
Abstract
Introduction
Peroxiredoxins (PRXs) are a newly characterized family of six antioxidant enzymes (PRX1–6) ubiquitously expressed in mammalian cells. In the brain, the expression of PRX isozymes shows distinct distribution patterns in different regions, cell types, and subcellular compartments, which may be related to their cell-specific functional roles (1, 11, 13, 34). All PRXs have the capacity of decomposing peroxides such as H2O2 and alkyl peroxides using thioredoxin (Trx) or other electron donors (30). However, in addition to their common peroxide scavenging activity, a unique feature of PRXs is that certain PRX isoforms may directly engage in divergent intracellular signaling pathways that govern specific biological processes, including proliferation, differentiation, gene expression, and apoptosis (14).
PRX2 is a relatively abundant neuron-specific PRX in brain (1, 13, 34). Increased expression of PRX2 is found in aging brain and in frontal cortex Alzheimer's disease, Parkinson's disease, and Down syndrome postmortem brains, suggesting that PRX2 is involved in the elevated neuronal antioxidant response under oxidative stress (30). Homozygous mice deficient in PRX2 show remarkably increased accessibility to oxidative stress-induced tissue damage, indicating a crucial role for PRX2 in endogenous antioxidant defense (19). Moreover, a direct neuroprotective effect by PRX2 against cell death has been demonstrated in cultures of PRX2-overexpressing neurons challenged by growth factor depletion or ischemic/oxidative injury (2). PRX2 catalyzes the reduction of peroxides through the oxidation of its two active cysteine (Cys) residues into the sulfenic acid form, which is then recycled back to a thiol using one of several cell-specific disulfide oxido-reductases, such as the highly efficient thioredoxin/thioredoxin reductase (Trx/TrxR) system (30). Because PRX2 utilizes Trx for recycling from its oxidized forms in the PRX2-catalyzed peroxide scavenging reaction, we have recently speculated that over-consumption of PRX2 under oxidative stress could deplete the reduced Trx in neurons, resulting in the activation of pro-death signaling molecules such as apoptosis signal–regulating kinase 1 (ASK1) that are normally kept at an inactive state by reduced Trx (33).
In this study, using a newly created PRX2 transgenic (Tg-PRX2) model, we have investigated the neuroprotective effect and the underlying mechanism of PRX2 against ischemic/reperfusion brain injury. Novel evidence is presented to demonstrate that PRX2 overexpression confers marked and prolonged neuroprotection by suppressing the ASK1/JNK-mediated mitochondrial apoptosis signaling pathway.
Innovation
Despite the importance of oxidative stress in the pathogenesis of ischemic/reperfusion brain injury (7, 21), numerous antioxidative drugs targeted at scavenging overproduced free radicals have failed in clinical trials. An improved understanding of the mechanisms that maintain intracellular redox homeostasis in neurons is therefore imperative to identify novel therapeutic targets for stroke management. The current study documents an active involvement of peroxiredoxin 2 (PRX2), an antioxidant enzyme, in the defense against neuronal death after ischemia/reperfusion. We demonstrate for the first time that neuronal overexpression of PRX2 potently reduced brain infarct and improved neurological outcomes evaluated up to 3 weeks after ischemia/reperfusion. Further study indicates that in addition to its commonly recognized role as an antioxidant enzyme, PRX2 is able to exert neuroprotection through a novel mechanism whereby this redoxsensitive molecule interferes with thioredoxin-apoptosis signal–regulating kinase 1 (ASK1) interactions and the activation of the ASK1-JNK pro-death signaling pathway. Taken together, our research results shed light on the pro-death signaling regulation by PRX2 in neurons under ischemia/reperfusion conditions. PRX2 may represent a potential target for stroke intervention.
Results
Characterization of Tg-PRX2 mice
Several independent lines of Tg-PRX2 mice were confirmed by polymerase chain reaction genotyping (Supplementary Fig. S1A; Supplementary Data are available online at
The offspring of breeding between heterozygous Tg-PRX2 mice showed genotypes in a ratio consistent with mendelian transmission without disproportional pre- or perinatal lethality. Determined at 8 and 12 weeks of age, respectively, there was no significant difference in either body weight or brain weight between Tg-PRX2H mice and wild-type (WT) littermates (Supplementary Fig. S2A). Overexpression of PRX2 did not significantly affect the expression levels of PRX1, PRX3, or PRX4 in the forebrain cortex of Tg-PRX2H mice (Supplementary Fig. S2B) and did not significantly change the numbers of microglia (Iba-1 positive) or astrocytes (GFAP positive) in proportion to neurons (Supplementary Fig. S2C–E).
Tg-PRX2 mice are highly resistant to focal cerebral ischemia
To determine the effect of PRX2 overexpression on ischemic brain injury, we performed either transient middle cerebral artery occlusion (30- or 60-min tMCAO) or permanent middle cerebral artery occlusion (pMCAO) in Tg-PRX2 mice and WT littermates. In all experimental paradigms, PRX2 overexpression decreased infarct volume in a dose-dependent manner at 48 h after MCAO (Fig. 1A–C). In particular, Tg-PRX2H mice showed greater neuroprotection against each ischemic condition compared to Tg-PRX2L mice (Fig. 1B). Regional cerebral blood flow as determined using laser Doppler flowmetry (Fig. 1D) or a laser speckle imager (Fig. 1E) was not significantly different between the transgenic and WT groups. Moreover, neither brain surface blood vessels in the MCA territory nor posterior communicating artery plasticity was significantly affected by PRX2 overexpression (Supplementary Table 1). Physiological parameters (blood pressure, core temperature, blood gases, etc.) did not differ significantly between the transgenic and WT groups (data not shown).

The protective effect of PRX2 against ischemic cell death was further confirmed by stereology-assisted quantification of NeuN-immunoreactive or DNA-fragmented cells on brain sections at 24 h after 60 min of tMCAO. tMCAO resulted in robust loss of NeuN-positive cells and induction of DNA fragmentation in both infarct core and infarct inner boundary regions in WT mice (Fig. 1F–H), whereas these changes were significantly attenuated in Tg-PRX2H mice. The high-expressing Tg-PRX2H line was used in all subsequent studies.
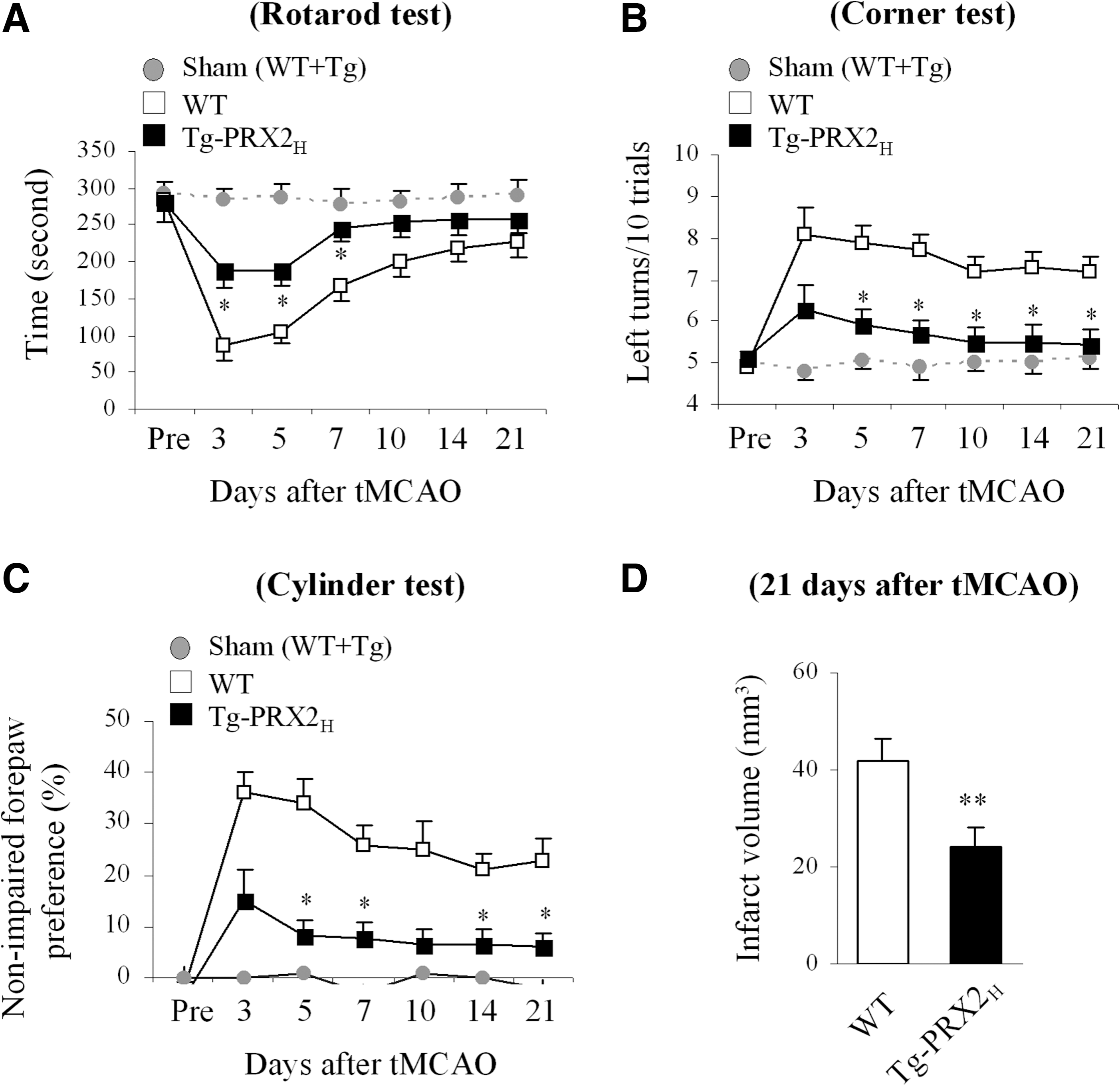
PRX2 overexpression improves long-term neurological recovery after tMCAO
To further determine the impact of PRX2 overexpression on neurofunctional outcomes after stroke, neurobehavioral assays including the rotarod test, corner test, and cylinder test were administered during the 21-day recovery period after 60-min tMCAO. Sham-operated mice displayed no significant difference in sensorimotor performance regardless of their transgenic phenotypes. Postischemic sensorimotor dysfunction was significantly improved in Tg-PRX2H mice compared with WT littermates up to 21 days after ischemia (Fig. 2A–C). At the end of the 21-day recovery period, infarct volume was measured to determine the long-term effect of PRX2 against ischemia. Tg-PRX2H mice showed significantly smaller infarct volume than WT mice (Fig. 2D), indicating that PRX2 overexpression conferred long-lasting neuroprotection against tMCAO instead of simply delaying ischemic injury.
PRX2 prevents ischemia-induced ASK1 activation by maintaining Trx at a reduced state
When catalyzing the reduction of peroxides, PRXs are oxidized at active cysteine residues to generate sulfenic acid (30) that is slowly recycled back to a reduced state by the Trx/TrxR system (24). However, under oxidative stress, the sulfenic acid may be overoxidized to generate cysteine sulfinic acid (Cys-SO2) and cysteine sulfonic acid (Cys-SO3) (24), which are thought to be irreversible (7). Oxidative modification of PRXs results in a reduction in antioxidative activity, and thus dramatically influences their cytoprotective capacity (20). Using an antibody recognizing overoxidized PRX (PRX-SO3), we showed that PRX-SO3 immunoreactivity was barely detectable in nonischemic brains, but was significantly augmented after tMCAO (Fig. 3A, B). To further determine the PRX2-specific oxidative modification, tissue lysates from ischemic brains were subjected to immunoprecipitation (IP) (31) using the PRX2 antibody followed by immunoblotting against PRX-SO3. Similar to total PRX-SO3, the PRX2-derived PRX-SO3 (PRX2-SO3) was elevated up to 6 h after tMCAO (Fig. 3A). The increases in PRX2-SO3 appeared to contribute to a large portion of total PRX-SO3 following tMCAO.
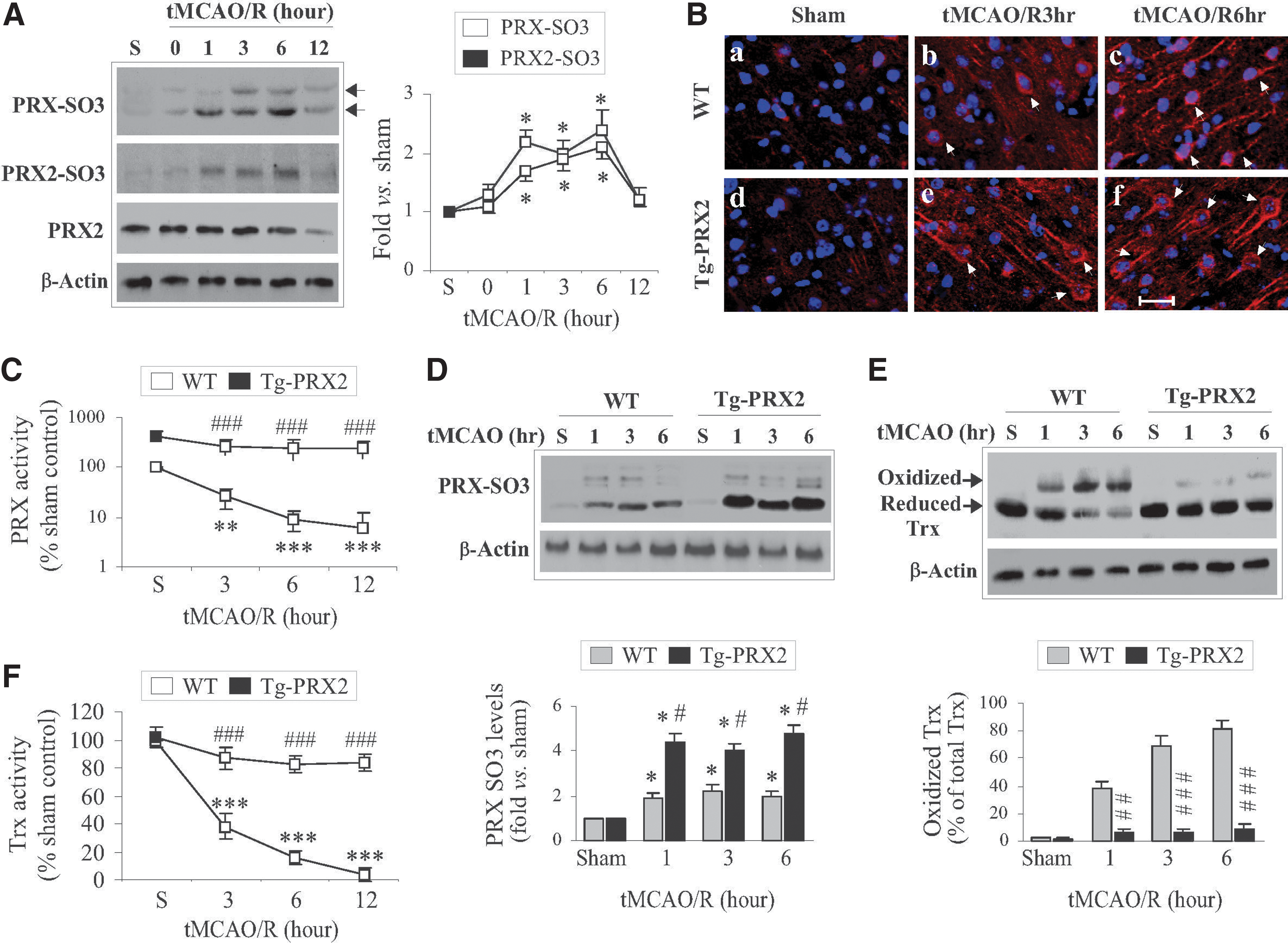
The results of PRX activity analysis revealed dramatic decreases in cellular PRX activity (an index for PRX antioxidant reserves) following tMCAO (Fig. 3C) in parallel with the increases in PRX2-SO3 (Fig. 3A), indicating a severe exhaustion of brain PRXs after ischemia. In contrast to WT mice, Tg-PRX2 mice showed approximately fivefold increases in PRX activity in nonischemic brain, and maintained PRX activity at significantly elevated levels after tMCAO (Fig. 3C). Interestingly, significantly higher levels of PRX-SO3 were observed after tMCAO in Tg-PRX2H mice compared to WT mice (Fig. 3C, D). These results suggest that exogenously overexpressed PRX2 actively participated in peroxide scavenging in ischemic brain, ultimately preventing the exhaustion of endogenous PRXs reserves after tMCAO.
Since Trx serves as a reducing enzyme both in PRX2-catalyzed peroxide-scavenging reaction and in recycling the oxidized PRX2 under oxidative stress (24), we hypothesized that exhaustion of PRX reserves after tMCAO could deplete the endogenous reduced Trx. We further hypothesized that depletion of reduced Trx might lead to ASK1 activation in ischemic neurons as Trx oxidation and subsequent dissociation from ASK1 are prerequisites for ASK1 activation under oxidative stress (33). Indeed, tMCAO resulted in marked accumulation of oxidized Trx but a decrease in reduced Trx in brain (Fig. 3E), which was accompanied by significant decreases in cellular Trx activity (Fig. 3F), an index of Trx reserves. In contrast to WT mice, tMCAO-induced oxidation of Trx and decreases in Trx activity were significantly attenuated in Tg-PRX2H mice (Fig. 3E, F), suggesting that PRX2 overexpression preserved reduced Trx from overconsumption after tMCAO.
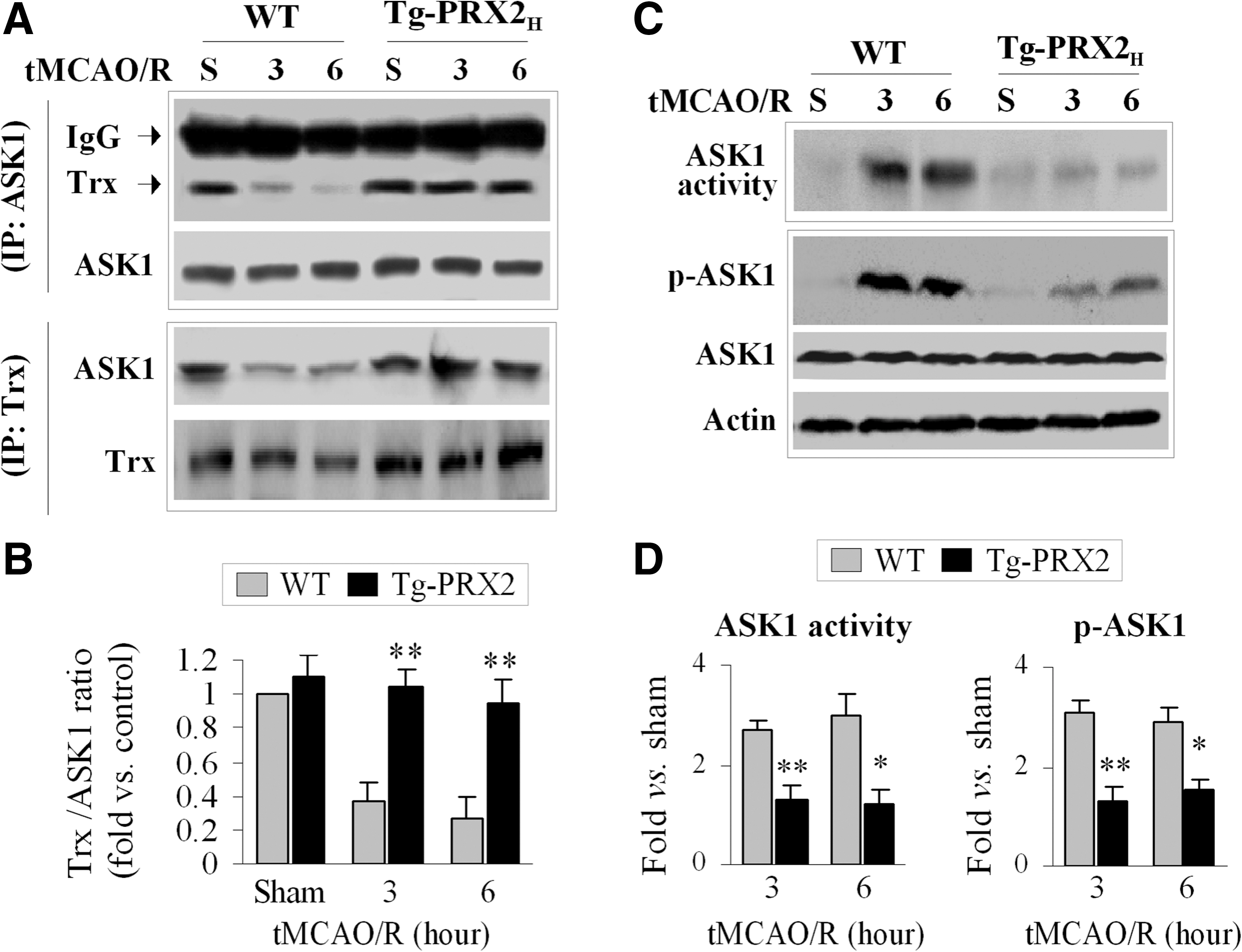
To examine the interactions between Trx and ASK1 after tMCAO, co-IP was performed using brain extracts from Tg-PRX2H mice and WT littermates. An association between Trx and ASK1 was readily detectable in sham control mice regardless of genotype (Fig. 4A, B). However, a marked dissociation of Trx from ASK1 occurred at 3 and 6 h after tMCAO in WT mice but not in Tg-PRX2H mice (Fig. 4A, B). Consistent with the co-IP results, tMCAO-induced activation of ASK1 (increased phosphorylation at Thr845 and increased ASK1 activity) was significantly attenuated in Tg-PRX2H mice compared to WT mice (Fig. 4C, D). Taken together, these results suggest that PRX2 suppresses ischemia-induced ASK1 activation by maintaining Trx at a reduced state.
The 2-C catalytic sites are essential for PRX2-afforded neuroprotection in cultured neurons
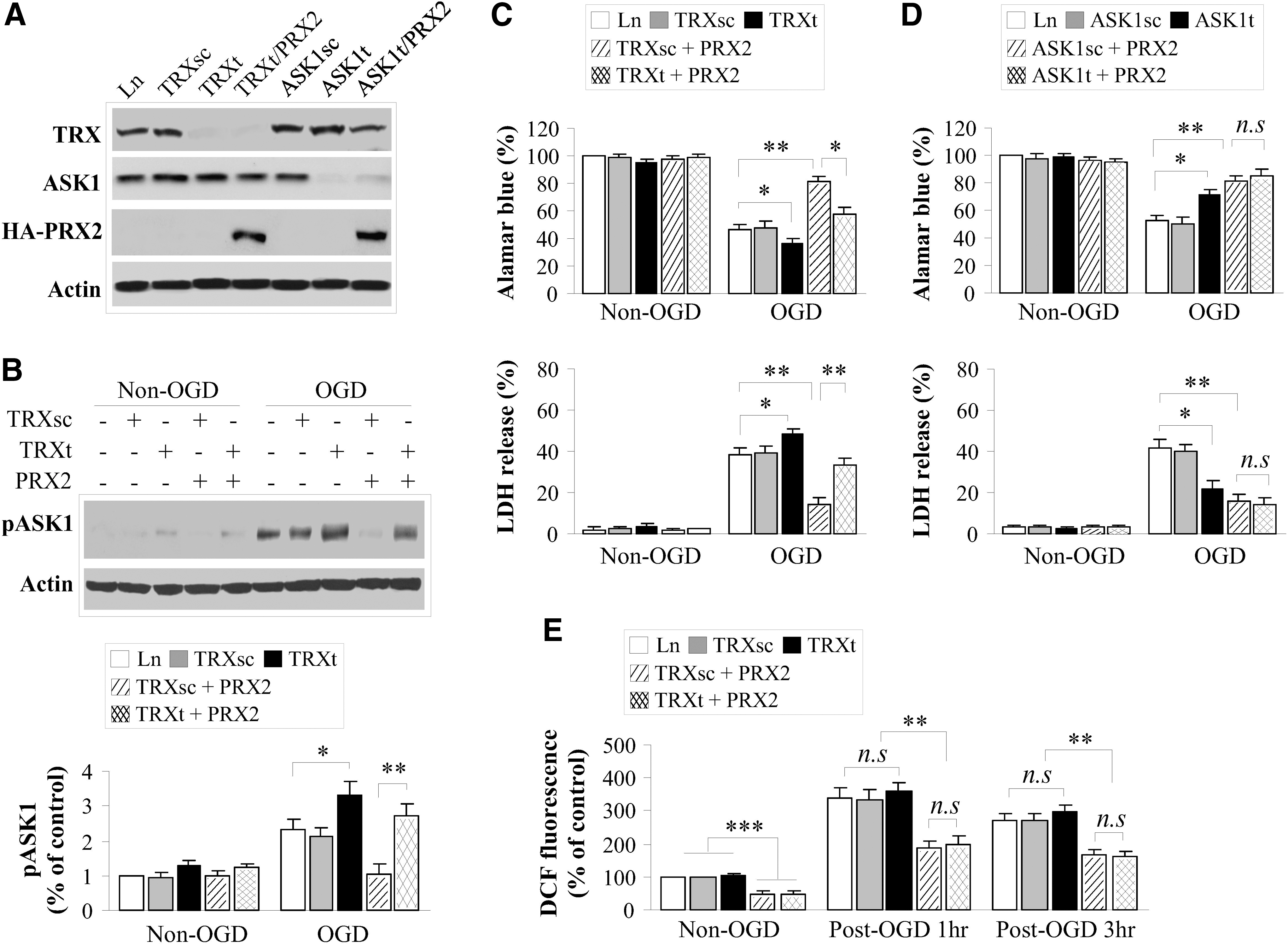
To test the hypothesis that the peroxide-scavenging activity possessed by PRX2 is essential for its neuroprotective effect via interaction with the Trx-ASK1 complex, we constructed lentiviral (Ln) vectors expressing either WT human PRX2 or its catalytically inactive mutants in which the two cysteine residues at amino acids 51 and 172 were mutated to alanine (PRX2C/A, Supplementary Fig. S3). Incubation of vectors in primary cultures of rat cortical neurons yielded highly efficient gene transfection of PRX2 and PRX2C/A, respectively (Fig. 5A). As predicted, overexpression of PRX2, but not PRX2C/A, markedly elevated the peroxide-scavenging activity in neurons before and after oxygen and glucose deprivation (OGD) (Fig. 5B). As a result, the otherwise diminished Trx activity following OGD was restored to near preischemic levels in PRX2-transfected but not in PRX2C/A-transfected neurons (Fig. 5C).

To determine the effect of PRX2 on the interaction between Trx and ASK1 after OGD, co-IP was performed. Consistent with the role of PRX2 in protecting Trx against the loss of reducing activity under oxidative stress, PRX2, but not PRX2C/A, overexpression inhibited OGD-induced dissociation of Trx from ASK1 and, consequently, ASK1 activation (Fig. 5D).
Using three independent assays for cell death/survival, we evaluated the neuroprotective effect of PRX2 transfection against OGD in primary rat neurons. Transfection of PRX2 but not PRX2C/A significantly increased neuronal survival in OGD-challenged cultures (Fig. 5E, F). In separate experiments, Ln transfection of PRX2 in primary cortical neurons derived from mouse embryos also significantly increased neuronal viability after OGD (Supplementary Fig. S4), indicating that PRX2 protects against ischemic injury in both rat and mouse neurons.
Critical role of the Trx-ASK1 signaling complex in mediating the neuroprotective effect of PRX2
To determine if the Trx-ASK1 signaling complex and PRX2 interact with different pro-death signaling pathways after ischemia, we performed lentivirus-short hairpin RNA (shRNA)-mediated knockdown of Trx and ASK1, respectively, in primary rat neuron cultures (Fig. 6A and Supplementary Fig. S5). In all transfection experiments, the scramble sequences or empty vector served as controls. As shown (Fig. 6B, C), Trx knockdown significantly enhanced OGD-induced ASK1 activation and neuronal death in cultures. Furthermore, Trx knockdown markedly impaired the ability of PRX2 to suppress OGD-induced ASK1 activation and neuronal death (Fig. 6B, C). Similarly, ASK1 knockdown was protective against OGD-induced neuronal death, whereas PRX2 overexpression did not confer further neuroprotection in ASK1-deficient neurons (Fig. 6D). These results suggest that the Trx-ASK1 signaling complex is a major target for PRX2-afforded neuroprotection against ischemic injury.
Assessment of intracellular ROS using the H2O2-sensitive dichlorofluorescein fluorescence assay showed that while PRX2 overexpression significantly reduced ROS before or after OGD, Trx knockdown had an insignificant effect on the levels of H2O2 in neurons with or without PRX2 transfection (Fig. 6E). Thus, the impaired neuroprotective effect by PRX2 in Trx-deficient neurons could not be attributed to enhanced oxidative stress due to Trx knockdown. Alternatively, these results emphasize the importance of Trx as a specific and redox-sensitive endogenous inhibitor for the ASK1-dependent signaling pathway.
Attenuation of the MKK4/JNK signaling cascade in Tg-PRX2H mice
We further determined the effect of PRX2 overexpression on the MKK/JNK pathway, the main signaling pathway downstream of ASK1 activation that contributes to ischemic neuronal death (8, 15, 18, 29, 37). Determined using Western blots, tMCAO robustly activated MKK4 (increased phospho-Ser257) and, to a lesser extent, MKK7 (phospho-Ser271/Thr275) in brain of WT mice; tMCAO also activated JNK (phospho-Thr183/Tyr185) and its specific target c-Jun (phospho-Ser63). In contrast to WT mice, Tg-PRX2H mice showed nearly complete attenuation in cerebral activation of the MKK4/JNK/c-Jun signaling cascade after tMCAO (Fig. 7A, B). Immunofluorescent staining for phospho-MKK4 and phospho-c-Jun on brain sections, which showed predominant neuronal localizations, confirmed the inhibitory effect of PRX2 overexpression on MKK4/JNK activation after tMCAO (Fig. 7C).

To determine if attenuation of MKK4/JNK has a major role in mediating the neuroprotective effect of PRX2, WT, or Tg-PRX2H mice were administered the JNK inhibitor SP600125 (10 mg/kg) before tMCAO. SP600125 treatment attenuated JNK activation (Fig. 7D) and significantly decreased infarct volume after tMCAO in WT mice (Fig. 7E), but failed to reduce infarct volume in the Tg-PRX2H mice (Fig. 7E). These results suggest that inhibition of MKK4/JNK signaling is an integral component of the mechanism underlying PRX2-afforded neuroprotection against ischemic brain injury.
Discussion
Currently, the only established therapy for ischemic stroke, intravenous administration of recombinant tissue plasminogen activator, benefits just a small fraction of patients because of its limited time window of efficacy. Moreover, thrombolytic reperfusion may augment oxidative stress and the risk of intracranial hemorrhage in the ischemic brain (16). Thus, it is imperative to develop alternative therapeutic strategies for ischemic/reperfusion injury, such as aiming to limit ROS-mediated neuronal damage (5). In this study, we demonstrate for the first time that neuronal overexpression of PRX2 significantly reduced brain infarct and improved neurological outcomes up to 3 weeks after ischemia/reperfusion. We further show that PRX2 down-regulates a redox-dependent mitochondrial pro-death pathway via enhancing Trx-ASK1 interaction and thus prevents the activation of the ASK1-JNK signaling cascade in ischemic neurons.
The cytoprotective effect of PRX2 has recently been observed in neuronal cultures under ischemia-like conditions (2), and this PRX2-afforded protection is thought to be attributed to its antioxidant property. Indeed, PRX2 actively participates in the peroxide-reducing reactions after ischemia/reperfusion, resulting in remarkably increased PRX2 over-oxidation but decreased PRX activity reserve in ischemic neurons. This observation is consistent with a previous study showing increased production of PRX-SO3 after brain ischemia (10). However, the neuroprotective effect of PRX2 against ischemic/reperfusion could not be fully explained by its antioxidant actions per se, as PRX2 overexpression only partially reduced the levels of ROS in ischemic neurons. Moreover, knockdown of Trx expression, which alone did not augment oxidative stress in neurons, almost completely abolished PRX2-afforded neuroprotection against ischemia, supporting the notion that PRX2 achieves a neuroprotective effect through a Trx-dependent mechanism. Our results thus suggest an additional action of PRX2 via direct engagement in Trx/ASK1-dependent apoptosis-regulatory signaling. The following model is proposed: PRX2 overexpression, by maintaining Trx in a reduced state, enhances the interaction between Trx and ASK1 that keeps ASK1 at its inactive form, thus preventing the activation of ASK1 and of the ASK1-dependent pro-death signaling pathway after ischemia (Fig. 8).
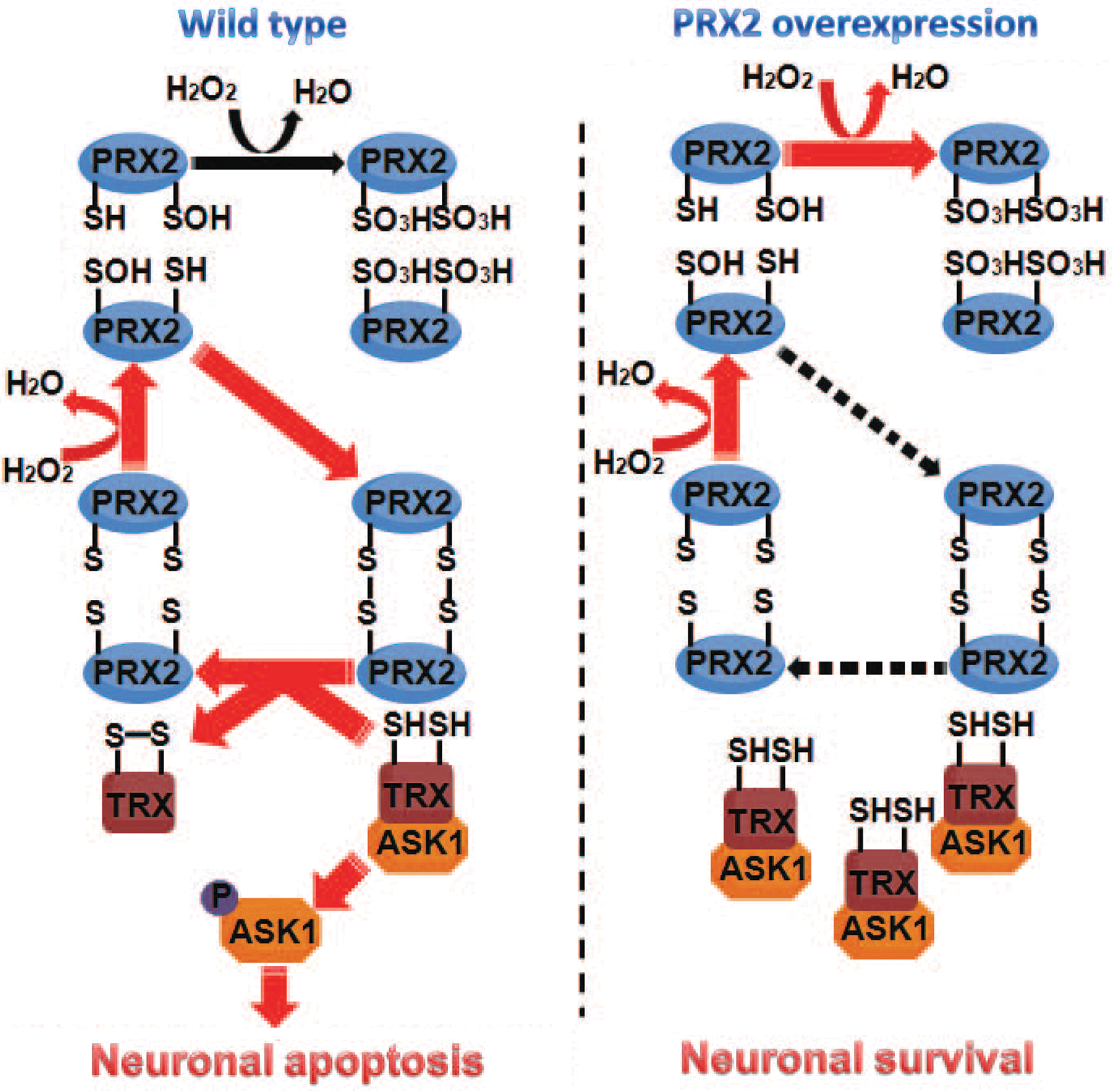
The peroxide-reducing activity of the active site Cys51 and Cys172 on PRX2 is essential for the functional interaction between PRX2 and the Trx/ASK1 complex. Cys51 on PRX2 is most accessible to oxidation under peroxide-imposed oxidative stress (41). Accordingly, under oxidative stress Cys51 is first oxidized to Cys51-SOH, which then is overoxidized to the irreversible form Cys51-SO2H or Cys51-SO3H; alternatively, Cys51-SOH forms a disulfide with Cys172 on another PRX2 molecule, and the disulfide can be reduced back to the active thiol form by the Trx/TrxR system (41, 42). Consistent with this concept, we found that mutation of Cys51 to alanine avoided any oxidation of PRX2 by hydrogen peroxide and that PRX2 with mutations at both Cys51 and Cys172 completely lost its ability to interact with the Trx/ASK1 complex or to inhibit ASK1 in ischemic neurons. Our results also show that overconsumption of PRX2 after ischemia/reperfusion led to depletion of reduced Trx in neurons, whereas PRX2 overexpression preserved Trx at its reduced state.
It has been well established that ischemia/reperfusion-induced overproduction of ROS causes neuronal demise by either directly targeting cellular macromolecules and/or triggering various pro-death signaling pathways (32, 36). Thus, in principle, antioxidant therapy should hold great promise in translation into an effective clinical treatment for stroke. However, such a therapeutic strategy, exemplified by the disufenton sodium (NXY-059) trials, so far has failed to produce beneficial effects in stroke patients (21, 35). Although many factors could contribute to the unsuccessful clinical trials, one issue worthy of consideration is that an untitrated suppression of ROS may deprive the normal physiological functions of free radicals and potentially result in unwanted effects. In contrast to classic antioxidant agents, PRXs possess a moderate ROS-scavenging property and yet are capable of potently inhibiting redox-sensitive pro-death signaling pathways that critically determine the fate of ischemic neurons. Several PRXs, including PRX1 and PRX2, are potentially inducible proteins in the brain (12, 17, 27). Conceivably, brain levels of PRX1 and/or PRX2 can be enhanced by PRX inducers or carrier-based protein delivery. Taken together, the results of this study demonstrating long-lasting neuroprotective effect by PRX2 warrant further investigation into PRX2's potential therapeutic value in experimental stroke, which would also include a poststroke treatment regimen.
While the current study focuses on PRX2, it does not exclude the possibility that PRX2 acts in conjunction with other PRX isoforms in neuroprotection. All mammalian PRX isoforms have been detected in the CNS and each isoform could have a unique neuroprotective role depending on its specific cellular distribution. For example, it is plausible that the glia-enriched PRX1 could influence stroke outcome by modulating astroglial and/or microglial responses (26). It would be interesting to determine if enhanced levels of both PRX2 and PRX1 offer additive neuroprotective effects against stroke. Moreover, future studies would also need to compare the neuroprotective effect of PRX2 in male versus female animals, as emerging evidence supports the important concept that there may be sex differences in response to neuroprotective agents in stroke models (23, 39).
In summary, transgenic overexpression of PRX2 confers long-term neuroprotection against ischemic/reperfusion brain injury in mice. This study also characterizes a potential mechanism underlying PRX2-afforded neuroprotection whereby PRX2 inhibits the ASK1-mediated pro-death signaling cascade in a Trx-dependent manner. Our results suggest that PRX2 may represent a potential target for stroke intervention.
Materials and Methods
Details beyond the descriptions here are provided in Supplementary Materials and Methods.
Models of cerebral ischemia
All animal experiments were approved by the Institutional Animal Care and Use Committee of Capital Medical University and University of Pittsburgh, respectively, and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male 8- to 12-week-old mice were randomly assigned to various experimental groups with the surgeon blinded to the genotypes of the mice. Focal cerebral ischemia was produced by intraluminal occlusion of the left MCA as described previously (3). To calculate infarct volume, brains were removed at either 2 or 21 days after MCAO, and subjected to 2,3,5-triphenyltetrazolium chloride (TTC) staining and MAP-2 immunohistochemical straining, respectively. Infarct volume was determined using MCID™ imaging with correction for edema.
To model ischemia-like conditions in vitro, primary cortical neuronal cultures were exposed to OGD for 60 min followed by reperfusion (37). Neuronal viability/death was quantified using the Alamar blue assay, lactate dehydrogenase release assay, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) (37).
Neurobehavioral tests
Neurobehavioral tests were performed before surgery and 3–21 days after MCAO by a researcher who was blinded to genotypes of the mice. The assessment for sensorimotor deficits consisted of three different tests: the rotarod test, the corner test, and the cylinder test. Rotarod and corner tests were performed as described previously (37). The cylinder test was adapted for use in mouse to assess forepaw use and rotation asymmetry. The mouse was placed in a cylinder 9 cm in diameter and 15 cm in height, and videotaped for 5 min. Videotapes were analyzed, and forepaw (left/right/both) use of the first contact against the cylinder wall after rearing and during lateral exploration was recorded. Nonimpaired forepaw (right) preference is expressed as the relative proportion of right forepaw contacts, which was calculated as: (right−left)/(right+left+both)×100 (6).
Construction of viral vectors
Ln vectors carrying either the human full-length (LnPRX2) or catalytically inactive mutant (LnPRX2C/A) PRX2 cDNA (also encoding a Ha tag) were constructed as described previously (9). To construct Ln vectors expressing shRNA against rat Trx or ASK1, the gene-specific targeting sequence (TRXt or ASK1t) or its counterpart scramble sequence (TRXsc or ASK1sc) was inserted into the transfer vector FSW under the control of the U6 promoter. Large-scale production, purification, and titration of the recombinant lentivirus was performed using a protocol described previously (38).
PRX and Trx activity assay
Peroxidase activity was determined as previously described (9). The PRX activity assay kit (Redoxica, Little Rock, AR) was used for the measurement according to the manufacturer's instructions. Trx activity was determined by the insulin disulfide reduxing assay as previously described (25). The data were expressed as percentage changes in PRX or Trx activity over control noninjured cell cultures or animals.
ASK1 kinase assay
Tissue or cell lysates were prepared under nondenaturing conditions as described. To assay for ASK1 activity, the lysates (150 μg total protein) were first subjected to ASK1 capture using the specific anti-ASK1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), and then incubated with recombinant myelin basic protein (MBP) in the presence of [γ-32P]ATP, and MBP phosphorylation was detected using autoradiogram. The data were expressed as fold changes in ASK1 activity over control noninjured cell cultures or animals.
Statistical analysis
Results are reported as the mean±standard error of the mean (SEM). The significance of difference between means was assessed by Student's t-test (single comparisons) or by analysis of variance with post hoc Bonferroni's/Dunn's tests (for multiple comparisons). A value of p<0.05 was considered statistically significant.
Footnotes
Acknowledgments
This work was supported by Chinese Natural Science Foundation Grant #30870854 (to X.J.), Chinese Ministry of Education Grant NCET-08-0625 (to XJ), and NIH Grants NS036736, NS043802, and NS045048 (to JC). Xiaoming Hu is supported by AHA grant 10POST4150028. We thank Carol Culver and Susan Giegel for their excellent editorial assistance.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.