
Abstract
Introduction
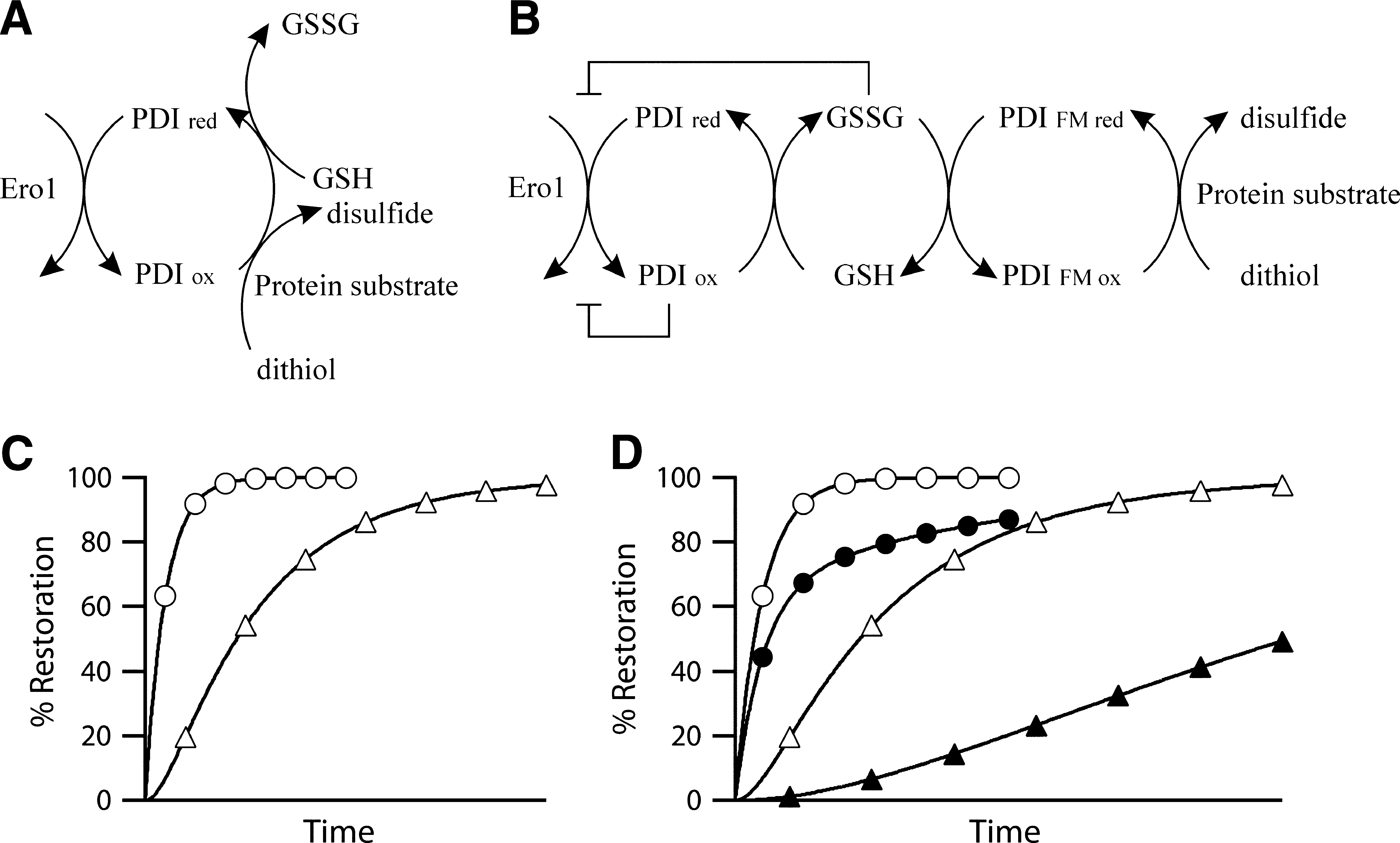
This review will focus on the first step in the process and, in particular, disulfide bond formation in the ER of mammalian systems. For more than 30 years low-molecular-weight compounds, in particular glutathione, were thought to play a primary role in disulfide bond of both mammals and other eukaryotes such as yeast. However, from 1998 onward the major route for oxidation in the ER was thought to be via the ER-resident sulfhydryl oxidase family member Ero1 (reviewed elsewhere in this Forum; 40) with protein disulfide isomerase (PDI) (21) as an intermediate (Fig. 1A). This organization mirrors the pathways subsequently found in the mitochondrial intermembrane space (42) and parallels can easily be drawn with the known pathways in the periplasm (25). However, it is known that premature termination of Ero1 is not lethal in Drosophila melanogaster (52), that its downregulation by RNAi enhances the lifespan of Caenorhabditis elegans (14), and that mice with both Ero1 isoforms disrupted show a relatively minor phenotype (59). While none of these systems may result in complete abolition of Ero1 activity, with concomitant issues arising regarding residual activity (Fig. 1), these results lead to the questions: What are the alternative routes for disulfide bond formation in these systems? and what is the physiologically normal route for disulfide bond formation in wild type animals, is it based on Ero1 or something else?
There are a variety of possible answers to the first question, all of which are based on the catalyzed or noncatalyzed use of low-molecular-weight oxidants to form disulfide bonds. These possible routes form the basis of this review.
Whatever the scenario, to answer either question, three requirements must be considered—thermodynamics, kinetics, and availability; that is, are the reactions thermodynamically favored based on the relative reduction potentials of the reactants? Are the kinetics, catalyzed or noncatalyzed, sufficiently fast to be physiologically relevant? Are the species available in sufficient amount to account for disulfide bond formation? For all of the possible low-molecular-weight oxidants these three questions must be addressed.
Molecular Oxygen
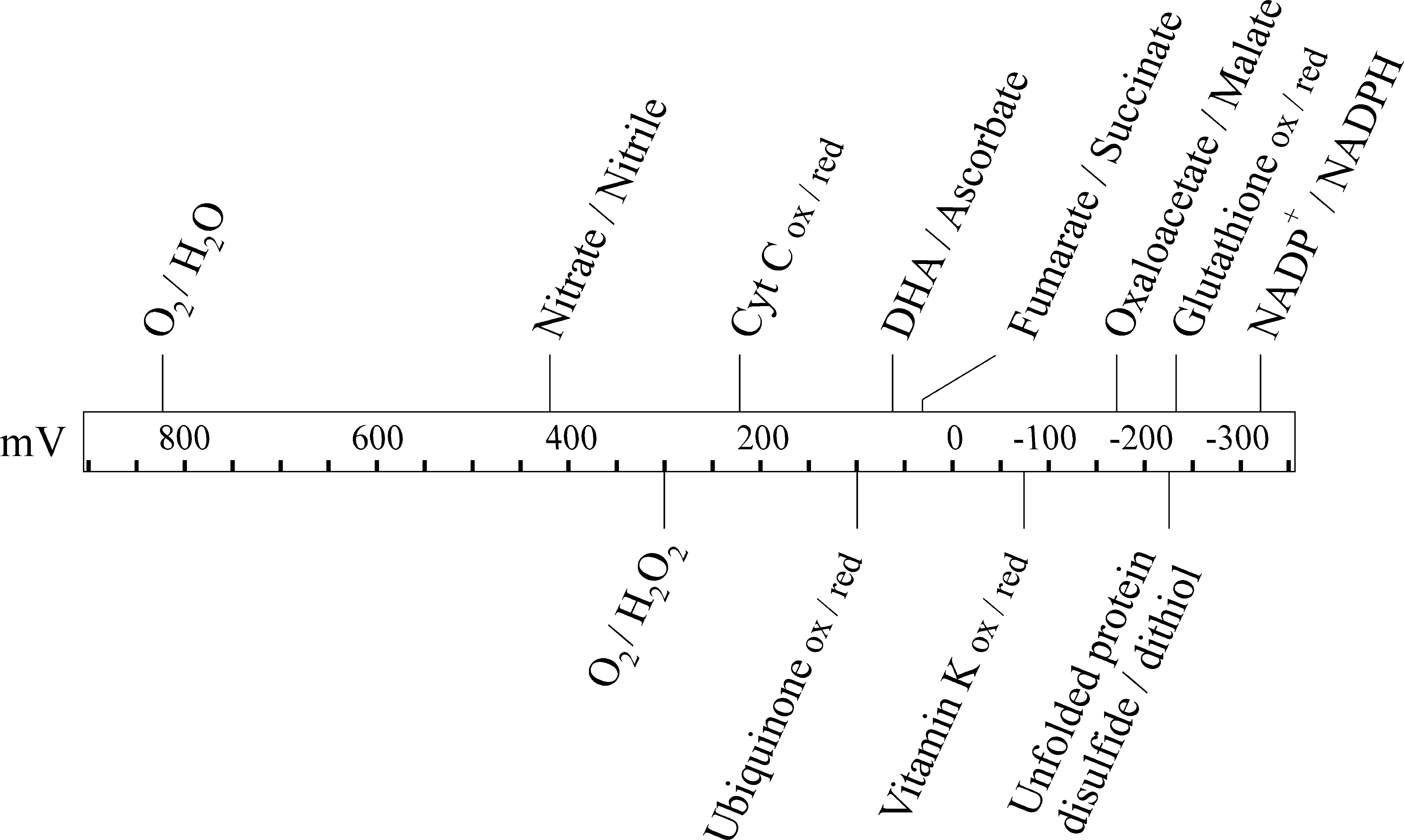
For aerobic organisms, or facultative anaerobes growing in aerobic conditions, molecular oxygen, O2, is the ultimate electron acceptor, that is, the net oxidant, in most biochemical pathways. As the derivation of the word oxidation suggests, oxygen is a potent oxidant with a reduction potential of 816 mV. This is more than a volt more oxidizing than the reduction potential for a disulfide/dithiol redox couple in an unfolded protein (Fig. 2). Hence, thermodynamically the oxidation by molecular oxygen of two cysteine side chains to form a disulfide bond is favored to the extent where it can be considered to be an irreversible reaction. The hydrogen peroxide produced as a byproduct in this reaction (Fig. 3A) can also be used to generate disulfide bonds (see Hydrogen Peroxide and Other Reactive Oxygen Species section). In vitro molecular oxygen is sufficient to oxidize cysteine side chains to form disulfide bonds; indeed this reaction forms the basis of most oxidative protein refolding on an industrial scale.
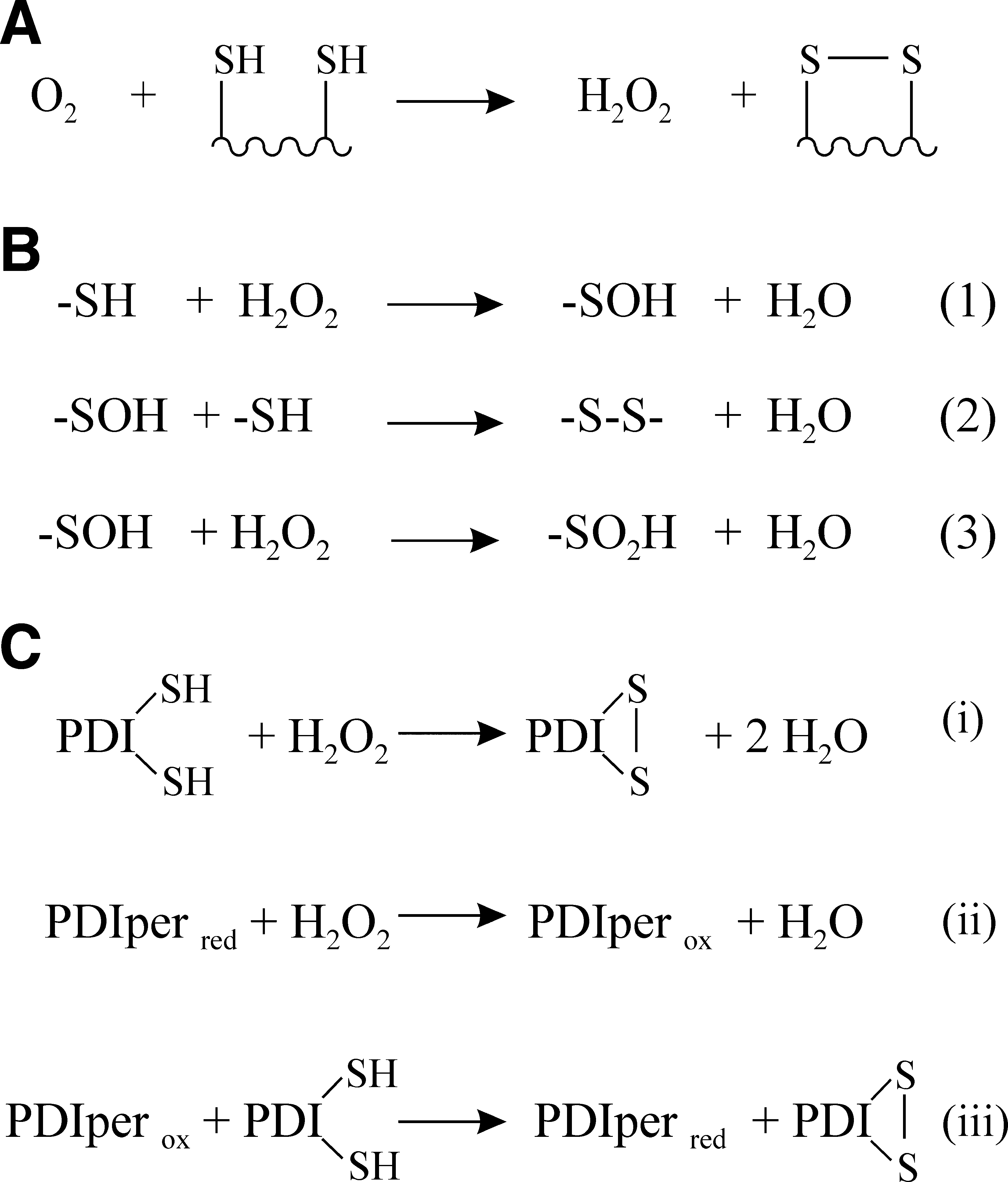
While thermodynamically favored, the noncatalyzed generation of disulfide bonds by molecular oxygen is kinetically too slow to be a significant physiological route in the ER. In vitro the reaction is catalyzed by the presence of transition metal ions, but in vivo the levels of these ions are highly regulated and most are present in complexes; for example, iron is stored in ferritin inside the cell (34). Hence the level of free metal ions is so low that metal ion–catalyzed oxidation by molecular oxygen will also not be a significant physiological route in the ER.
There are multiple enzymes that catalyze the oxygen-dependent formation of disulfide bonds including Ero1. In addition to two Ero1 family members, mammals have two other sulfhydryl oxidases that enter the ER, quiescin sulfhydryl oxidase (QSOX) 1 and 2. Both of these are transmembrane proteins, though QSOX1 has a splice variant that is a soluble secreted protein, and both contain an ERV/ALR sulfhydryl oxidase domain and a thioredoxin-like domain. This combination of domains makes QSOX family members very efficient at catalyzing disulfide bond formation in vitro (28). QSOX1 is the best studied and is thought to be a post-ER secretory pathway protein based on V5-tagged protein localization studies (12). Hence, it is unclear whether it can significantly contribute to disulfide bond formation in the ER under physiological conditions. Furthermore, while its overexpression in yeast does allow viability of an ero1-deleted strain, the growth of this strain is significantly slower than that of a strain rescued by overexpression of an ER-resident sulfhydryl oxidase Erv2p (12). Consistent with this observation, a genetic interaction has been found between QSOX1 and Ero1 in Drosophila, with the proposal that QSOX1 plays a redundant role in disulfide bond formation in the Notch signaling protein (52).
QSOX2 has been described as being a nuclear and outer plasma membrane protein (55), but the intracellular localization shown, in a network on one side of the nucleus, is reminiscent of ER staining. Furthermore, QSOX2 contains an RXR motif in its C-terminal cytoplasmic region and such motifs are responsible for ER localization of other proteins involved in oxidative protein folding, for example, TMX4 (43). It is therefore plausible that QSOX2 may contribute significantly to oxidative folding in the ER via catalyzing the molecular oxygen–dependent oxidation of a dithiol to a disulfide.
The third issue that requires addressing relates to availability. At standard temperature and pressure there is around 240 μM dissolved oxygen in a typical aqueous solution. While the amount of oxygen in the air is fairly constant at 21%, the amount of oxygen available to a eukaryotic organism may vary considerably depending on the environment it is in. Furthermore, in multicellular organisms there are multiple oxygen gradients (24). These include the following: (i) the gradient between arterial and venal blood and gradients within tissues, for example, the liver that has a twofold difference in oxygen levels across it; (ii) the position of each cell with respect to the closest blood vessel and oxygen consumption of adjacent cells; and (iii) oxygen consumption within cells leading to intracellular oxygen gradients. While it is effectively impossible to measure oxygen levels in the ER of specific cells in a multicellular organism it is likely that oxygen levels will be several orders of magnitude greater than that required to sustain disulfide bond formation. Indeed, oxygen consumption required for protein synthesis per se using aerobic respiration far exceeds that possibly required for oxygen-dependent disulfide bond formation.* Hence, if there is enough oxygen to synthesize the protein, then there is sufficient for it to be directly involved in the sole route for oxidative folding.
Hydrogen Peroxide and Other Reactive Oxygen Species
Reactive oxygen species (ROS), as the name suggests, are either derived from or contain oxygen and are highly chemically reactive compounds. A range of ROS exist; the main biologically relevant ones are superoxide, hydroxyl radicals, hypochlorite, and peroxides such as hydrogen peroxide. Of these hydrogen peroxide is the best studied with respect to oxidative folding of proteins.
The hydrogen peroxide–dependent oxidation of a dithiol to a disulfide is a two-step process (Fig. 3B). Both reactions are thermodynamically favored and effectively irreversible. As long as the peroxide concentrations are not so high as to result in overoxidation of the cysteine sulfenic acid intermediate to a sulfinic acid (Fig. 3B, reaction 3), this reaction should efficiently generate protein disulfide bonds with minimal side reactions. Indeed, this was recently shown to be the case for hydrogen peroxide–dependent oxidative folding in vitro (26).
While thermodynamically favored and kinetically faster than oxygen-dependent oxidation, the second order rate constant for the formation of disulfide bonds in folding protein substrates by hydrogen peroxide is only 5.0 M−1s−1 (26). This implies that the noncatalyzed reaction is too slow to be a physiologically significant route for oxidative folding. The active site of PDI can also directly be oxidized by peroxide, but this is only circa twofold faster (26) and is similarly probably too slow to be a physiologically major route for disulfide formation.
In the last few years two other distinct mammalian ER–located catalyzed routes for the peroxide-dependent formation of disulfide bonds have been reported. These are dependent on the enzymes GPx7, GPx8, and PrxIV (38, 49 –51, 60). While all three enzymes have been shown to act as PDI peroxidases and hence catalyze the peroxide-dependent formation of disulfide bonds (Fig. 3C), the methodologies used to characterize them have been very different. Hence, it is difficult to make conclusive cross-comparisons as to which is more likely to be the more physiologically significant route.
GPx7 and GPx8 are members of the glutathione peroxidase family with GPx7 being a soluble protein and GPx8 being transmembrane. Despite their name they are not glutathione-dependent peroxidases, but rather they are members of the thioredoxin peroxidase subfamily (38) and hence in the ER they are best viewed as PDI peroxidases; that is, they catalyze the peroxide-dependent oxidation of the active site of PDI family members (Fig. 3C). In vitro characterization has shown that either protein, in conjunction with PDI, is able to catalyze very efficiently peroxide-mediated oxidation of folding proteins to their native state (38). The rate-limiting step in this reaction is the oxidation of GPx7 or GPx8 by peroxide, with GPx7 reacting circa 10-fold faster with peroxide than the active site of PDI. Both proteins also interact with Ero1α in vivo and GPx7 stimulates oxygen consumption by Ero1α in vitro. The simplest model that emerges is that they have evolved to more efficiently utilize any hydrogen peroxide generated by Ero1 during its catalytic activity.
PrxIV is a soluble ER-resident peroxiredoxin. At the simplest level the mechanisms of action of PrxIV mirror those of GPx7 or GPx8. However, PrxIV forms a decameric complex and the reaction of PrxIV with peroxide results in a more complex pattern of intra- and intermolecular disulfide bonds (49, 50) than can occur in GPx7 or GPx8. To date much more in vivo work has been reported on PrxIV (49 –51, 60) than on GPx7 or GPx8. PrxIV is able to rescue an Ero1 temperature-sensitive mutant in the yeast Saccharomyces cerevisiae (60) suggesting it is able to modulate oxidative folding in vivo, possibly by directly catalyzing peroxide-mediated oxidation. Like other peroxiredoxins, the reaction of PrxIV with peroxide is extremely rapid (49, 51). The oxidized form of PrxIV can transfer disulfide bonds via PDI to folding substrates (51, 60), but the overall rate of this in vitro (60) is similar to that of the GPx7- and GPx8-mediated reaction (38) due to the slower reduction of PrxIV by PDI. This apparently slow transfer of disulfide bonds between PrxIV and PDI, and the ease of overoxidation of PrxIV to a catalytically inactive state (49), causes some conceptual difficulties for a direct catalytic role for PrxIV in peroxide-mediated oxidative folding. In addition, PrxIV is a nonessential gene product, though knockout mice show testicular atrophy (23), implying it is dispensable for oxidative folding. Furthermore, PrxIV deletion does not modulate the recovery of the resting redox state of the ER after treatment with dithiothreitol (DTT) (53). Other peroxidredoxin family members are involved in redox-dependent signaling and/or have redox-dependent molecular chaperone activity (7). Either of these could be consistent with the published in vivo data for PrxIV.
While further elucidation of the physiological roles of GPx7, GPx8, and PrxIV is required, including parallel in vitro and in vivo characterization, it is clear that both the thermodynamic and kinetic questions can be plausibly answered for peroxide-mediated disulfide bond formation. The third question, “are the species present in sufficient amounts to account for disulfide bond formation?” is more difficult to answer. The intraluminal concentration of peroxide is unlikely to be high due to its reactivity toward a wide range of biological molecules, but if it can be generated sufficiently fast so as to allow for the maximal rate of disulfide formation required [100,000 disulfide bonds per second (11) or 50% restoration of GSSG levels within 10 s of DTT washout (4)] then it could be a physiologically significant route.
Three sources are usually mentioned when discussing possible sources of peroxide in the ER: (i) the action of sulfhydryl oxidases, (ii) the mitochondrial respiratory chain, and (iii) NADPH-dependent oxidases (10, 60). Each of these needs to be considered in turn.
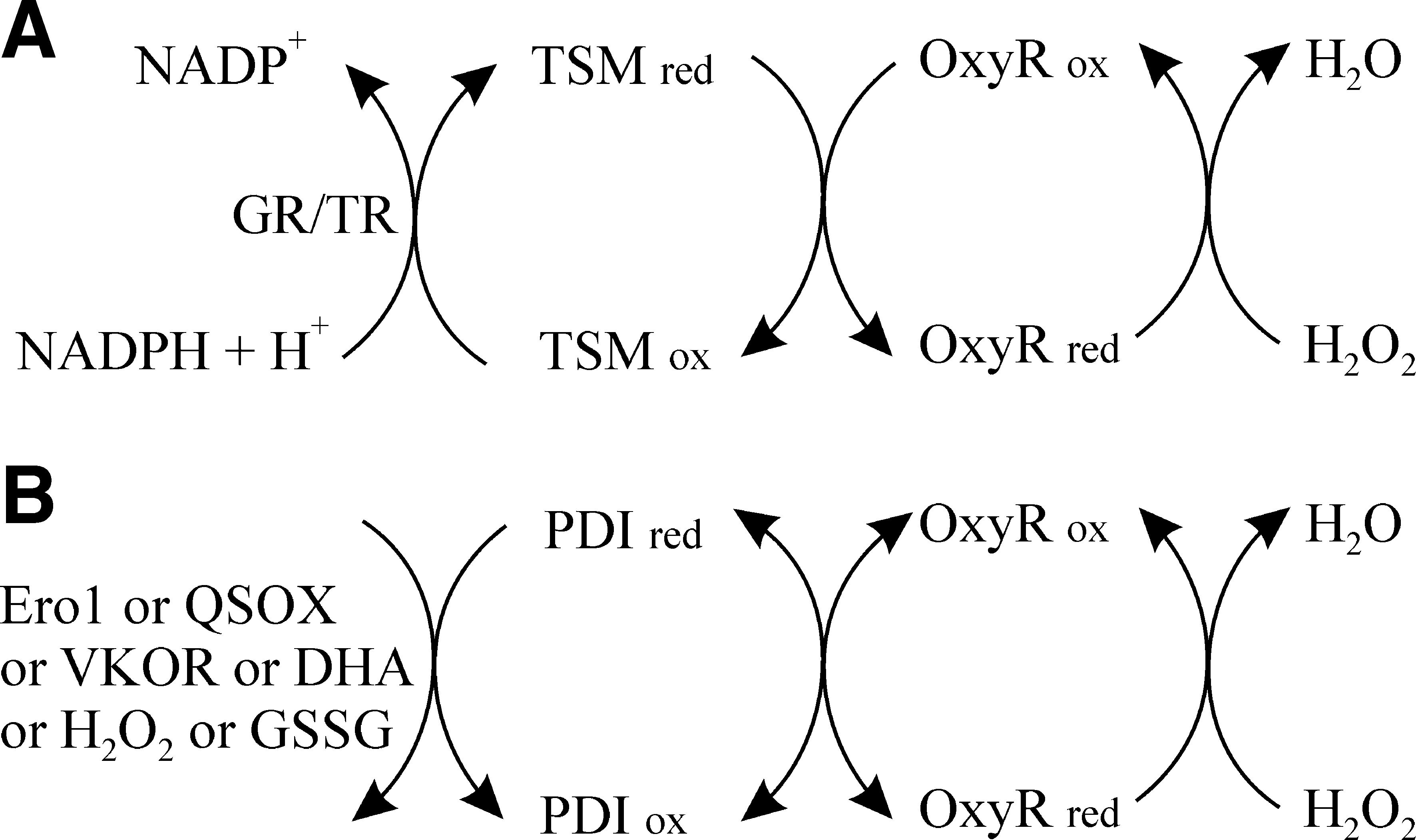
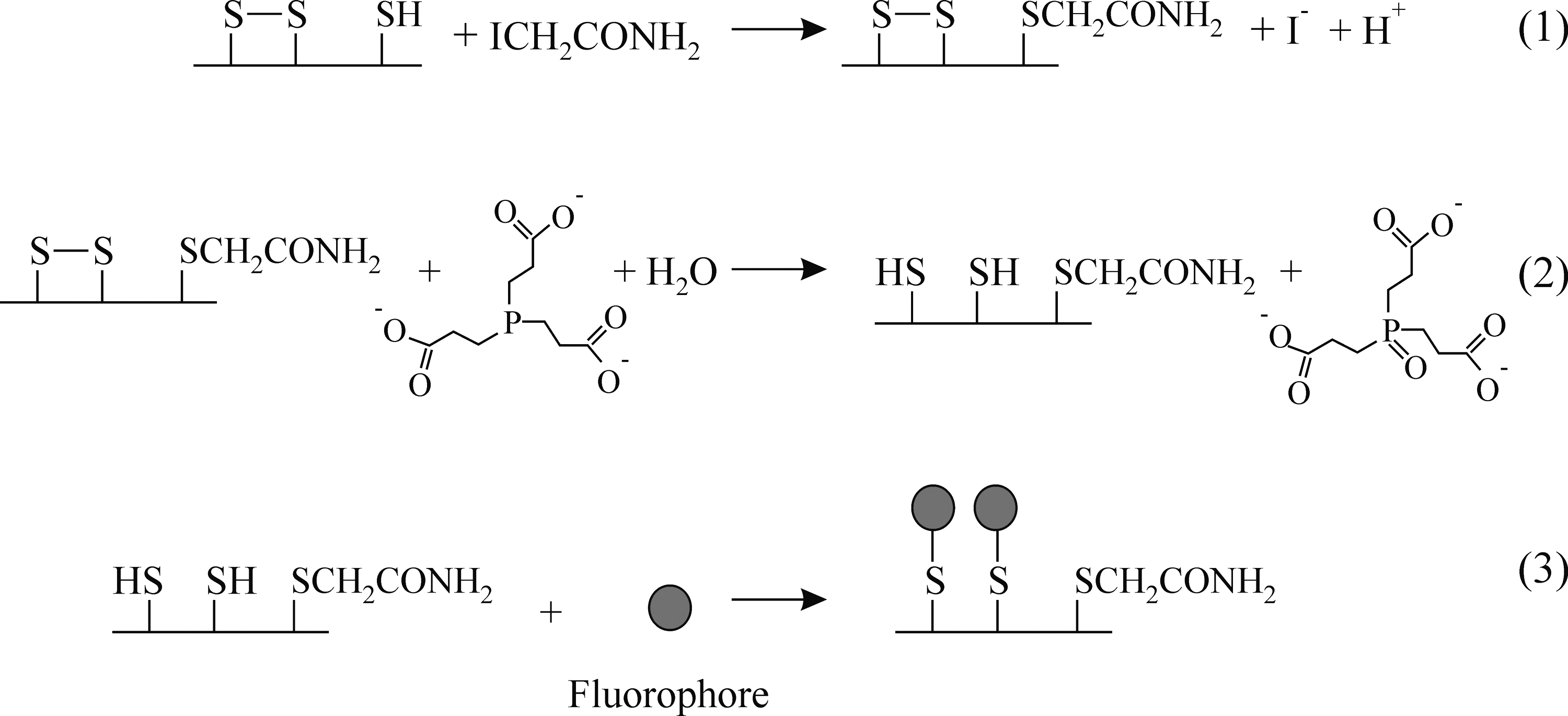
In vitro, when using molecular oxygen, Ero1 family members generate one molecule of hydrogen peroxide per disulfide bond made (20), as per other sulfhydryl oxidase family members. However, in vivo Erv1p, the yeast mitochondrial sulfhydryl oxidase can transfer electrons to cytochrome c (47), obviating the need to generate potentially toxic hydrogen peroxide. While many of the ER-associated cytochromes have their active sites located on the cytoplasmic side of the membrane, there are a number which are located on the luminal side of the membrane and it is plausible that Ero1 could similarly transfer electrons to cytochromes in vivo rather than to molecular oxygen. Under such a scenario Ero1 would not make peroxide as a byproduct. The only published evidence that Ero1 makes peroxide in vivo (18) comes from the use of the HyPer system. However, this probe may be sensitive to the redox state of PDI as well as to the presence of peroxide (Fig. 4) and hence it is unclear whether Ero1 generates peroxide in vivo under physiologically normal conditions.
A second possible source of hydrogen peroxide for oxidative folding in the ER is the mitochondrial respiratory chain. This is supported by studies showing the physical and functional cross-talk between the ER and mitochondria, including the localization of human Ero1α at mitochondrial-associated membranes [e.g., see Refs. (9, 19)] and by a report on regulation of the protein disulfide proteome by mitochondria (57). While conceptually appealing to be a significant route for oxidative folding in the ER this model requires the specific transfer of large amounts of a chemically reactive species from one membrane-bound compartment to another without triggering peroxide-dependent intracellular signaling—something which is implausible without a dedicated transport system. Further questions arise due to (i) the presence of mitochondrial glutathione peroxidases that consume peroxide; (ii) multiple reports on mitochondrial catalase expression that do not show deleterious effects [e.g., see Ref. (15)]; (iii) the Fenton reaction, the breakdown of hydrogen peroxide to generate highly reactive hydroxide radicals, is known to occur at the ER membrane (32); (iv) the methodology used to determine mitochrondrial regulation of the disulfide proteome (57) does not provide evidence that this is a major route for disulfide formation in the ER (Fig. 5) and nor was it claimed to be.
The third discussed source of peroxide for oxidative folding in the ER is the NADPH-dependent oxidases, the NOX family (27). These are involved in a variety of biological responses including the regulation of cellular pH (via mediating proton currents), antimicrobial defense, signaling cascades, and gene expression. With the possible exception of Nox4, the human NOX family members are unlikely to contribute significantly to peroxide-dependent disulfide bond formation in the ER for two reasons. First, they do not generate peroxide, rather they generate superoxide. Superoxide can potentially be used directly to make disulfide bonds, or can be converted via the action of superoxide dismutase into peroxide. However, this does not seem a plausible route for evolution to have adopted as a major route to make disulfide bonds given the high reactivity of superoxide toward other biochemical species and by the lack of an ER-resident superoxide dismutase. Second, all except Nox4 show a narrow tissue distribution (27). In contrast, Nox4 has a broader tissue distribution and it is thought to directly produce peroxide rather than superoxide (35), though the mechanism for this is unclear. However, the tissue distribution of Nox4 is neither ubiquitous nor does its expression correlates well with PDI expression or protein secretion. While there are other human proteins that have NAD(P)H-dependent oxidase activity and generate peroxide (Duox family members) or catalyze protein disulfide-thiol exchange (ENOX family members), these are not ER associated nor are they ubiquitously expressed.
One further source of peroxide in the ER is the action of L-gulonolactone oxidase, the terminal enzyme in the synthesis of ascorbate or vitamin C. This enzyme uses molecular oxygen to oxidize L-gulono-1,4-lactone to L-xylo-hex-3-gulonolactone that spontaneously converts to L-ascorbate. For each molecule of L-gulonolactone oxidized, one molecule of hydrogen peroxide is generated. While this is potentially a major route for peroxide generation in the ER of many eukaryotes, several species, including humans, do not express a functional L-gulonolactone oxidase.
In summary, while thermodynamically and kinetically hydrogen peroxide is a strong candidate to play a significant role in disulfide bond formation in the human ER, there are as yet no clear and confirmed in vivo sources that generate sufficient peroxide.
Ascorbate Derivatives
Ascorbate is a major component of redox biology. It is a cellular antioxidant that has two higher oxidation states (Fig. 6A): semidehydroascorbate (SDA) and dehydroascorbate (DHA). The reduction potential of the ascorbate/DHA couple is sufficiently oxidizing (Fig. 2) for DHA-dependent oxidation of a dithiol to be effectively irreversible.
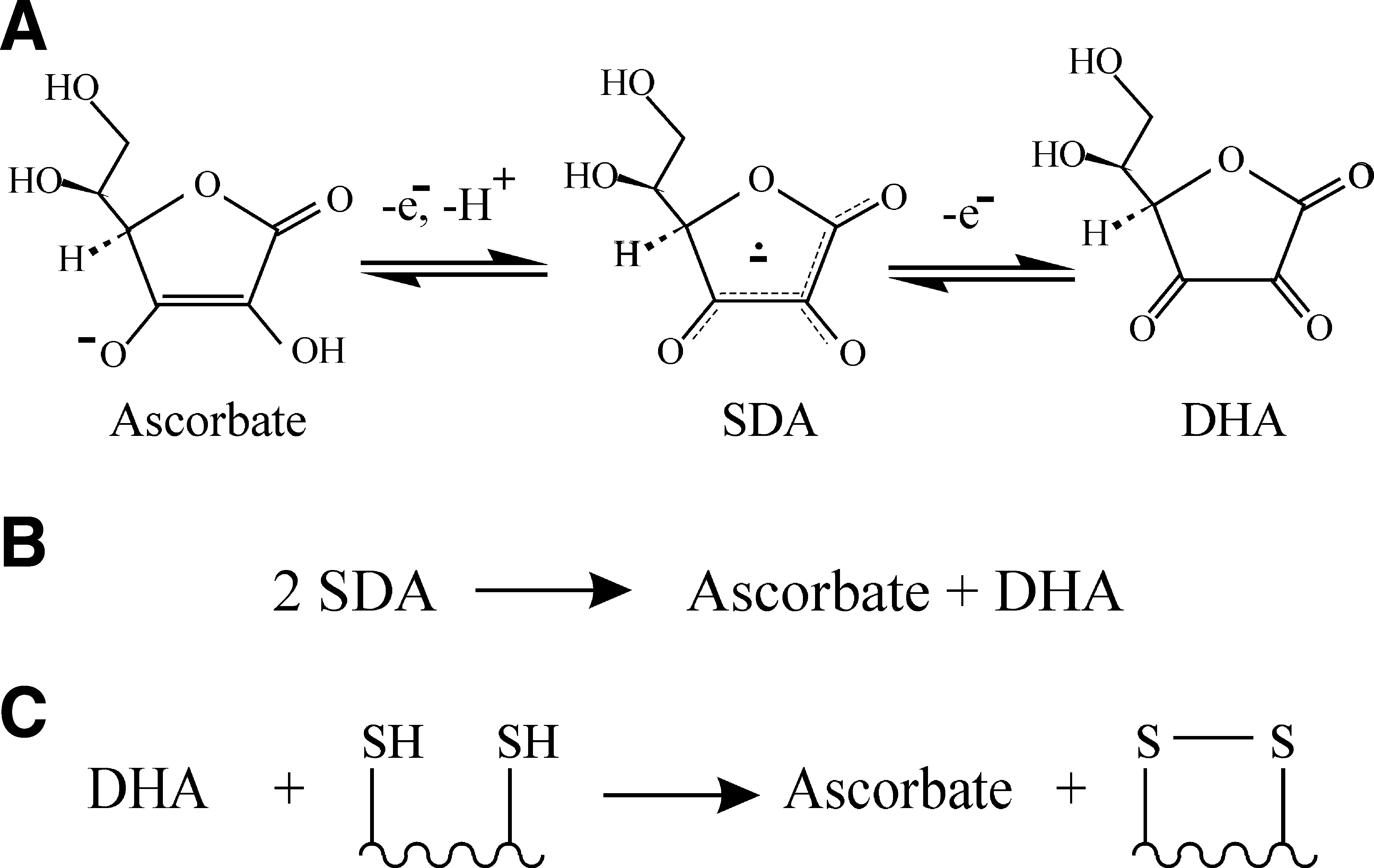
DHA was used in many of the original experiments for in vitro oxidative refolding, but then largely overlooked for nearly four decades. It was thought that DHA-dependent disulfide bond formation would be a PDI-catalyzed reaction [e.g., see Ref. (6)], but a recent study has shown that in vitro DHA is able to oxidize dithiols in unstructured peptides and proteins up to sixfold faster than it can oxidize the active site of PDI (Fig. 6B) (44). The rate constant for the noncatalyzed oxidation of an unfolded protein was reported to be over 20-fold faster than oxidation of the same protein by oxidized glutathione or hydrogen peroxide (26, 44). Hence it is possible that noncatalyzed DHA-dependent oxidation may be sufficiently fast to be a significant pathway for disulfide bond formation in the ER. Whether it is rapid enough depends on local DHA concentrations.
While ascorbate is known to be present in millimolar concentrations inside the cell (13), DHA is more difficult to quantify especially in the ER. However, DHA is likely to be present at low concentrations especially since it is sensitive to hydrolysis. Hence the same issue arises for DHA as for hydrogen peroxide; is the rate of localized DHA production sufficiently high for it to be a significant pathway for disulfide bond formation?
There are multiple possible routes for DHA production. The simplest, the formation of DHA during the noncatalyzed antioxidant action of ascorbate, is unlikely to generate sufficient DHA to support a significant route for disulfide bond formation. While plants have ascorbate oxidases that utilize oxygen to oxidize ascorbate to DHA (16), the only related proteins in mammals—ceruloplasmin, hephaestin, and coagulation factors V and VIII—have well-characterized extracellular functions. However, human hephaestin has two C-terminal cytoplasmic RXR transmembrane protein ER-localization motifs, while the closely related hephaestin-like protein does not, suggesting the potential for differential localization. Other enzymes use ascorbate as a direct electron acceptor and so also generate DHA. These include the ER-resident copper monoxygenases dopamine β-hydroxylase, Moxd1, and peptidyl-glycine α-amidating monoxygenase, all of which generate one molecule of DHA via the formation of SDA (33, 36, 56) each catalytic cycle. Finally, DHA can be produced as a minor byproduct of other ascorbate-dependent ER-resident proteins such as prolyl hydroxylase and lysyl hydroxylase, both of which generate up to one DHA every 25 catalytic cycles (37).
So, like peroxide, DHA is thermodynamically and kinetically a strong candidate to play a significant role in disulfide bond formation in the ER, but there are as yet no clear and confirmed in vivo sources for its production in sufficient quantities.
Vitamin K
Like DHA, vitamin K has a long history in disulfide bond formation but has been largely overlooked with respect to its possible action in the ER. Vitamin K is not a single compound, but rather a class of closely related compounds (Fig. 7A). Vitamin K2 (menaquinone) is one of the two primary electron acceptors from the enzyme DsbB, a key player in disulfide bond formation in the bacterial periplasm (25), while Vitamin K3 (menadione) has been widely used as a strong oxidant in studies on the effects of overoxidation on disulfide bond formation.
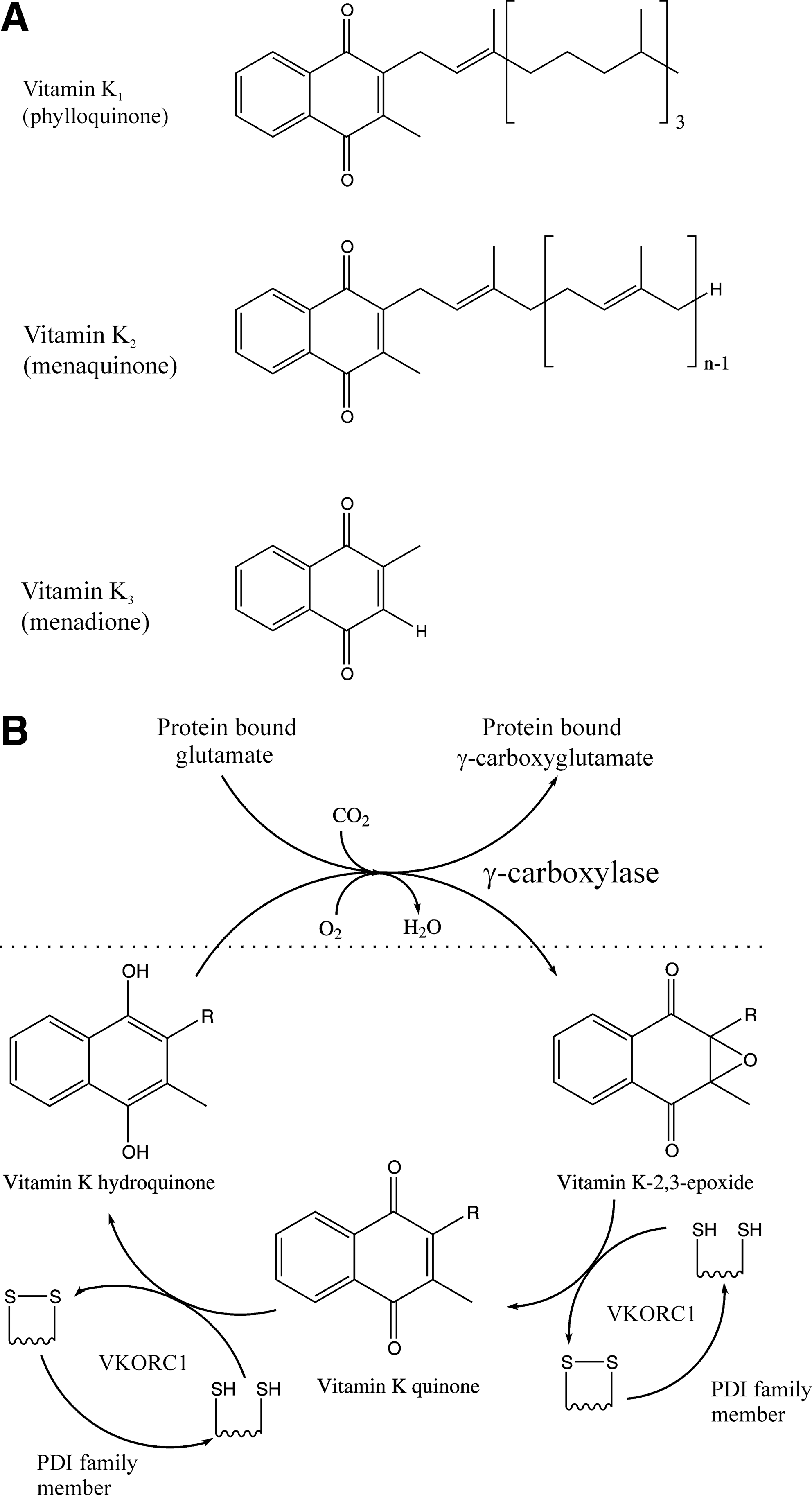
There have been few studies on the kinetics of noncatalyzed vitamin K–dependent disulfide bond formation, probably due to the instability of the vitamin K hydroxyquinone reduced states and the poor solubility of the biologically relevant forms—both vitamin K1 (phylloquinone) and menaquinone contain extended aliphatic chains. It is possible that the noncatalyzed reaction is sufficiently fast to accommodate significant disulfide bond formation. However, this is unlikely due the presence of the bacterial catalysts that use vitamin K, DsbB, or the recently discovered bacterial vitamin K–dependent oxidoreductase (VKOR) to catalyze periplasmic disulfide bond formation (17).
Mammals express two VKOR family members, VKORC1 and VKORC1L1. Both are ER-resident transmembrane proteins. VKORC1 has a function in the catalytic cycle of γ-carboxylation of some secreted proteins (Fig. 7B) including blood clotting factors. This catalytic cycle generates disulfide bonds, with PDI family members being thought to be the primary in vivo reductant (46). Hence γ-carboxylation is linked to disulfide bond formation in the ER. However, γ-carboxylation is a quantitatively minor posttranslational modification compared with disulfide bond formation. Consistent with this, VKORC1 knockout mice show no phenotype until birth, but then have early postnatal lethality due to severe bleeding (48), that is, a phenotype linked to γ-carboxylation and not to disulfide bond formation. VKORC1L1 has to date been poorly studied, but a recent publication demonstrates that it has a vitamin K–dependent intracellular antioxidant function (54). If either enzyme could function as the bacterial homologue, that is, not coupled to γ-carboxylation, then this could potentially generate a significant route for disulfide bond formation in the ER; however, there is currently no evidence for this.
Glutathione
Glutathione is a tripeptide, γ-glutamylcysteinylglycine, that can exist in the reduced state (GSH) or an oxidized disulfide linked dimeric state (GSSG). The ratio and concentrations of GSH and GSSG determine the redox potential of a variety of intracellular compartments, with the ER being more oxidizing than the cytoplasm due to higher levels of GSSG. The potential role and regulation of glutathione in disulfide bond formation in the ER is a complex subject that requires its own review (2).
Here it is sufficient to say that glutathione probably plays a major role in native disulfide bond formation in the lumen of the ER both oxidizing and reducing PDI family members (Fig. 1B) with complex and species-dependent kinetics (31). However, GSSG is not a net oxidant; rather the disulfide bond gets transferred from GSSG to the species which it oxidizes. Hence GSSG can be thought of a transfer intermediate and GSH/GSSG as a critical redox buffer system. Consistent with its importance, the glutathione-dependent redox potential of the ER is regulated and is very rapidly restored if perturbed (4).
Other Low-Molecular-Weight Species
Other low-molecular-weight species could potentially have a role in disulfide bond formation in the ER due to their redox active biophysical properties. These include ubiquinone, tocopherols/tocotrienols (vitamin E), xanthine, and uric acid. These may show species specificity; for example, urate oxidase is nonfunctional in humans and other primates, as per L-gulonolactone oxidase (see Hydrogen Peroxide and Other Reactive Oxygen Species section). While ubiquinone is one of the major electron acceptors from DsbB in bacterial disulfide bond formation (25), there is little evidence currently for a significant role for any of these species in disulfide bond formation in the ER. It is also possible that other redox couples could be utilized; for example, the 5-lactone/glucose and fumarate/succinate redox couples are both sufficiently oxidizing to be thermodynamically linked to disulfide bond formation and both involve the transfer of two electrons and two protons. Finally, some organisms use derivatives of the chemical species discussed previously. For example, yeast synthesizes D-erythroascorbic acid (22) that differs from ascorbate by the chirality at one position, while some protozoa including trypanosomes use trypanothione—two molecules of glutathione linked together by a polyamine linkage—as a major redox species (30).
Conclusions
While there are multiple plausible routes for disulfide bond formation in the ER, it is unclear which are major routes and which are minor under physiological and nonphysiological conditions. Just because a route exists does not necessarily mean that it is physiologically significant.
Ero1 can be disrupted without lethality in several organisms and the in vitro kinetics of Ero1-catalyzed disulfide bond formation [e.g., see Ref. (5)] are too slow to account for the observed in vivo kinetics. However, the disruptions do not necessarily abolish Ero1 activity (Fig. 1) and differences between in vitro and in vivo conditions make cross-correlations of relative kinetics complex. It could be that Ero1 is the normal physiological route for the majority of disulfide bond formation in the ER of wild-type and Ero1 disrupted mice, but that other minor pathways exist. Alternatively it could be that Ero1 forms a minor pathway. In such a scenario there could either be one other route being the major source of disulfide bond formation or there may be multiple possible pathways all of which contribute significantly to disulfide bond formation under physiologically normal conditions. With the generation of the Ero1 disrupted mice it should be possible, through the use of inhibitors of specific pathways or combinations of knockouts together with transcriptomic and proteomic analysis and biochemical studies, to identify the pathways that allow disulfide bond formation to occur in these mice. These can then potentially be cross-checked in wild-type systems to see whether they are the major physiological route or a minor route that steps in when the major route is compromised.
Finally, two things should be remembered. First, that being an essential (or nonessential gene) product may say little about whether the protein is the major physiological route for the bulk of proteins that fold in the ER. Second, oxidation is not the rate-limiting step in native disulfide bond formation; rather, isomerization of nonnative disulfides is rate limiting (21). Hence a significant reduction in the rate of oxidation may result in no change in the rate of native disulfide formation. The field grows in complexity and it is only by combining genetic, in vivo and in vitro biochemical data using multiple substrates along with knowledge of the tissue distribution, kinetics, thermodynamics, and quantities of the possible species involved that we can answer the question regarding the physiologically relevant route(s).
Footnotes
Abbreviations Used
*
Six oxygen molecules are required to generate circa 29 ATP molecules and circa 5 ATP molecules are required per amino acid polymerization, that is, approximately one oxygen molecule per amino acid polymerization event (1, ![]() ). In contrast, one oxygen molecule is required to generate two disulfide bonds.
). In contrast, one oxygen molecule is required to generate two disulfide bonds.