). In humans, the predicted probability for the aberrant transcript to occur naturally is relatively high, at ∼87%, but is potentially secured from spliceosome assembly by the formation of a secondary mRNA structure (73). The intronic G>C mutation increases this probability to ∼90%–95%, thereby increasing the occurrence of the aberrant transcript (73, 93). However, residual expression of the normal transcript has been observed in the affected skeletal muscles (73, 93). Potentially, this residual expression might be sufficient for basal cellular ISC requirements, consistent with patients exhibiting normal muscular functions at rest (58, 59, 73), and may also explain the mosaic pattern of ISC deficiency among muscle fibers (73, 93).
On the other hand, the tissue-specific distribution of the normal and aberrant transcript is likely to be a pathognomonic feature due to spliceosome variance inherent to the tissue type, or tissue-specific expression of partner proteins that affects alternate splicing (127). More recently, a report described a pair of siblings heterozygous for the G>C intronic mutation and a missense mutation in the other allele that resulted in a nonfunctional protein (73). The clinical features are atypical of HML symptoms, showing more severe muscle wastage and evidence of cardiomyopathy (73). The additional pathology in the compound heterozygous patients is consequential to the expression of non-functional protein that further exacerbates basal Iscu expression. Interestingly, further to the tissue specificity, no hematological defects were observed in both forms of HML (58, 59, 73), as would be expected in disorders of defective ISC synthesis (e.g., XLSA/A).
Summary and Future Perspective
Research in the past decade has unraveled a number of mitochondrial proteins that play key roles in the metabolism of iron within the mitochondrion, such as Ftmt, Mfrn1/2, frataxin, Glrx5, and members of the ABC superfamily of mitochondrial transporters (ABCB6, ABCB7, and ABCB10), just to name a few. These discoveries have provided researchers in the field of iron metabolism with a better understanding of the regulatory controls that exist in the cell and the mitochondria, which work to maintain a tight regulation of iron metabolism. Indeed, in many cases, mutations in these mitochondrial molecules lead directly to human diseases ranging from slight anemia to devastating neurological and cardiological disorders such as FA.
Undoubtedly, there are still pieces missing from the puzzle of mitochondrial iron metabolism. Such pieces include (i) the identification of the molecule that mediates the communication between the mitochondrion and the cytosol; (ii) whether this iron communicator is associated with a mitochondrial iron product such as heme or ISC's; (iii) the precise involvement of frataxin in the regulation of mitochondrial iron processing; and (iv) whether frataxin functions in one or more of the four partially overlapping models proposed herein. Further research in this intriguing field is still required, but accumulating evidence to date has unquestionably identified the mitochondrion as a site of intense iron metabolic activity, where the modulation of mitochondrial iron requirements can dictate the processing of iron at the whole-cell level.
Footnotes
Acknowledgments
D.R.R.'s laboratory was supported by grants from the National Health and Medical Research Council of Australia, the Muscular Dystrophy Association USA, Muscular Dystrophy Association New South Wales, and Friedreich's Ataxia Research Alliance Australia.