
Abstract
A cell's “redox” (oxidation and reduction) state is determined by the sum of all redox processes yielding reactive oxygen species (ROS), reactive nitrogen species (RNS), and other reactive intermediates. Low amounts of ROS/RNS are generated by different mechanisms in every cell and are important regulatory mediators in many signaling processes (redox signaling). When the physiological balance between the generation and elimination of ROS/RNS is disrupted, oxidative/nitrosative stress with persistent oxidative damage of the organism occurs. Oxidative stress has been suggested to act as initiator and/or mediator of many human diseases. The cerebral vasculature is particularly susceptible to oxidative stress, which is critical since cerebral endothelial cells play a major role in the creation and maintenance of the blood–brain barrier (BBB). This article will only contain a focused introduction on the biochemical background of redox signaling, since this has been reported already in a series of excellent recent reviews. The goal of this work is to increase the understanding of basic mechanisms underlying ROS/RNS-induced BBB disruption, with a focus on the role of matrix metalloproteinases, which, after all, appear to be a key mediator in the initiation and progression of BBB damage elicited by oxidative stress. Antioxid. Redox Signal. 15, 1305–1323.
Introduction
Oxidative stress: Cellular and physiological background
ROS are produced by a great variety of mechanisms and originate from many sources. Exogenous sources include exposure to di-oxygen, ozone (a powerful oxidizing agent that can oxidize biological components directly), ionizing and nonionizing irradiation, UV radiation, air pollutants, cigarette smoke, industrial contaminants, the invasion of pathogens, and food that may contain various kinds of oxidants, including peroxides, aldehydes, oxidized fatty acids, and transition metals (89). Endogenously, two ROS-generating systems exist: a) the leakage of activated oxygen from mitochondria during oxidative phosphorylation; and b) oxygen-metabolizing enzymatic reactions such as xanthine oxidase, cytochromes P450, NADPH oxidases, myeloperoxidase, and nitric oxidase synthase (140).
ROS can basically be subdivided into two groups: a) The radical group containing superoxide ion radical, hydroxyl radical, nitric oxide radical, peroxyl and alkoxyl radicals, and one form of singlet oxygen; and b) nonradical compounds comprising a large variety of substances such as hypochlorous acid, hydrogen peroxide, organic peroxides, aldehydes, ozone, and oxygen (89). The terms reactive oxygen species, reactive nitrogen species, oxygen-derived species, pro-oxidants and oxidants are often used synonymously in the literature. In the following, oxidative stress and nitrosative stress will be referred to as oxidative stress only.
Longstanding medical interest has focused on the deleterious effect of oxidative stress on the cerebral microvasculature. The compromised functional integrity of the BBB leads to increased permeability of cerebral microvessels, which contributes to the pathology of a number of neurological diseases with inflammatory characteristics (reviewed in Ref. 140). A list of major prooxidant enzymes in cerebral endothelial cells (cEC) is shown in Figure 1.

Reactive oxygen species
The superoxide anion (·O2 −) is formed by the univalent reduction of triplet-state molecular oxygen (3O2). Its intracellular generation is mediated by enzymes such as NAD(P)H oxidases and xanthine oxidase or nonenzymatically by redox-reactive compounds such as the semi-ubiquinone compound of the mitochondrial electron transport chain or in auto-oxidation reactions. ·O2 - is relatively unreactive with a half-life time of about 10−3 sec, compared to the highly reactive hydroxyl radical (·OH) (half-life time of about 10−10 sec) and has a rather poor ability to cross biological membranes. ·O2 - can enzymatically and nonenzymatically be converted to hydrogen peroxide (H2O2) which, in turn, may either be converted into water by the enzymes catalase or glutathione peroxidase (GSHPx), or, in the presence of reduced transition metals (ferrous or cuprous ions), to the hydroxyl radical ·OH (34).
·O2 - is capable of acting directly on cEC, thereby modulating the permeability of the BBB. It has been suggested that ·O2 - is produced during the firm adhesion of monocytes to cEC, which triggers cytoskeletal rearrangements allowing infiltration of monocytes into the brain (170).
Reactive nitrogen species
NO is formed by three different mammalian types of NO synthases: the constitutive isoforms endothelial NO synthase (eNOS/NOS3) and neuronal synthase (nNOS/NOS1), and the inducible isoform (iNOS/NOS2). NO plays an important role in the regulation of cerebral blood flow and neuronal cell viability; however, excessive production of NO by iNOS and nNOS in the brain participates in neurotoxicity (162).
Actually, NO is a weak oxidant and, by itself, is hardly capable of inducing oxidative stress. However, as shown in a rodent brain perfusion model, various NO redox forms (NO·, NO+, NO-) exert different permeability effects on the BBB. While NO· causes only modest BBB disruption, a combination of NO·/NO- or NO·/NO-/NO+ elicits severe BBB damage, as reflected by 14C-sucrose uptake (18).
By interaction of NO· and ·O2 -, formation of the highly noxious peroxynitrite (ONOO-) occurs. The protonated form of ONOO- is a powerful oxidizing agent and exerts deleterious effects in biological systems by causing the oxidation of DNA and the nitration of aromatic amino acid residues in proteins (e.g., forming 3-nitrotyrosine) (34, 89). At physiological pH, ONOO- may decompose through the intermediate formation of its conjugated acid ONOOH, which may either form the very reactive radicals NO2 - and ·OH or may react in biological conditions with carbon dioxide (154).
BBB dysfunction elicited by NO and NO-derivatives is well documented (129, 156). Transcriptional upregulation of eNOS and iNOS, as well as increased production of 3-nitrotyrosine, appears to be characteristic of BBB breakdown and cerebral edema following cerebral trauma (120). iNOS-deficient mice showed significantly reduced disruption of the BBB following experimental bacterial induction of meningitis compared to wild-type mice, which was associated with a lack of ONOO- and 3-nitrotyrosine formation (180). The purine metabolite uric acid proved to be a powerful scavenger of ONOO- in inflammatory brain disease (83). In a mouse model of active experimental autoimmune encephalomyelitis (the mouse correlate of human multiple sclerosis), exogenously administered uric acid was shown to act at two levels. It reduced BBB damage and penetrated the compromised BBB to block ONOO--mediated tyrosine nitration and apoptotic cell death in inflamed neuronal tissue (67).
Targets of oxidative damage
Numerous studies have demonstrated that ROS can attack virtually all biological molecules. The major targets are lipids, proteins, and DNA and are discussed in more detail below.
Lipids
Free radicals attack the phospholipid components of cellular membranes, leading to alterations of membrane permeability and lipid fluidity, as well as modified lipid–protein interactions (58, 191). The damage to lipids, termed lipid peroxidation, is a chain reaction. It is initiated by any primary free radical that has sufficient reactivity to abstract a hydrogen atom from a reactive methylene group of a polyunsaturated fatty acid. The formation of the initiating complex is followed by a bond rearrangement. The lipid radical then takes up oxygen to form the peroxy radical, which can combine with each other and even attack proteins.
The breakdown of lipid peroxides generates radicals that propagate lipid peroxidation and also nonradical fragmentation products such as malondialdehyde, alkanals, 2-alkenals, and 4-hydroxyalkenal, many of which are biologically active. The highly reactive 4-hydroxynonenal (4-HNE) is the biologically most significant class of peroxidation products. The main aldehydes that have been found in peroxidizing biological samples are malondialdehyde, hexanal, propanal, and 4-HNE (26, 28, 37, 38).
Increased plasma levels of malondialdehyde and nitrates/nitrites were found to be associated with hypoxic/ischemic encephalopathy (97). In vitro, H2O2-induced oxidative stress was shown to induce lipid peroxidation and membrane dysfunction in cerebral endothelial cells, which could be reduced by decomposition of free radicals by nitronyl nitroxides (17). Further, in an in vitro BBB model, 4-HNE was shown to increase the barrier permeability, obviously by decreasing the content of reduced glutathione in cEC (113). A genotoxic effect of 4-HNE was reported by Karlhuber et al. (82), showing that treatment of cultured cEC with 4-HNE resulted in chromosomal aberrations and the formation of micronuclei.
Proteins
ROS-mediated oxidation of proteins can result in protein crosslinking and increased proteolytic degradation, inactivation, or altered cellular functions. Numerous modifications of amino acid side chains may occur, including hydroxylation of aromatic groups and aliphatic amino acid side chains, nitration of aromatic amino acid residues, nitrosylation of sulfhydryl groups, sulfoxidation of methionine residues, chlorination of aromatic groups and primary amino groups, and conversion of some amino acid residues to carbonyl derivatives (26, 145). Methionine residues and cysteine residues of proteins are particularly sensitive to oxidation by ROS but, unlike oxidation of other amino acid residues, the oxidation of these sulfur amino acids is reversible. Protein damage is mainly caused by ·OH, RO·, and nitrogen-reactive radicals, yielding protein oxidation products such as aldehydes, keto compounds, and carbonyls. During the last couple of years, increasing attention has been paid to the importance of protein oxidation in aging, supported by the observation that levels of oxidized proteins increase with age (144, 146). This age-related accumulation of oxidized proteins may reflect age-related increases in rates of ROS generation, decreases in antioxidant activities, or losses in the capacity to degrade oxidized proteins. As mentioned above, one of the major adducts during protein oxidation is produced by the interaction between ONOO- and other nitrogen reactive radicals with the amino acid tyrosine, forming 3-nitrotyrosine. This adduct can easily be detected in tissues and plasma and is used as a marker for oxidative damage of proteins (130). The increased production of 3-nitrotyrosine contributes to BBB disruption. For instance, in Aß42 peptide-injected rat hippocampus, the selective inhibition of iNOS reduced 3-nitrotyrosine levels and diminished BBB leakiness to IgG (139).
DNA
Some ROS produce a multiplicity of modifications in DNA, such as release of free bases from DNA and generation of strand breaks with various sugar modifications and damage to the DNA repair system. Not all ROS can cause damage. ·O2 − and H2O2 are normally not reactive towards DNA. However, upon metal catalyzation, H2O2 and ·O2 might be converted to ·OH known to readily attack DNA. A wide spectrum of oxidative modifications, which yield oxidation products such as 8-hydroxydeoxyguanosine (8-OHdG) (reviewed in (89)), or 8 (or 4-, 5-)-hydroxyadenine, thymine peroxide, thymine glycols, and uracil glycol, are elicited by ROS. In addition, peroxynitrite can easily cause DNA damage similar to that obtained when ·OH is involved. The biological consequences of many of the oxidative products are known. For example, thymine glycol interferes with DNA replication and is potentially lethal to cells, while 8-oxoG is readily bypassed by the DNA polymerase and is highly mutagenic. Unrepaired 8-oxoG will mispair with deoxyadenosine (dA), leading to an increase in G (guanine) to T (thymine) transition mutations (138).
Cytotoxic and genotoxic effects of oxidative stress were demonstrated using cultured cEC (21). Aglycemic culture conditions exacerbated the cytotoxic effects of oxidative insult, as evidenced by increased apoptosis and decreased mitotic activity of cEC (21).
Endothelial and Epithelial Tissue Barriers: Biological Systems Vulnerable to Oxidative Stress
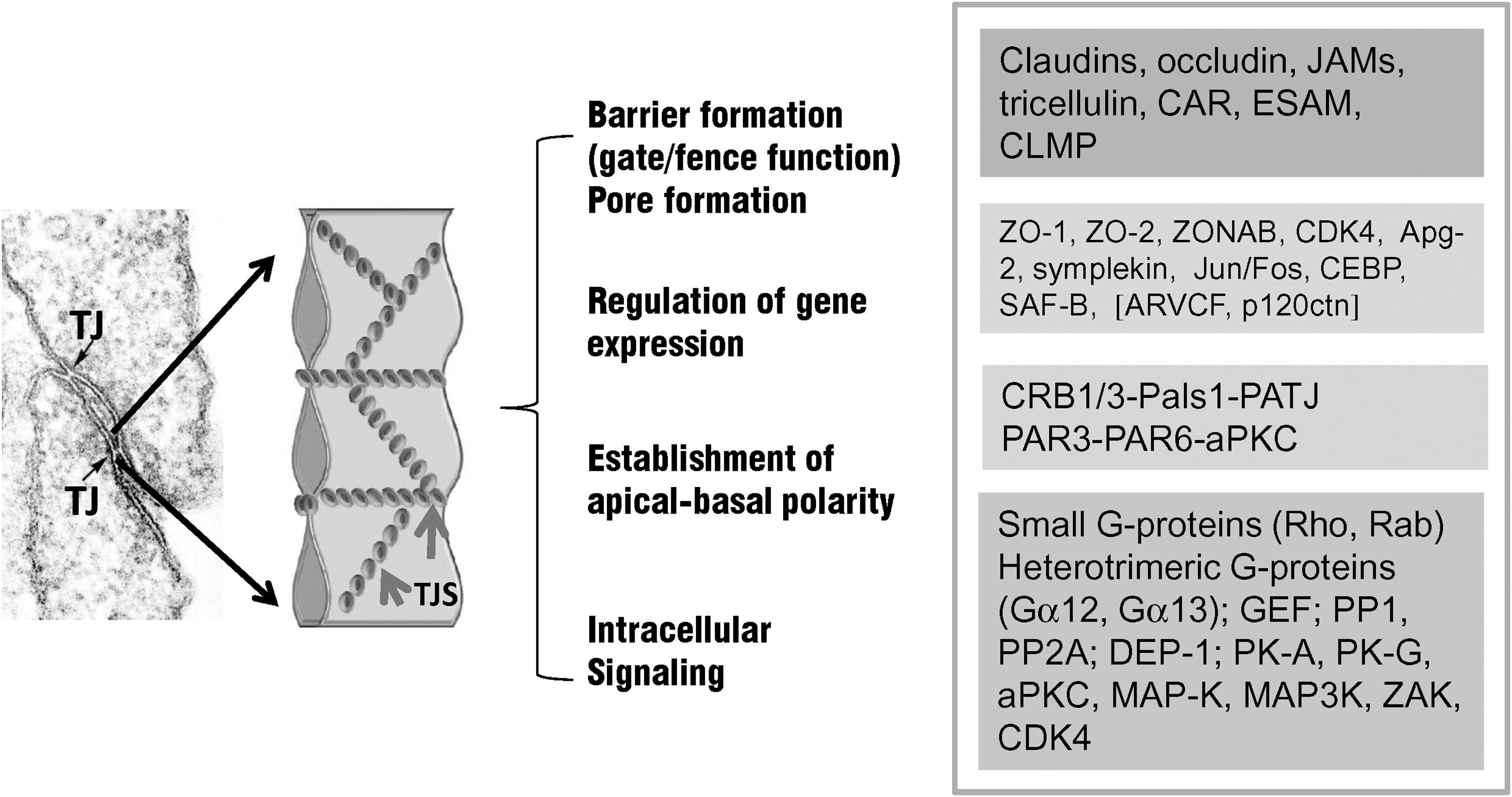
All epithelial and endothelial tissue barriers, such as the vertebrate BBB, the blood–retina barrier, or the blood–cerebrospinal fluid barrier, rely on the establishment of particularly tight intercellular contacts (tight junctions, TJs) between neighboring cells. TJs are the most restrictive intercellular junctions located at the apical border of the lateral membrane where they fulfill two main tasks: They act as a permeability barrier in the paracellular space and as a fence that separates apical and baso-lateral domains within the plasma membrane of epithelial and endothelial cells. In due course, a distinct apical-basal polarity is created, allowing the vectorial transport of ions and macromolecules across the cell. The structural features of TJs resemble all other occluding or adhesive junctions acting between epithelial and endothelial cells (i.e., a single- or multi-pass transmembrane component is connected to cytoplasmic proteins, commonly referred to as peripheral junctional proteins). The extracellular domains of the integral junctional proteins of two opposing cells interact in a homophilic or heterophilic manner to create a paracelluar “seal” (25a, 94, 166).
Two groups of proteins are involved in the formation, function and modulation of TJs: a) integral membrane proteins that bridge the intercellular space, and b) peripheral proteins, which assemble at the cytoplasmic surface of the junction site where they interact directly or indirectly with the transmembrane components (6, 35).
Integral membrane proteins found at endothelial TJs include the tetraspan proteins claudins (125a) and occludin (17a), which create two extracellular loops, exposing their N- and C-termini to the cytosol. The junctional adhesion molecules (JAMs) (13a) and the endothelial cell-selective adhesion molecule (ESAM) (66), which belong to the immunoglobulin superfamily due to their Ig-like domains located at the extracellular portion of the proteins, represent the major single span proteins contributing to the endothelial BBB (13, 66, Fig. 2). ESAM mediates homophilic adhesion between endothelial cells and was shown to bind to the scaffolding protein MAGI-1 (membrane-associated guanylate kinase with inverted domain structure 1) (177). ESAM is specifically expressed in endothelial cells and platelets and is involved in platelet aggregation and leukocyte transmigration across the endothelial wall (147,178).
So far, a plethora of peripheral proteins located at TJs have been discovered, the role of which has just partly been unravelled. Proteins localizing at the cytoplasmic surface of TJs comprise adaptor or scaffolding proteins, signaling molecules, and transcriptional regulators (6, 11, 35, 52, 57). The zonula occludens (ZO) proteins, ZO-1 and ZO-2, were shown to localize to the cytoplasm and to the nucleus in epithelial and endothelial cells (165). These proteins exert a dual role, being structural components at the junctional site and regulators of gene expression in epithelial and endothelial cells (52a).
The vertebrate BBB is one of the key interfaces between blood and neural tissue of higher vertebrates, including mammals. TJs of the BBB restrict the paracellular movement of ions and polar solutes between the blood and brain and impede the transport of macromolecules to the central nervous system (CNS) and vice versa (1, 36, 141), thereby TJs contribute to the creation and maintenance of a specific intracerebral homeostatic milieu. cEC are in close contact with other non-neural and neural cell types (pericytes, astrocytes, neurons, and microglia) and the extracellular matrix (ECM), a complex commonly referred to as the “neurovascular unit” (63). Disturbance of TJs by oxidative stress leads to impairment of the BBB, compromising the CNS (95, 104). Apparently, a finely tuned interplay of the cells constituting the BBB is important to cope with oxidative insult. In this context, astrocytes were found to be less susceptible to oxidative stress compared to cECs, which may protect and support the maintenance of the BBB during oxidative insult (21). In addition, astrocytes are involved in the regulation of neuronal glutathione homeostasis and, in this way, may contribute to neuronal protection during oxidative stress (176). The role of pericytes, which are intimately attached to the cerebral capillary wall, has been studied extensively (for review, see Ref. 96). Although their supportive role in BBB function is generally accepted, pericytes were shown to exhibit sustained contraction upon ischemia-induced oxidative stress, which impairs the restoration of the cerebral microcirculation and negatively affects the survival of neuronal tissue (184).
Selected Pathological Conditions Eliciting Oxidative Stress
Tumors
Alterations in oxygen metabolism appear to be a common feature of tumor cells, although the genetic profile of each type of tumor varies considerably. In normal cells, oxygen is used to produce energy (ATP) through oxidative phosphorylation. During the last couple of years, intriguing evidence has accumulated suggesting that cancer cells activate glycolysis (in the presence of O2) to cope with their increased energy demand. As a consequence, part of the oxygen is available for generation of ROS, particularly ·O2 − and H2O2. This metabolic switch from oxidative phosphorylation to aerobic glycolysis has repeatedly been observed in cancer cells (48). Accumulation of ROS and oxidative stress play an important role in carcinogenesis and is subject of many comprehensive reviews (87, 105, 127, 163).
In addition to carcinogenic agents, the activation of several oncogenes and mutations in the tumor suppressor gene p53 were found to increase the cellular production of ROS (91, 109, 167).
The critical role of ROS in tumor biology has prompted the elaboration of novel approaches for antioxidant drug treatments in cancer therapy (redox chemotherapeutics). In addition, induction of oxidative stress by pro-oxidant agents is emerging as an attractive anticancer strategy. Some anticancer agents produce H2O2 which is known to be an efficient inducer of cell death in cancer cells (3, 117). Knockout of antioxidant defense enzymes was shown to increase oxidative damage promoting age-related cancer (59).
In recent years, a particular focus has been directed towards the role of ROS in brain tumors, particularly gliomas (astrocytomas/glioblastomas, ependymomas, oligodendrogliomas), the largest group of primary brain tumors. Oxidative DNA damage (8-oxoguanine) is high in some glioblastoma multiforme (GBM) cell lines and many GBM tumors. Also, the incidence of HNE-immunopositive tumor cells in astrocytic and ependymal tumors parallels the increasing grades of malignancy. Again, highest levels of HNE were found in tumor cells of GBM (78).
A significant decrease of antioxidant enzyme activity such as glutathione reductase (GRx) and superoxide dismutase (SOD) activity was observed in most types of brain tumor cases, leading to the assumption that the degree of decline in antioxidant levels may indicate severity of malignancy in brain tumors (132).
Downregulation of endothelial TJ proteins and impairment of the BBB in glioma was reported severalfold (72, 101, 181). Particularly, expression of claudin-1, −3, −5, and occludin appears to be affected in microvessels of human glioblastoma and astrocytoma. Sufficient evidence suggests that VEGF (vascular endothelial growth factor) also contributes to BBB disruption with subsequent edema in brain tumors (106).
Diabetes and diabetic retinopathy
Diabetes in general is characterized by pancreatic beta-cell dysfunction. Oxidative stress produced by islet-infiltrating immune cells has been considered to contribute to the destruction of beta cells. Diabetes is usually accompanied by both, production of free radicals and impaired antioxidant defenses (reviewed in Refs. 39, 108, 179).
Under diabetic conditions, chronic hyperglycemia gradually deteriorates beta-cell function, and exacerbates insulin resistance, a process called “glucose toxicity.” Oxidative stress activates the JNK pathway, which is likely to be involved in pancreatic beta cell dysfunction and insulin resistance (81). Increased levels of ROS contribute to insulin resistance, as demonstrated by Houstis et al. using two cellular models (68).
The main source of free radicals is glucose, which is converted into reactive ketoaldehydes and to superoxide anion radicals. In addition, the hyperglycemic environment leads to nonenzymatic glycosylation of numerous proteins forming advanced glycation end products (AGEs) that accumulate in diabetic plasma and tissues (142). A well-characterized receptor for AGEs was shown to be present on monocytes, macrophages, and neutrophils (27, 42). AGEs alone trigger minimal ·O2 -/H2O2 production by neutrophils, but upon chemoattractant stimulation the production of ROS becomes markedly increased. This process seems to be related directly to glycemic control involving protein kinase C (PKC), NADPH oxidase, and the calcium-independent phospholipase A(2) (iPLA2) (5, 54, 64). However, recent evidence suggests that diabetic cells are capable of developing a mechanism of self-defense by increasing their antioxidant content, which results in high levels of glutathione and overexpression of a large set of genes encoding antioxidant proteins such as hemeoxygenase-1 and the transcription factor Nrf2 (nuclear erythroid-related factor-2/NF-E2–related factor 2) (98).
Diabetes causes severe pathologic aberrations in the retina, collectively termed as “diabetic retinopathy” (RP). Diabetic RP affects vascular and nonvascular cells of the retina. Vascular damage ranges from increased vascular permeability and leukostasis (i.e., abnormal attraction and adhesion of leukocytes to the vascular wall) to capillary occlusion and degeneration. Increased expression of inflammation-related proteins, including iNOS, COX-2, ICAM-1, caspase 1, VEGF, and NF-κB, and increased production of ROS and inflammatory cytokines is characteristic of early stages of diabetic RP (85). Antioxidants have been found to inhibit the development of inflammatory changes in retinas of diabetic animals (92, 93). For detailed reviews on diabetic RP, see Frey and Antonetti (43a), and for inflammation of cerebral barriers see Coisne and Engelhardt (25a).
Disruption of the BBB is an early event in the development of diabetic RP. Again, VEGF was shown to play a key role in the pathogenesis of diabetic retinopathy by inducing the PKC-dependent phosphorylation and subsequent degradation of occludin (61, 119).
Glucocorticoids reduce retinal endothelial cell permeability and stimulate the synthesis and dephosphorylation of occludin (40). In an in vitro model hydrocortisone was shown to increase barrier properties in cEC by inducing enhanced expression of occludin (43). In primary retinal endothelial cells, glucocorticoids increased the transcription of occludin and claudin-5 through a transactivation mechanism that does not involve direct promoter binding by the glucocorticoid receptor (41).
Ischemic stroke and reperfusion injury
The immediate loss of blood supply to the cerebral tissue is commonly referred to as ischemic stroke. Microvascular responses to ischemia are very rapid and highly coordinated, although they vary topographically with the degree of ischemic injury (32). Within 1 to 2 hours following middle carotid artery occlusion, initial loss of the primary microvessel permeability barrier occurs, producing transudation of small molecules, fibrinogen, and plasma (32). The pathogenesis of ischemic stroke involves various mechanisms, among which oxidative injury and inflammatory processes appear to be the most critical events. ROS are substantially involved in the pathological pathways of cerebral ischemia and reperfusion (99). In an initial phase, ROS production by nNOS and eNOS type I and type III is activated, while at a later stage increased activity of iNOS in glia and neutrophils may be observed (182). Actually, all cells of the neurovascular unit are able to activate one or more enzyme systems (including NOS, enzymes of the mitochondrial respiratory chain, xanthine oxidase, and NADPH-dependent oxidases) to contribute to the intra- and extracellular formation of ROS (153). ROS may potentiate the activation of stress-related transcription factors, including hypoxia inducible factor-1 and NF-κB (20, 114, 159). In turn, the expression of cytokines and adhesion molecules in cerebral endothelial cells is induced, which further facilitates activation and recruitment of inflammatory cells to the infarcted site (185).
Overexpression of radical-scavenging enzymes was shown to protect against stroke, and animals lacking such enzymes were more susceptible to cerebral ischemic damage (50). Migration of monocytes across a monolayer of rat cEC was reduced by scavenging ·O2 - with SOD and by blocking the ROS-producing enzyme xanthine oxidase (XO) with allopurinol (170). The gp91phox containing NADPH oxidase contributes to ROS formation by glia cells, fibroblasts, vascular endothelial cells, and most of all by leukocytes (53, 169). In leukocytes, NADPH oxidase is responsible for the respiratory burst in which the cell generates toxic amounts of superoxide anions. In NADPH oxidase-knockout (gp91phox–/–) mice, middle cerebral artery occlusion-induced BBB disruption and lesion volume were shown to be largely diminished compared with those in wild-type mice (80).
A critical role of ROS in cerebral injury accompanying ischemic stroke is also reflected by the fact that even delayed treatment with antioxidants and free radical scavengers could be neuroprotective in such pathological states (50, 168, 173). However, in aged rats, antioxidant treatment (i.e., suppression of NADPH oxidase activity) even increased BBB permeability to sucrose following ischemic stroke, suggesting that age adds a further level of variability to the treatment of oxidative brain damage (84).
Increased expression of metalloproteinases and inflammatory cytokines and reduced levels of occludin and claudin-5 were observed in ischemic rat brains. The loss of TJ proteins was reflected by BBB disruption, as evidenced by increased Evans blue extravasation into the brain (73). A decrease in ZO-1 and occludin protein levels, and an increase in tyrosine phosphorylation of occludin, was suggested to be critical for the disruption of TJs and dysfunction of the BBB after cerebral ischemia (79). Prevention of occludin phosphorylation by the Src tyrosine kinase inhibitor PP2 inhibited BBB leakage and decreased the infarct volume following transient focal ischemia in rats (155). Moreover, selective inhibition of the EP1 subtype of prostaglandin E2 receptors in a mouse model of focal cerebral ischemia, produced by middle cerebral artery occlusion, decreased occludin phosphorylation and reduced BBB disruption, pointing towards an additional mechanism underlying the deleterious effects of prostaglandin E2 in the setting of cerebral ischemia (44).
Mediators Through Which Oxidative Stress Acts on BBB Function (ROS, Cytokines, Leukocytes)
The disruption of epithelial and endothelial barriers seems to be a crucial step in the acquisition of inflammation after oxidative insults (133).
Endothelial cells may be both a source and target of ROS. ROS attack lipids and proteins of their target cells, leading to the release and activation of cytokines and proteases that propagate the vascular damage. The most studied cytokines related to inflammation in acute ischemic stroke are tumor necrosis factor-α (TNF-α), the interleukins (IL), IL-1β, IL-6, IL-20, IL-10, and IL-17, and transforming growth factor (TGF)-β (reviewed in Ref. 99).
IL-17A was demonstrated to induce NADPH oxidase- or XO-dependent ROS production. The resulting oxidative stress activated the endothelial contractile machinery, which was accompanied by downregulation of occludin. Blocking either ROS formation or myosin light chain phosphorylation prevented IL-17A-induced BBB disruption (70).
Inflammatory cytokines were shown to induce the expression of various cell adhesion molecules (CAMs) on endothelial cells (174, 175). The appearance of P-selectin, intercellular adhesion molecule-1 (ICAM-1), and E-selectin on microvessel endothelium, together with their counter-receptors (e.g., P–selectin glycoprotein-1 (PSGL-1) and the ß2-integrin CD18) on PMN (polymorphonuclear) leukocytes was shown to accompany the initial movement of inflammatory cells into the ischemic region (32). ROS treatment of pulmonary endothelial cells induced the phosphorylation of the focal adhesion kinase pp125FAK, a cytosolic tyrosine kinase implicated in the oxidant-mediated adhesion process (171). In addition, expression of chemokines, such as the monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1α (MIP-1α) in cerebral cells, further supports leukocyte entry and increased oxidative damage (115, 116, 175).
TNF-α plays a pivotal role in various types of inflammatory brain diseases (8, 22, 45, 46, 186). ·O2 − appears to be a primary mediator of TNF-α-induced oxidative stress in the cerebral vascular endothelium. An important cellular source generating proapoptotic ·O2 − in response to TNF-α in cerebral vascular endothelium is the xanthine oxidase/reductase system (126). XO and XDH (xanthine dehydrogenase) are interconvertible forms of the same enzyme (123), known as xanthine oxidoreductase (XOR). Normally this enzyme is present in vivo as an NAD-dependent cytosolic dehydrogenase (XDH), incapable of ROS production. Following conversion of XDH to XO by sulfhydryl oxidation or limited proteolysis, the enzyme produces ·O2 - and H2O2 (122). One of the primary roles of XOR is the conversion of hypoxanthine to xanthine and xanthine to uric acid (Fig. 3).
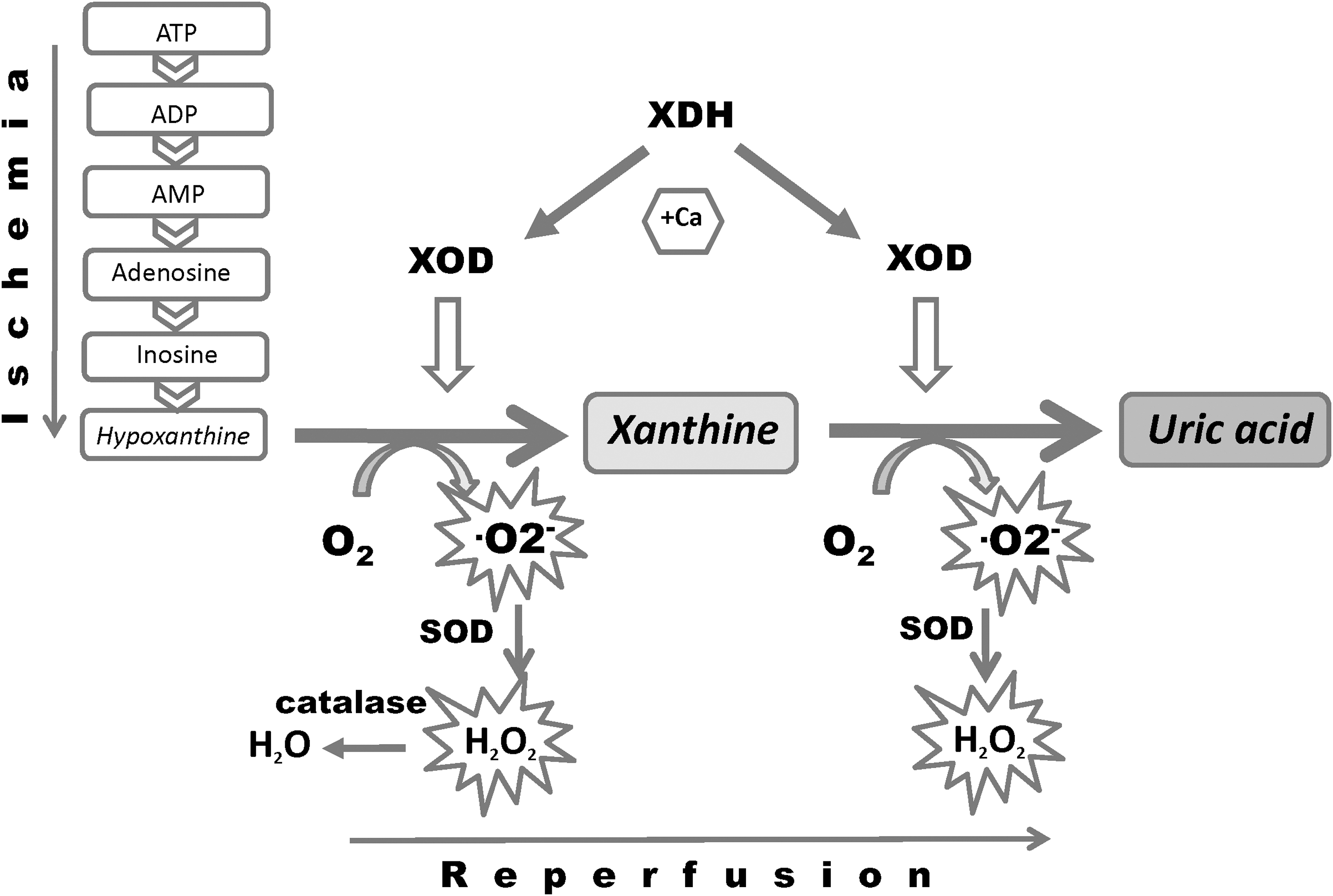
In hypoxic endothelial cells, XO is phosphorylated, at least in part, via casein kinase II and the stress-activated p38 MAP kinase which seems to be necessary for the hypoxia-induced enzymatic activation. XO was shown to be released into the blood stream in human ischemia-reperfusion injury (157). Interestingly, it has been proposed that circulating XO binds to sulphated glycosaminoglycans on the surface of endothelial cells (2). This endothelial cell-bound XO continues to produce oxidants, which can further trigger endothelial dysfunction and may contribute to organ injury also in remote organs (157).
ROS selectively activate signaling cascades involving RhoA, PI3 kinase, and protein kinase B (PKB/Akt) to increase monocyte migration across an in vitro BBB model. Thereby, rearrangements of the actin cytoskeleton and spatial redistribution and disappearance of occludin and claudin-5 take place (143). Hypoxia-induced cytoskeletal alterations contributing to BBB disruption were also reported by Hicks et al. (65). The authors showed that hypoxic stress leads to redistribution of moesin and VASP (vasodilator-stimulated phosphoprotein), two proteins involved in actin polymerization in BBB endothelial cells (65). Oxidative stress also induces the expression of distinct Toll-like receptors in cerebral endothelial cells in vitro, which leads to downregulation and delocalization of occludin and claudin-5 and a concomitant increase in permeability (121).
Redox-Sensitive Transcriptional Regulation
During oxidative stress, the proper function and integrity of the BBB depends on various redox-sensitive systems, which are active at different ROS levels (Fig. 4). Redox-sensitive signaling proteins and downstream transcription factors act in concert to maintain the intracellular prooxidant–antioxidant homeostasis (128). The redox-sensitive thioredoxins (Trxs) constitute a family of enzymes which, together with other redox-sensitive systems, play important roles in maintaining the intracellular redox balance. Many recent publications have shown that redox regulation by Trxs is critically involved in the pathogenesis of various oxidative stress-associated disorders (90, 128). In the nucleus, Trxs interact physically with the ubiquitously expressed bi-functional protein Redox-factor-1 (Ref-1/Ape1/HAP1/APEX1) (16, 160). Ref-1 is involved in DNA repair but also acts as transcriptional activator by reducing oxidized cysteine residues within the DNA-binding domain of a number of transcription factors, such as activator protein-1 (AP-1), nuclear factor κB (NF-kB), p53, early growth response protein-1 (Egr-1), activating transcription factor/cAMP-response element–binding protein (ATF/CREB), hypoxia-inducible factor (HIF)-1α, paired box-containing proteins (Pax), and many more. The reduced form of Ref-1 is restored by Trx in a redox cycle (reviewed in Refs. 124, 160). In addition, Ref-1 suppresses the intracellular production of ROS by negatively regulating the activity of the Ras-related GTPase Rac1 (131). In phagocytic cells, Rac proteins have been previously shown to regulate ·O2 − production through the neutrophil NADPH oxidase system (19). Similar to the NADPH oxidase complex in phagocytic cells, an FAD-binding protein regulated by Rac1 may regulate ROS levels in nonphagocytic cells such as fibroblasts (150).
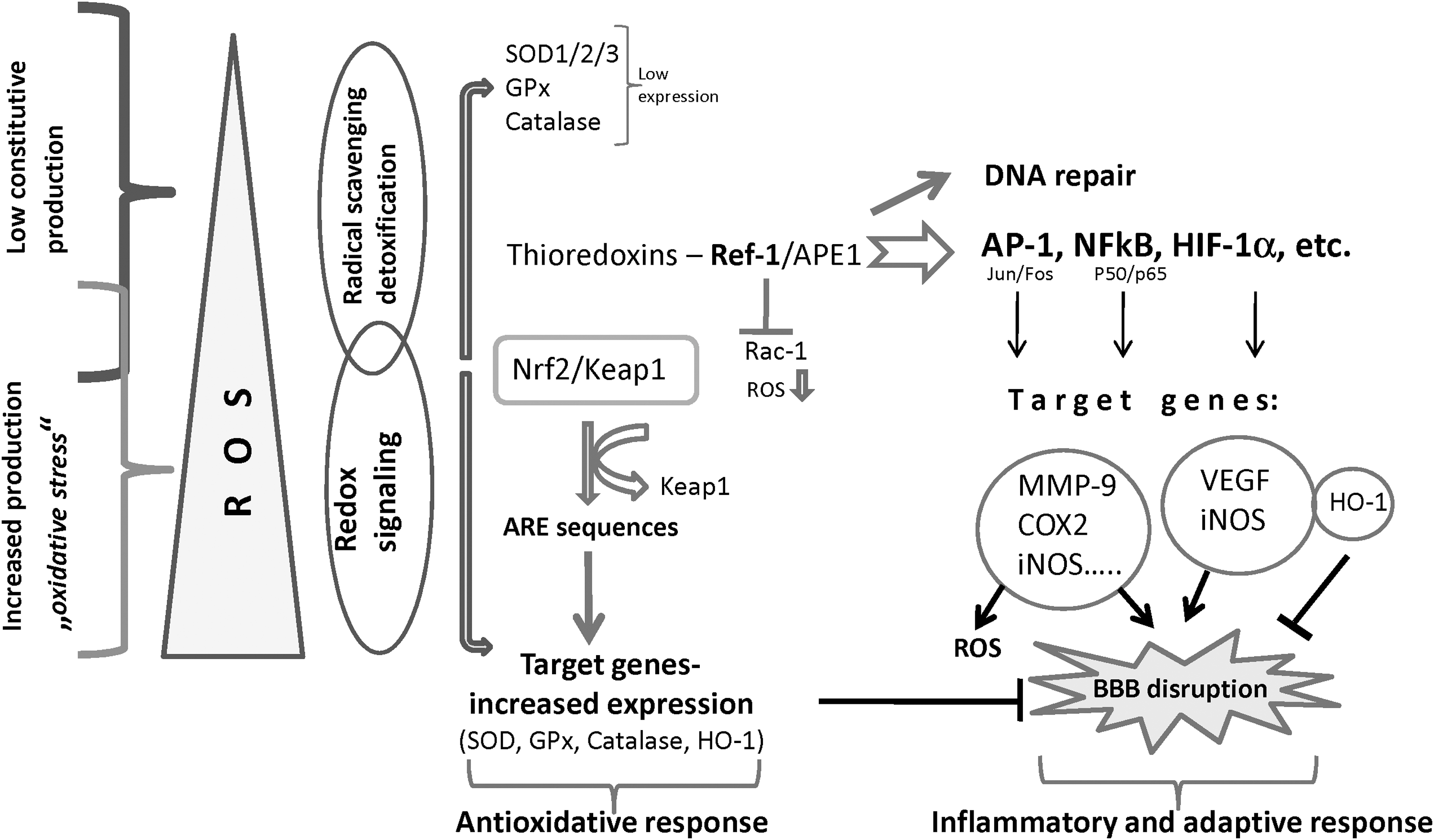
Both in vivo and in vitro studies demonstrated that the elevation of intracellular ROS induces a rapid and transient upregulation of Ref-1 protein expression, leading to an increase in cell resistance toward oxidative stress and DNA damage (55, 131). Activation of Ref-1 may also be obtained by a process independent of de novo protein synthesis. This involves the cytoplasmic–nuclear redistribution of Ref-1 during oxidative stress or upon physiologic increase in intracellular ROS production (131). Transgenic mice overexpressing Trx1 show an extended life span and are relatively resistant to ischemia-mediated brain damage (128). Also, intravenously administered Trx was shown to reduce oxidative cerebral damage in mice following transient focal ischemia (62).
Oxidative Stress and Metalloproteinases
A number of proteases, including neutral proteases (caspases), serine proteases, tissue-type and urokinase-type plasminogen activator (tPA, uPA), two families of metalloproteinases (matrix metalloproteinases, MMPs), and the adamlysins (ADAMs), are involved in the vascular response to oxidative insult. In this main section, we will focus on the role of MMPs which appear to be key mediators in the pathogenesis of cerebral oxidative damage and concomitant BBB dysfunction.
Biology of MMPs
MMPs are proteolytic enzymes (zinc-dependent endopeptidases) which are secreted as zymogens. To be active, these zymogens have to be cleaved by other MMPs or proteases such as tPA or uPA (149, 172). For instance, MMP-3 was shown to activate proMMP-1, −7, −8, −9, and −13, while MMP-2 can activate proMMP-1, −9, and −13 (29). Oxidation and S-nitrosylation are two protease-independent processes that can activate MMPs (23).
A detailed study of the complex regulatory mechanisms of MMP-9 expression and the role of multiple co-activators regulating MMP-9 transcription was reported recently (190). The authors showed that MMP-9 gene expression depends on binding of the sequence specific activators AP-1 and NF-κB to their corresponding cis-elements on the MMP-9 promoter. In addition, members of three classes of coactivators are essential for MMP-9 expression, including the CREB-binding protein/p300 (CBP/p300), the p300/CBP-associated factor (PCAF), the coactivator-associated arginine methyltransferase 1 (CARM1), and the glucocorticoid receptor interacting protein-1/steroid receptor coactivator-2 (GRIP-1/SRC-2).
Since MMPs are capable of degrading all kinds of ECM proteins, they play a central role in tissue repair and ECM remodeling during development, morphogenesis, and angiogenesis. Besides being responsible for ECM degradation processes, MMPs are involved in the cleavage of cell surface receptors and the release of apoptotic ligands (shedding) and in cytokine inactivation (23).
Based on their substrate specificity, cellular localization and protein domain structure, four to five major classes of MMPs may be defined, including collagenases, stromelysins and matrilysins, gelatinases, and membrane-type metalloproteinases (MT-MPs). Gelatinases, stromelysins, and MT-MPs are the main MMPs found in the brain. Although MMPs show particulate substrate preferences, there is much overlap in substrate specificity between subgroups.
The protein structures of MMPs share a basic common pattern of propeptide and catalytic domains (86). The matrilysin MMP-7 has the simplest domain structure, with the propeptide domain and a zinc-containing catalytic domain, although it degrades most ECM components. MMP-14, −15, and −16 contain an additional transmembrane domain, while the stromelysins (MMP-1, −3, and −10) are characterized by a hemopexin domain. The gelatinases (MMP-2 and −9) have a unique fibronectin binding domain, allowing them to bind to basement membranes. MMP-2 and −9 (alternatively referred to as gelatinase-A, and -B) specifically digest type IV collagen, laminin, and fibronectin. The latency state of MMPs is provided by the common pro-peptide.
MMPs are inhibited by four endogenous inhibitors, termed tissue inhibitors to the metalloproteinases (TIMPs). The balance between MMPs and TIMPs is critical for cell survival. TIMP-1 is a 28 kD molecule that forms a complex with MMP-9, whereas TIMP-2 complexes with MMP-2 (at higher concentrations), although TIMPs are able to inhibit all of the MMPs. TIMP-3 is unique in that it is bound to the cell surface where it inhibits preferentially MMP-3 and the tumor necrosis factor-alpha converting enzyme (TACE) (29).
MMPs and vascular diseases
Expression of MMPs in the adult brain is very low but many MMPs are rapidly upregulated in response to injury. MMP-2, MMP-3, and MMP-9 are the main soluble MMPs produced in the CNS (23).
The role of MMPs in cerebral diseases, including ischemia/reperfusion and stroke, has been a focus of intensive research during the last decade. There is general agreement that MMPs are critically involved in the initiation and progression of ischemic/hypoxic cerebral insults preceding BBB disruption (23, 107). Results derived from numerous in vivo and in vitro studies concerning the role of MMPs in cerebral oxidative stress have been summarized in a series of comprehensive reviews (23, 29, 33, 47, 75, 86, 107, 136, 187).
Activation of MMPs in response to oxidative stress
The role of MMPs in oxidative stress-induced cerebral diseases is complex, and various proposed mechanisms have been based upon the knowledge derived from ischemic/hypoxic insult and inflammatory processes in other tissues. There is evidence that MMP-2, which is constitutively expressed by astrocytes, becomes activated upon the formation of a ternary complex involving MMP-2, TIMP-2, and MT1-MMP. This provides the basis for a first and local event of proteolysis, leading to basal lamina degradation and BBB damage. In addition, expression and release of pro-MMP-9 is augmented as a response to the increase of ROS and inflammatory molecules released by activated astrocytes and due to infiltrating inflammatory cells (leukocytes, monocytes/neutrophils, macrophages) (23) (Fig. 5).
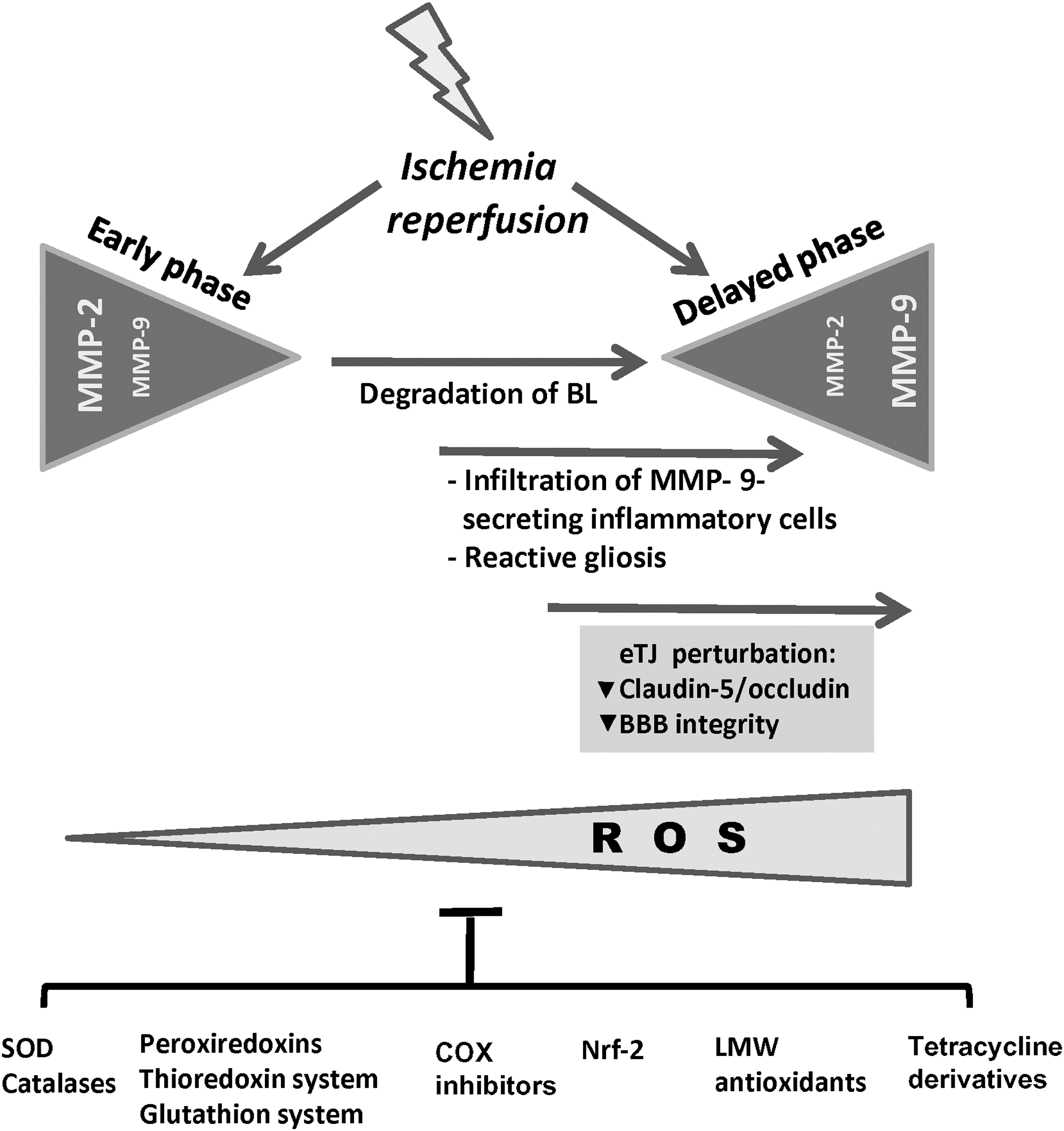
During hypoxic/ischemic and inflammatory conditions, all major cell types of the neurovascular unit, including neurons, cEC, reactive astrocytes, and microglia, express MMPs, either through induction by inflammatory cytokines (TNF-a, IL-6, IL-1ß, endothelin-1) or growth factors (23, 102, 151).
Evidence that ROS mediate BBB disruption through activation of MMPs
Free radicals can regulate expression and activity of MMPs both directly and indirectly. Direct regulation may occur through oxidation or S-nitrosylation of MMPs, resulting in the unmasking of the propeptide (without cleavage of the pro-domain) and subsequent activation of MMPs (56, 112). In addition, ROS-induced inflammatory cytokines (e.g., IL1-ß, IL-6, TNF-α) may activate signaling pathways (including MEK1-Erk, MAPK P38, and the phosphatidylinositol 3 (PI3)-kinase), resulting in the increased binding of transcription factors (NF-kB and AP-1) to redox-sensitive cis-regulatory elements in the MMP-9 promoter. Thereby, the transcription of MMP-9 is regulated (69) (Fig. 6).
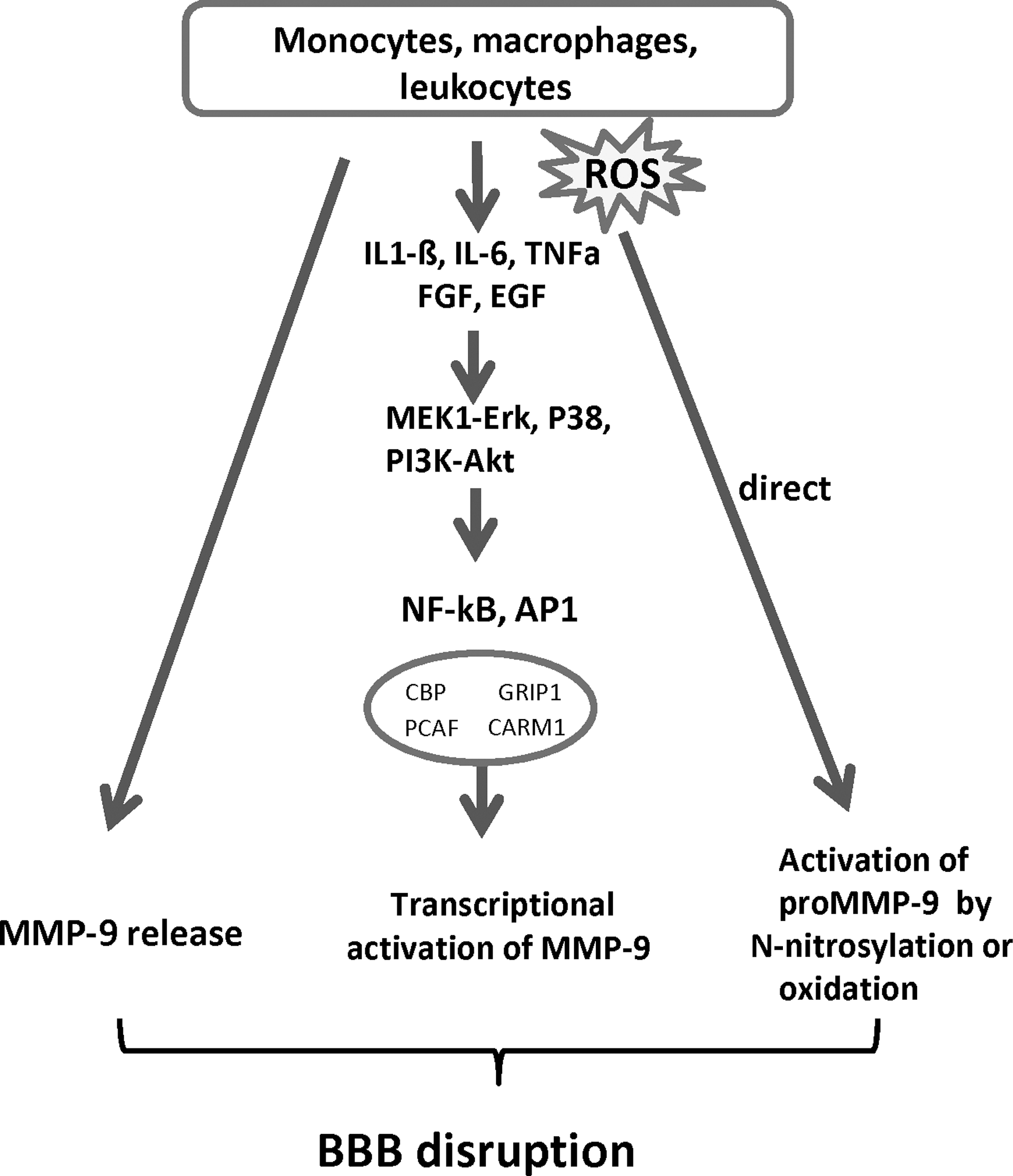
Results from a mouse transient focal cerebral ischemia model revealed that during reperfusion, pro-MMP-9 was increased more in SOD1-deficient mice than in their wild-type counterparts. In addition, active MMPs were found to co-localize with ·O2 − at the level of capillary walls and astrocytic processes in ischemic brain regions. In contrast, transgenic mice overexpressing SOD1 showed decreased lesion size and edema along with decreased activity of MMP-9, compared with wild-type littermates (118).
ROS were also found to increase BBB permeability and monocyte migration in vitro in a protein tyrosine kinase (PTK)-dependent manner (60). The authors showed that ROS not only increased activation of MMP-1, −2, and −9, which paralleled the degree of degradation of basement membrane components, but also enhanced tyrosine phosphorylation of tight junction-related proteins (60).
Evidence regarding the involvement of NO in the induction and activation of MMPs is still controversial. A dose-dependent, biphasic regulatory effect of NO on the activity of MMPs (MMP-9, −1, and −13) secreted from murine macrophages was described recently (135). However, a detailed study on the mechanism of NO-mediated activation of MMPs has shown that NO is incapable of directly activating proMMP-9, while a modest activation was found through S-nitrosylation by the N-donor S-nitrosocysteine (110).
The role of MMP-9 in BBB/TJ disturbance during oxidative stress
Accumulating evidence suggests that activation of MMP-9 appears to be a key step in the impairment of BBB function in focal cerebral ischemia (23, 29, 33, 86, 99). Following transient and permanent middle cerebral artery closure, the significant increase of proMMP-9 and induction of MMP-9 activity correlated with an increase of BBB permeability, which further confirms the important role for MMP-9 in the early formation of vasogenic edema after transient focal cerebral ischemia. Synthetic MMP inhibitors restore the early integrity of the BBB but are ineffective in the later BBB opening (137).
The infiltration of neutrophils in the infarcted tissue is a major source of MMP-9 (see also Fig. 6). In this context, MMP-9 was shown to be prepackaged in neutrophils and released under neuroinflammatory conditions (4, 49). MMP-9 deficient mice showed diminished BBB disruption and decreased inflammation following ischemic conditions (4, 152).
Early inhibition (day 1) of MMP-9 was shown to reduce infarction of day 14 in an ischemia/stroke model. However, the benefit was lost when the treatment was delayed until day 3 (187), suggesting that the effect of MMP-inhibition strongly depends on the timing of treatment in relation to the stage of brain injury.
It is important to mention that MMPs may have a different role during delayed phases after stroke. Because MMPs modulate vascular ECM, they may mediate beneficial plasticity and remodeling during the stroke recovery phase. If treatment with MMP inhibitors is administered too late (i.e., at a timepoint when neurovascular remodeling in the infarcted area has already started) recovery is impaired and tissue damage is exacerbated.
Compelling evidence has suggested that MMPs are involved in BBB disruption by acting directly on TJ-related proteins (183). Exposure of mice to prolonged hypoxia was demonstrated to increase MMP-9 activity and cerebral vascular permeability. Concomitantly, expression of occludin was decreased, resulting in disrupted continuity of occludin and zonula occludens-1 (ZO-1) staining in endothelial cells. Inhibition of MMP activity reduced vascular leakage and attenuated the disorganization of TJs (9). Treatment with normobaric hyperoxia could attenuate MMP-9-mediated early BBB disruption following ischemic stroke in rats (103). Treatment of brain slices or isolated microvessels with purified MMP-9 resulted in specific degradation of occludin (103). Significant degradation of occludin and claudin-5 was observed in cEC as a response to overexpression of MMP-9 (25). The observed alterations of TJ proteins correlated with increased BBB permeability to fluorescein isothiocyanate-dextran tracers (25). Further, live cell imaging has shown that transendothelial migration of monocytes coincides with the local disappearance of occludin and subsequent endothelial gap formation, which could be prevented by administration of a broad-spectrum inhibitor of MMPs (134).
Defense Mechanisms of the Brain and the BBB Against Oxidative Stress
Due to the impermeability of the BBB, antioxidative treatment of intraneural tissue is particularly difficult. Antioxidants should protect the endothelial barrier itself and must be able to transmigrate through the BBB to reach the brain parenchyma. A comprehensive survey of neuroprotective agents, supposed to be effective in the treatment of ischemic stroke, has been published previously (51).
Under normal physiological conditions, all cells are equipped with an elaborated antioxidant system for protection against reactive metabolites (for review, see Ref. 124). Basically, the antioxidant system can be classified into two major groups: enzymes and low molecular weight antioxidants. The enzymes include a limited number of proteins, SOD, catalase, GSHPx, GRx, and peroxiredoxins. All of the corresponding genes are under the control of Nrf2 (77, 99). Under normal conditions, Nrf2 is sequestered in the cytoplasm and associated with its repressor Kelch-like protein (Keap1), an actin-binding protein. On exposure of cells to oxidative stress, Nrf2 dissociates from Keap1 and translocates into the nucleus. Nrf2 binds to a recognition sequence, referred to as antioxidant response element (ARE), located in the upstream promoters of many genes encoding antioxidant proteins (88).
Activation of Nrf2 represents a promising treatment strategy to ameliorate cerebral oxidative insult (31, 76). In a rat model, the naturally occurring alkylating agent, sulforaphane, protected the brain from ischemic oxidative damage. The systemically administered sulforaphane was shown to enter the brain, as determined by increased mRNA and protein levels of the Nrf2-responsive gene heme oxygenase 1 (188). Even delayed administration of sulforaphane efficiently reduced the loss of TJ-related proteins (occludin, claudin-5) and preserved the BBB in a rodent model of brain injury (189). Further, short sulforaphane treatment of astrocytes resulted in long-lasting elevation of endogenous antioxidants and increased resistance to superoxide-induced cell damage (14).
Low molecular weight molecules comprise indirectly acting antioxidants (e.g., chelating agents) and directly acting antioxidants (e.g., scavengers and chain breaking antioxidants). The latter are extremely important in combating against oxidative stress. Common nonenzymatic antioxidants include the vitamins A, C, and E, glutathione, lipoic acid, mixed carotenoids, coenzyme Q10, bioflavonoids, antioxidant minerals (copper, zinc, manganese, and selenium), folic acid, and vitamins B1, 2, 6, and 12. Only a few of these molecules, such as glutathione, are synthesized by the cell itself. The majority, including ascorbic acid, lipoic acid, polyphenols, and carotenoids, are derived from dietary sources.
As outlined above, a considerable number of compounds are available which are potentially effective in reducing or preventing oxidative damage to neural cells in vitro. However, their successful application in vivo has been hampered due to the impermeability of the BBB. Therefore, sufficient knowledge of the molecular and functional characteristics of the BBB is essential for the successful design of systemically applied antioxidants.
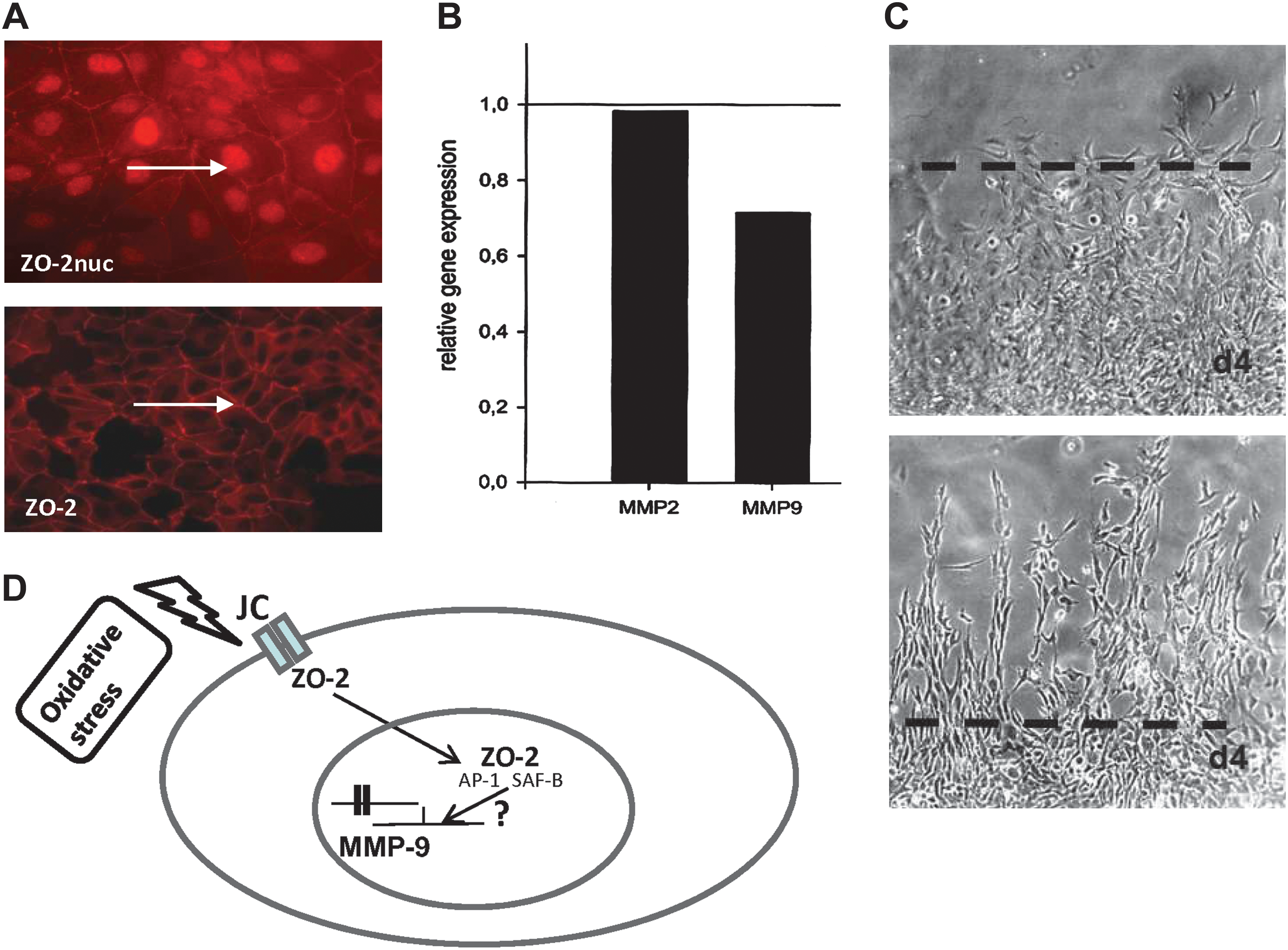
So far, much progress has been made concerning the elucidation of the molecular components of the intra- and intercellular network of tight endothelial and epithelial cell–cell contacts (Fig. 7) (see also Ref. 25a). As already outlined above, major integral proteins of TJs (occludin, claudin-5) are affected by oxidative stress (17a, 125a). In addition, peripheral junctional proteins, such as the multidomain ZO proteins appear to be major targets of oxidative insult (52a). The involvement of ZO proteins in oxidative stress response is of particular interest since ZO-proteins (ZO-1 and ZO-2), are "dual residency" proteins, capable of shuttling between the junctional site and the nucleus (for further details, see Ref. 11). Their role in transcriptional regulation has been shown several-fold (7, 15, 158). In vitro studies have demonstrated that nuclear translocation of ZO-2 in epithelial cells is induced upon environmental stress (heat shock, toxic insult) (165). Moreover, increased nuclear accumulation of ZO-2 was found in cEC in vitro subjected to oxidative stress (H. Bauer, unpublished observation). In addition, experimentally-induced nuclear overexpression of ZO-2 in cEC led to the downregulation of MMP-9 transcription (H. Bauer, unpublished observation) (Fig. 8). MMP-9 is a major mediator of oxidative damage in the brain and MMP9-deficient mice show reduced BBB disruption following ischemic conditions (4, 112). Although further studies are needed, it may be speculated that the nuclear targeting of ZO proteins represents a novel mechanism that is involved in a signaling cascade, leading to alterations of gene expression and probably BBB protection in response to oxidative stress.
Conclusion
Considerable medical and experimental evidence has suggested a critical role for ROS and MMPs in the pathophysiology of cerebral oxidative damage and BBB disruption. There is evidence that activation of the cellular antioxidant defense, either through enzymatic or nonenzymatic means, can provide some protection against BBB dysfunction.
Much of the information concerning the role of oxidative stress in BBB disruption has emerged from observations and experiments in vitro or in vivo, using co-culture systems mimicking the BBB or defined animal models. So far, several compounds were shown to protect the BBB in vitro and to reduce the loss of TJs at the BBB in brain microvessel endothelial cells. However, in practice, most of the drugs used in these experimental systems failed to provide neuroprotection during and following oxidative insult in clinical trials.
The BBB contributes to oxidative cerebral damage in two ways: First, the BBB itself is a target of ROS-mediated cellular damage (lipid peroxidation, protein/DNA modifications, junctional alterations) and, second, the BBB impedes the transport of many systemically administered antioxidative compounds.
Therefore, any drug design that enables BBB transport will depend on new knowledge of the molecular and functional biology of brain capillary endothelial cells in vivo and in vitro. Additional work will have to focus on the role of transcriptional regulators inducing endogenous antioxidant gene expression in cerebral tissue. Because cEC are a critical component responsible for normal BBB function, changes in intracellular signaling of BBB endothelial cells in response to an imbalance in the oxidative stress/antioxidant status should be a critical focus of future BBB research. In particular, one of the challenges will be to further expand our understanding of the physiological and molecular mechanisms underlying the interplay of ROS and MMPs in oxidative brain injury. This knowledge should help to develop new ideas for designing and elaborating effective medical treatment approaches to prevent or reduce the deleterious outcome of oxidative cerebral insults.
Footnotes
Acknowledgments
This work was supported by the Austrian FWF P-18743 and by research grants from the Paracelsus Private Medical University (PMU) Salzburg, Austria. H. Bauer and H.C. Bauer HC are participants of the NEUROBID consortium (EU 7th FP).