
Abstract
Introduction
Unfortunately, one of the greatest challenges for research scientists is to diminish the continuing accumulation of distortion and half truths that are reported in the popular media regarding the health benefits of certain foods or food supplements, including flavonoids. The use of food or food supplements is not new, but interest in their use has increased dramatically because of perceived health benefits that are presumably acquired without unpleasant side effects (52). This is especially true in cancer prevention and treatment. Flavonoids are touted to exert anti-inflammatory (147), anticancer (38), and antioxidant (2) effects in vitro. However, whether flavonoids can produce these effects in vivo has been disputed (116, 143, 147). Therefore, identifying the specific signal transduction pathways, gene, protein, and transcription factor targets, and mechanisms explaining the purported anticancer activity of specific dietary factors and in particular, flavonoids, might provide effective alternatives or additions to traditional methods of cancer prevention (i.e., chemoprevention) or cancer treatment (i.e., chemotherapy).
Our work has focused on clarifying the effects of various dietary components, especially flavonoids, on cancer cell proliferation and tumor growth, discovering mechanisms to explain the effects, and identifying the specific cellular and molecular targets of these compounds [reviewed in (91)]. Flavonoids are the most common family of polyphenolic compounds with approximately 8000 individual compounds identified (117). Flavonoids found in vegetables, cereals, legumes, fruits, and beverages such as wine, teas, and coffees have been subdivided into 14 different categories based on their chemical structure. These categories include the chalcones, dihydrochalcones, aurones, flavones, flavonols, dihydroflavonols, flavanones, flavanols, flavandiols, anthocyanidins, isoflavonoids, biflavonoids, and proanthocyanidins (22). A more recent review (34) categorizes flavonoids into only six major subclasses (flavanols, flavanones, flavones, isoflavones, flavonols, and anthocyanins) based on the structure of the three rings. Flavonoids suppress the growth of many different types of cancer cells through a variety of proposed mechanisms. This nonspecificity is compounded by the fact that thousands of flavonoids exist, and, therefore, their study has provided a very rich area of research. Only a few cases of computational work focusing on flavonoids exist (77, 121, 134). The use of conventional methods to select the best single or group of best chemicals for identifying compounds that are effective in treating or preventing a disease such as cancer is difficult. Computational strategies for determining protein targets of flavonoids have not yet received a great deal of attention. Over the last 3 years, our laboratory has utilized computational strategies that include virtual screening, shape-similarity screening, and molecular docking to identify the potential protein targets of flavonoids and other phytochemicals (91).
To identify the potential targets of a variety of flavonoid compounds and to carefully study their mechanism of action, we created a flavonoid database of 2620 compounds (24). We are continuing to enlarge the database by adding more flavonoid compounds. Based on this database, we have obtained results for every compound and its potential off-target proteins by using shape-similarity screening (24). We have more than 4.1 million records for a variety of protein targets and 374,000 records for the specific kinase targets. These results can be easily queried by compound identification or by protein type. Thus, our basic strategy for identifying specific molecular targets of flavonoids and other phytochemicals is to use supercomputer technology combined with protein crystallography, molecular biology, and experimental laboratory verification (Fig. 1). Protein kinases and their target transcription factors are our most common strategic targets for drug screening. Initially, we began with the virtual screening (105) of various ligand databases such as the Zinc is not commercial (ZINC) free database of commercially available compounds (
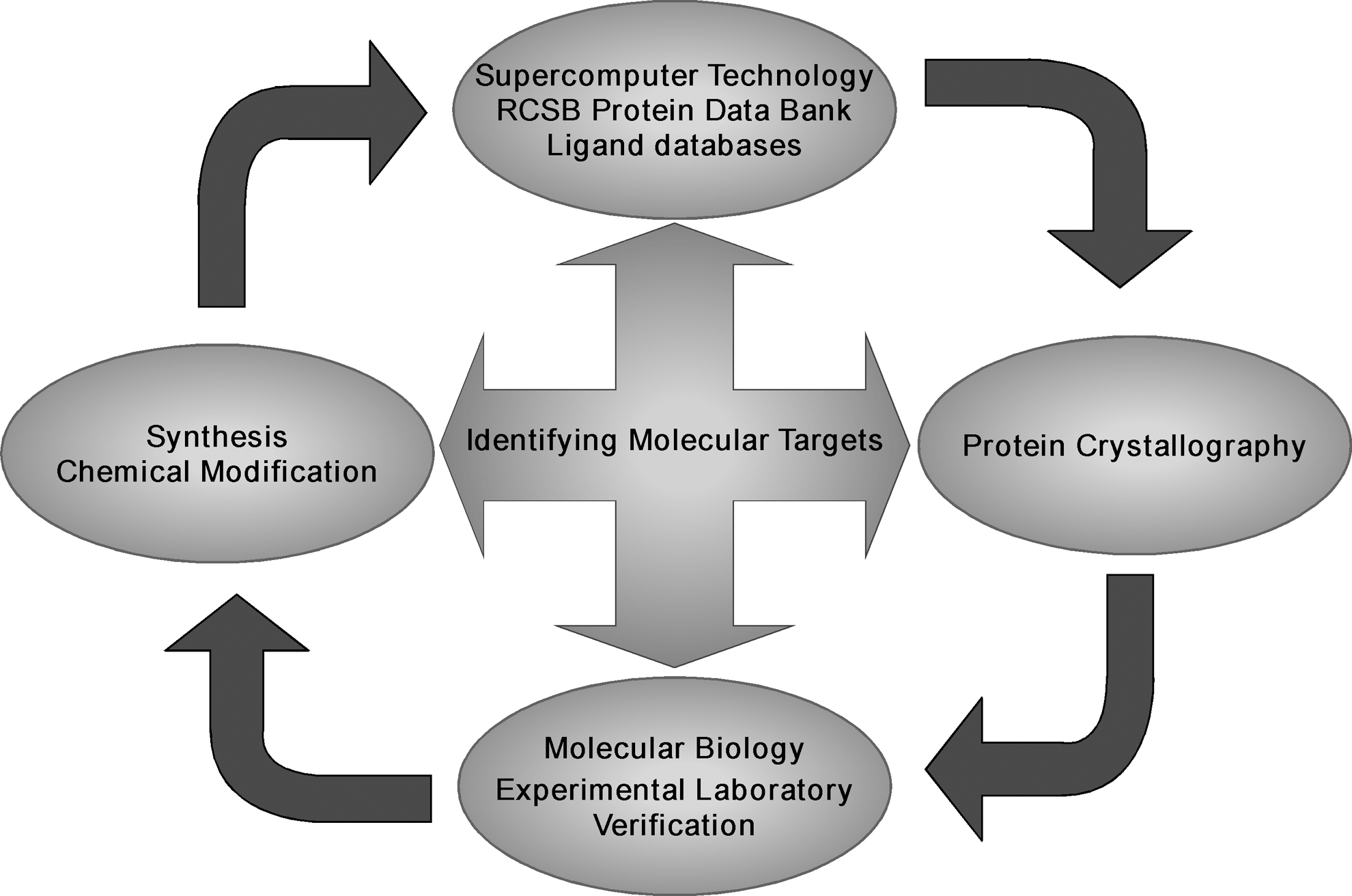
Using this strategy, we have studied the anticancer activities and clarified the role and molecular targets for various flavonoids found in our everyday diet. The remainder of this article will highlight our major findings with regard to the specific molecular mechanisms and protein-binding targets of a select few of these dietary flavonoids as effective anticancer agents. The main emphasis will be on the protein targets that have been identified for these compounds thus far.
Protein Targets of Flavonoids
Flavonoids are very effective antioxidants in vitro (31, 57). However, the contribution in vivo has been suggested to be negligible due to lack of bioavailability and metabolism (43, 99, 143). The cellular effects of flavonoids may be more related to their effects on signal transduction rather than general antioxidant effects (143). A variety of indirect anticancer effects have been reported, including inhibition of cancer cell proliferation and induction of apoptosis (3, 72, 118, 125), decreased inflammation (25, 113, 126), inhibition of tumor invasion and angiogenesis (3, 76), and stimulation of phase II detoxification enzyme activity (79, 140). While flavonoids have been associated with reduced cancers in animal models (4, 23, 27, 41, 42, 48 –50, 56, 60, 67 –71, 75, 85 –90, 93, 95 –98, 130, 131, 139, 148, 149), human studies are less convincing (1, 44, 45, 47, 58). More clinical trials will be necessary to determine whether specific flavonoids can be utilized to prevent or treat cancer.
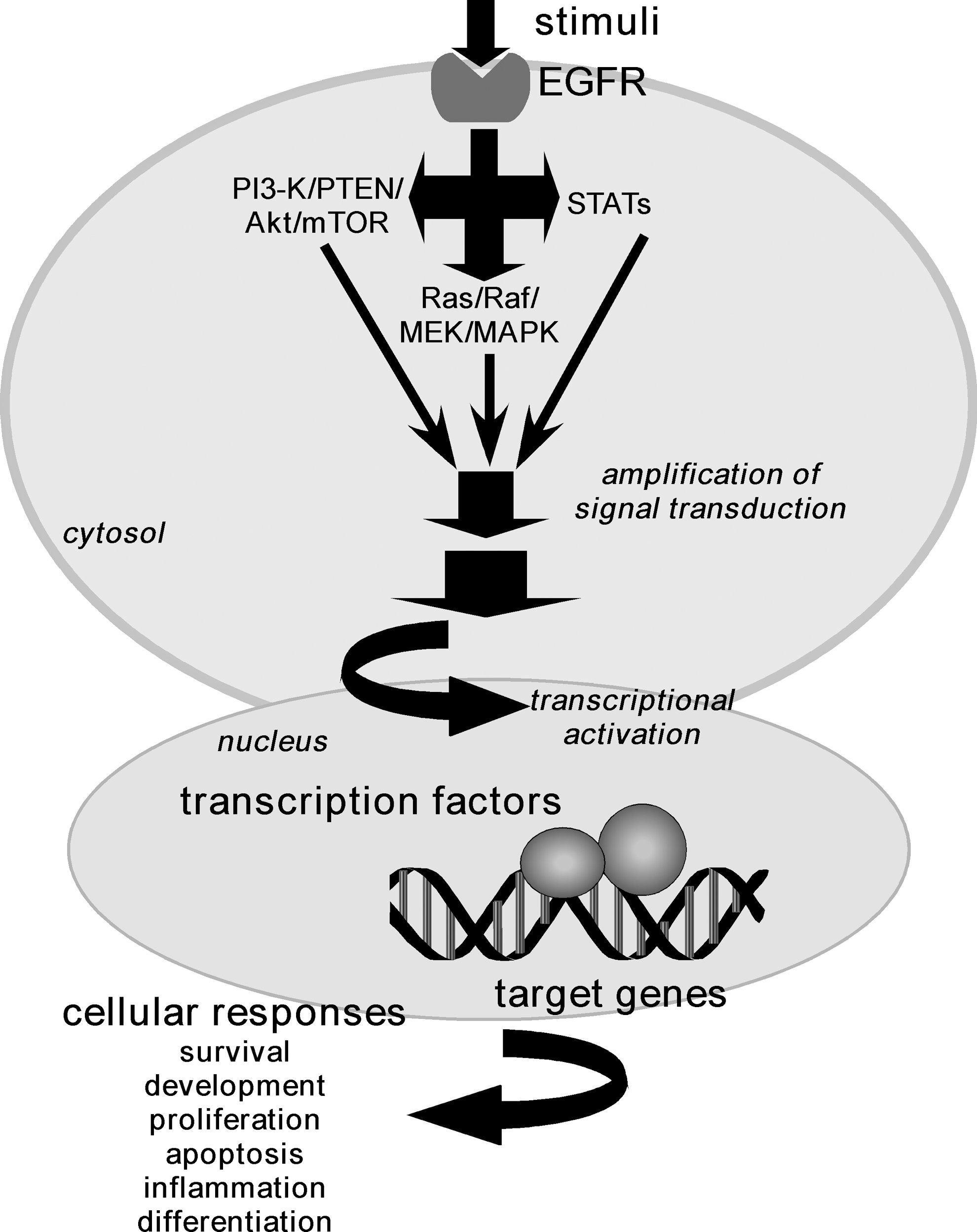
A major emphasis of much of our work over the last decade has been the clarification of molecular and cellular mechanisms and targets that are essential in cancer prevention, and especially skin cancer prevention. The prevailing idea these days is that cancer might be prevented (17) or treated by using small molecules, such as flavonoids, to target specific and, perhaps, multiple cancer genes, signaling proteins, and transcription factors. Due to its long length, the promotion stage of cancer development might be the phase at which diet could have the greatest impact. Our work thus far has revealed that many of the flavonoid compounds appear to target multiple downstream effectors of the human epidermal growth factor receptor (HER)/erbB family. The inappropriate phosphorylation and activation of this family of tyrosine kinase receptors has been observed in many cancers (32) and is associated with resistance to traditional cancer therapies. In addition, many of the epidermal growth factor receptor (EGFR) downstream protein effectors are also abnormally expressed in various cancers (141). Activation of the EGFR primarily triggers the Ras/Raf/MEK/MAPK pathway, the PI3-K/PTEN/Akt/mTOR pathway, and the signal transducer and activator of transcription (STAT) signaling cascade (6, 37, 39, 63, 65) (Fig. 2).
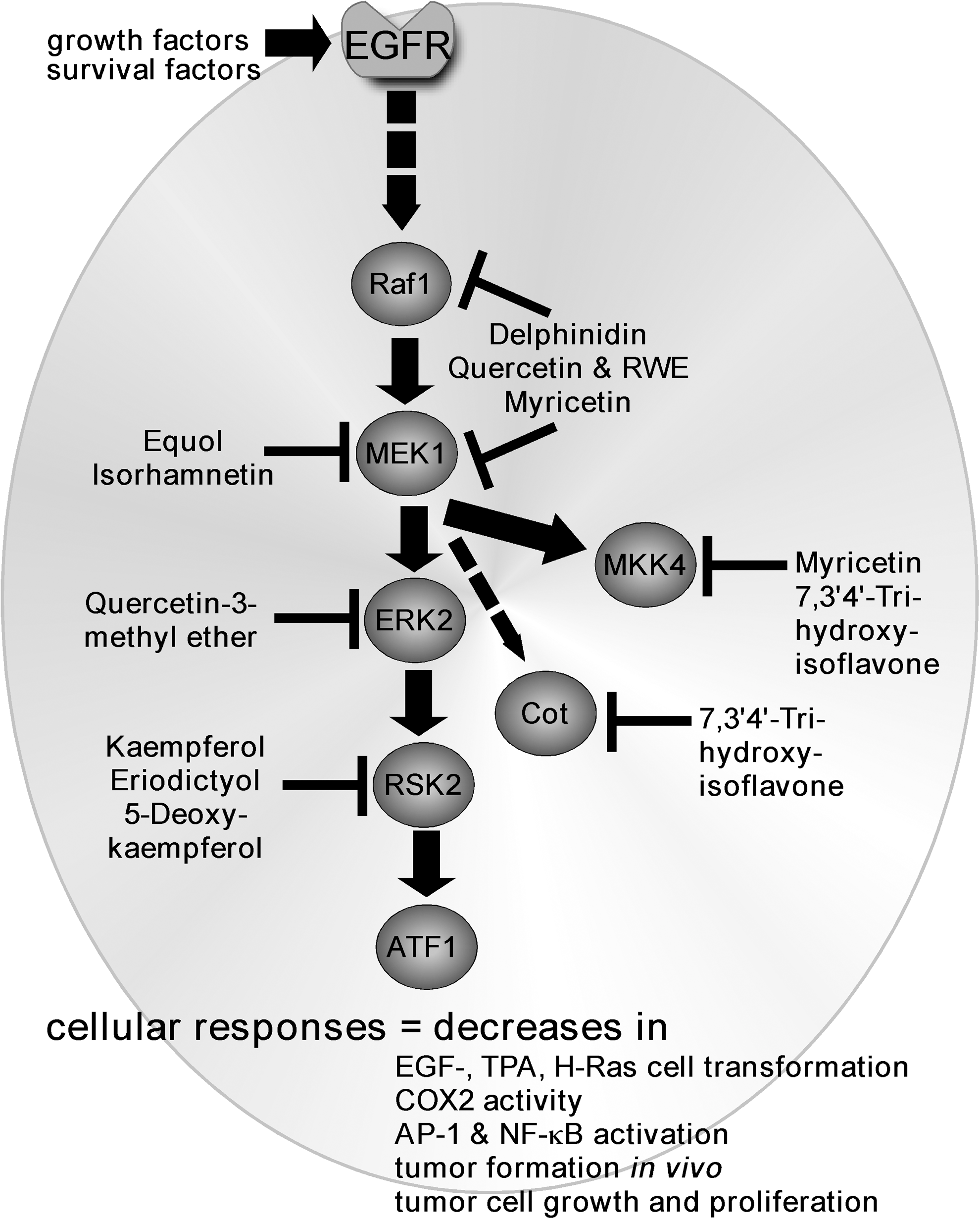
Inhibition of Ras/Raf/MEK/MAPK signaling by flavonoids
Neoplastic transformation of cells and inflammation are considered major events contributing to carcinogenesis. The EGFR is activated by growth factors, such as epidermal growth factor (EGF). The binding of EGF results in the activation and phosphorylation of EGFR on its tyrosine residues, which ultimately triggers the activation of Ras. Ras activates Raf, which phosphorylates mitogen-activated protein kinase/ERK kinase (MEK)1/2 that subsequently activates mitogen-activated protein (MAP) kinases or MAP kinase kinase (MKK) signaling. Downstream effectors of the MAP kinase and MKK cascades include extracellular signal-regulated kinases (ERKs), p90 ribosomal S6 kinases (RSKs), and c-Jun N-terminal kinases (JNKs), respectively (Fig. 3). Ras and/or Raf are constitutively activated in various cancer cell lines, and MEK plays a critical role in transmitting signals that are initiated by various tumor promoters, such as EGF or 12-O-tetradecanoylphorbol-13-acetate (TPA). Constitutive activation of MEK1 causes cell transformation and blocking MEK kinase activity can suppress transformation and tumor growth in vivo (35, 129).
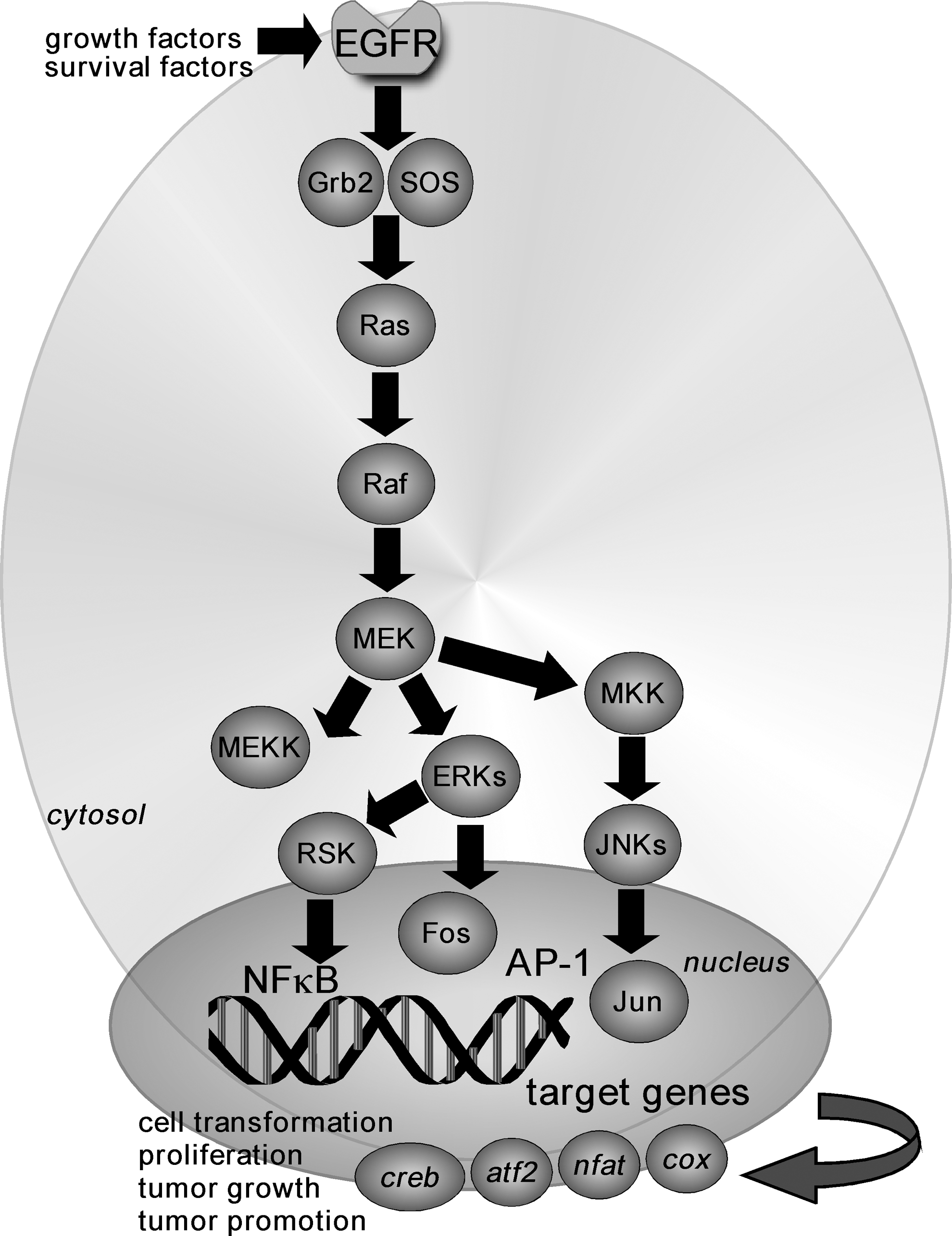
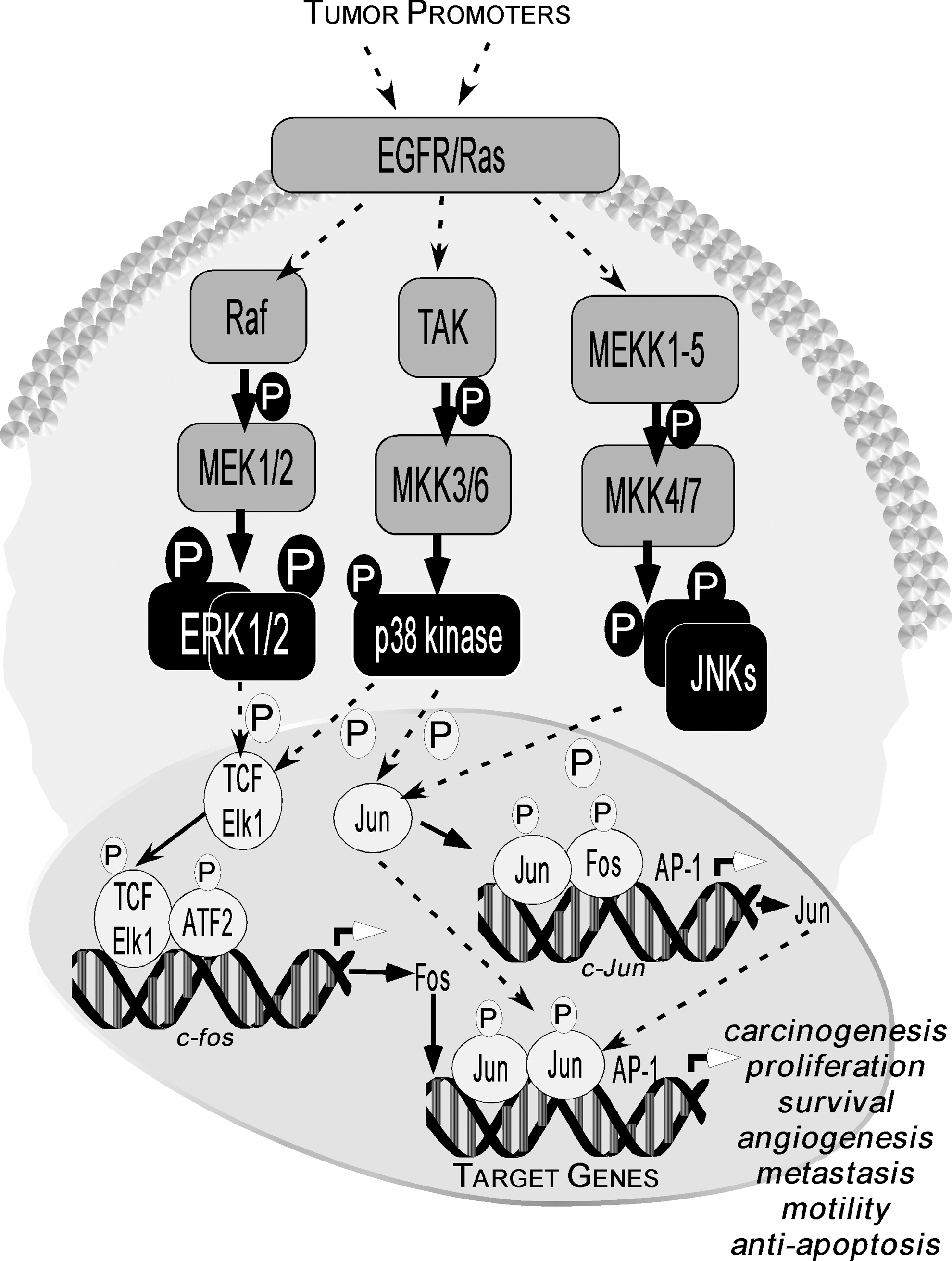
Furthermore, activation of the Ras/Raf/MEK/MAPK pathway results in the triggering of the activator protein-1 (AP-1) pathway (Fig. 4). Members of the AP-1 family of transcription factors are frequently regulated at the transcriptional and post-transcriptional levels by MAP kinases. AP-1 complexes are necessary not only for cell-cycle progression in several cell systems (80, 110) but also for cell transformation that is induced by a variety of oncogenes, including src, ras, and raf (114, 135). Members of the AP-1 family of transcription factors are usually classified into two subfamilies, namely the Jun (c-Jun, JunB, and JunD) (5, 122, 123) and the Fos (c-Fos, FosB, Fra-1, and Fra-2) (33, 104, 111, 152) families. Homodimerization of Jun proteins or heterodimerization of Jun and Fos proteins of the two subfamilies (120) or other transcription factors, including the activating transcription factor 2 (ATF2), cAMP response element-binding (CREB), nuclear factor of activated T-cells (NFAT), or SMAD proteins (7, 112, 153), provides the complexes with the ability to recognize specific DNA sequences that are known as tetradecanoyl phorbol acetate-responsive elements or AP-1 sites. In addition, phosphorylation of histone H3 occurs concurrently with transcriptional activation of the immediate-early response genes (e.g., c-jun, c-fos, and c-myc) (101). We reported that the EGF-induced phosphorylation of histone H3 at Ser10 up-regulates c-fos and c-jun transcriptional activity (28). Activation of these pathways can lead to increased proliferation, angiogenesis, metastasis, survival, motility, and decreased apoptosis (108), and these pathways appear to be preferred targets for many of the flavonoid compounds (Fig. 4).
Many of the flavonoid compounds that we have examined in our laboratory appear to favor MEK as a protein target (Fig. 5; Table 1), although most of the flavonoids target multiple proteins to exert their effects. A common characteristic among the interaction of various flavonoids with MEK is an adenosine triphosphate (ATP) noncompetitive inhibition. Delphinidin (Table 1) is an anthocyanidin (i.e., plant pigment) that is found in many fruits, especially berries and pomegranate. This compound attenuated EGF- or H-Ras-induced cell transformation and reduced cyclooxygenase 2 (COX2) expression in JB6 P+ epidermal skin cells by directly binding to and suppressing Raf and MEK kinase activities (70). The binding of delphinidin with Raf1 or MEK1 was indeed noncompetitive with ATP, which was similar to other MEK inhibitors. The inhibition of Raf and MEK by delphinidin also resulted in the subsequent attenuation of TPA-induced phosphorylation of MEK, ERKs, and RSK and decreased the activation of AP-1 and nuclear factor-kappaB (NF-κB) transcription factors induced by TPA (70).
Cdk, cyclin-dependent kinase; ERKs, extracellular signal-regulated kinases; G3BP1, Ras-GTPase-activating protein SH3 domain-binding protein 1; GRP78, glucose-regulated protein 78; IGF-IR, insulin-like growth factor-I receptor; MEK, mitogen-activated protein kinase/ERK kinase; MKK, MAP kinase kinase; PI3-K, phosphatidylinositol 3-kinase; PKCɛ, protein kinase Cɛ; RSK, p90 ribosomal S6 kinase; SH2, Src homology 2; STAT, signal transducer and activator of transcription; ZAP-70, zeta chain associated protein of 70 kDa.
Quercetin (Table 1) is a flavone (i.e., flavonol) compound that is found at high levels in various foods, including grapes and red wine. Quercetin reportedly suppressed the proliferation of cancer cells but did not affect normal cells, and it also reduced TPA-promoted mouse skin cancer (132). We reported that red wine extract (RWE) or quercetin reduced TPA-induced neoplastic JB6 epidermal skin cell transformation, which was associated with dose-dependent decreases in AP-1 and NF-κB activation (92). Results of pull-down assays indicated that RWE or quercetin directly bound with either Raf1 or MEK1 and reduced TPA-induced phosphorylation of ERKs and RSK. Even though RWE or quercetin suppressed Raf1 kinase activity, their effect on MEK1 was much more potent. In addition, quercetin had a stronger inhibitory effect than PD098059, a well-known pharmacologic inhibitor of MEK. In silico protein docking results suggested that quercetin formed hydrogen bonds with the backbone amide group of Ser212 on MEK1, which is a key interaction for stabilizing the inactive conformation of the activation loop of MEK1 (92).
Myricetin (Table 1) has a structure that is very similar to quercetin, differing only by the addition of a single hydroxyl group. This compound is a major flavone (i.e., flavonol) that is found in numerous foods, including onions, berries, and grapes as well as red wine (46, 51). It has been reported to inhibit two-stage skin tumorigenesis (109), reduce A549 lung cancer cell growth (100), and attenuate invasion associated with decreased matrix metallopeptidase (MMP)-2 protein expression and enzyme activity in colorectal carcinoma cells (78). These results suggest that myricetin might be an effective chemopreventive agent, but its specific molecular targets have not been identified until recently. Others and we have found that myricetin binds with various protein kinases both competitively and noncompetitively with ATP to exert its anticancer activities. We demonstrated that myricetin suppressed TPA- or EGF-induced transformation of JB6 P+ cells coinciding with a decreased activation of c-Fos or AP-1 (93). Myricetin directly bound to and inhibited MEK1 kinase activity noncompetitively with ATP, resulting in decreased phosphorylation of MEK downstream targets, ERKs or RSK. Notably, similar to quercetin, myricetin attenuated H-Ras-induced cell transformation more effectively than the well-known MEK inhibitor, PD098059 (93). We later reported that myricetin bound to and suppressed ultraviolet B (UVB)-induced Raf kinase activity also in an ATP noncompetitive manner (69). The suppression of Raf kinase activity was associated with both decreased UVB-induced wrinkle formation and epidermal thickening that is mediated through MMP-9 (69).
The isoflavones are a subfamily of flavonoid compounds that not only act as antioxidants but also behave similar to weak estrogen-active compounds and, thus, are referred to as phytoestrogens (53). These compounds are believed to be the major components in soy that are responsible for the reported biological effects (36). Enzymes in the colon convert soy isoflavones, such as daidzein, to numerous metabolites, including equol (66, 73), 6,7,4′-trihydroxyisoflavone [6,7,4′-THIF (81)], and 7,3′,4′-trihydroxyisoflavone [7,3′,4′-THIF (81, 82) Table 1]. Epidemiological and animal studies suggest that isoflavones might decrease cancer risk. In contrast, tumor-promoting effects were also observed in some human and animal studies as well. Thus, clarifying the underlying molecular targets and mechanisms of the activities of soy compounds such as daidzein and its metabolites, equol, 6,7,4′-THIF and 7,3′,4′-THIF is significant and critical in understanding the apparent contradictory effects of soy consumption. We found that the metabolites of daidzein were more effective chemopreventive agents than the parent daidzein compound. However, their protein targets are unique.
Equol (Table 1) was shown to protect against UV-induced skin cancer in a hairless mouse model (142). We compared the effects of equol and daidzein on TPA-induced AP-1 activity and transformation of JB6 P+ cells, and results indicated that equol, but not daidzein, targeted the MEK/ERK/p90RSK/AP-1 signaling pathway. It strongly suppressed MEK kinase activity, resulting in the suppression of c-Fos activation and AP-1 transactivation and reduced cell transformation (71). Equol, but not daidzein, attenuated TPA-induced phosphorylation of ERK1/2, p90RSK, and Elk, but not phosphorylation of MEK or JNKs. However, equol inhibited MEK1, but not Raf1, kinase activity. Importantly, equol specifically bound to MEK noncompetitively with ATP to suppress MEK activity (71). These results revealed a molecular basis for the anticancer action of equol and may partially account for the reported chemopreventive effects of soybean. The other metabolites of daidzein neither inhibited nor interacted with MEK or Raf and will be discussed later.
Isorhamnetin (Table 1) is another plant flavonol that is found in various fruits and medicinal herbs. We reported that isorhamnetin inhibited EGF-induced neoplastic cell transformation of JB6 cells and suppressed anchorage-dependent and -independent growth of A431 human epithelial carcinoma cells (75). Isorhamnetin attenuated EGF-induced COX2 expression in JB6 and A431 cells and reduced A431 tumor growth and COX2 expression in a xenograft mouse model. Isorhamnetin effectively suppressed EGF-induced phosphoryaltion of ERKs, RSK, p70S6 kinase, and Akt. These effects were explained by binding assay results, indicating that isorhamnetin directly binds to MEK1 in an ATP-noncompetitive manner to suppress its activity.
Downstream effectors of MEK activity include the ERK and RSK proteins. These kinases are also popular direct flavonoid targets (Fig. 5). Quercetin-3-methyl ether, a naturally occurring compound that is present in various plants, including the edible prickly pear cactus, reportedly has potent anticancer activity. We found that quercetin-3-methyl ether could control the growth of breast cancer cells that were sensitive or resistant to the receptor tyrosine kinase inhibitor, lapatinib (95). Quercetin-3-methyl ether appeared to act mainly by inducing cell-cycle arrest and apoptosis in either cell type (95). We also found that quercetin-3-methyl ether inhibited proliferation of mouse skin epidermal JB6 P+ cells in a dose- and time-dependent manner by inducing cell-cycle G2/M phase accumulation and decreasing AP-1 activation and ERKs phosphorylation (94). The net result was suppression of TPA-induced JB6 neoplastic cell transformation. Pull-down assays revealed that quercetin-3-methyl ether directly binds with ERKs, perferring ERK2, to exert its potent chemopreventive activity (94).
Ribosomal S6 kinase 2 (RSK2) is a member of the p90RSK (RSK) family of proteins. It is a widely expressed serine/threonine kinase that is phosphorylated and activated by ERKs and phosphoinositide-dependent kinase 1 (PDK1) in response to growth factors, and its activation enhances cell survival. RSK2 is an important regulator of tumor promoter-induced cell transformation, and its high expression increases proliferation as well as anchorage-independent transformation of JB6 Cl41 skin cells and foci formation in NIH3T3 cells (26). The RSK2 protein level is markedly higher in cancer cell lines as well as in cancer tissues compared with nonmalignant cell lines or normal tissues. We have solved and reported the crystal structure for the NH2-terminal kinase domain [NTD; (102)] and the COOH-terminal kinase domain [CTD; (103)] and used these structures to identify novel inhibitors of this oncogenic kinase.
Kaempferol (Table 1) is a flavonol that is present in various natural sources, especially in onion leaves (832.0 mg/kg) (107). We found that kaempferol inhibited proliferation of malignant human cancer cell lines, including A431 epithelial carcinoma, SK-MEL-5 and SK-MEL-28 melanomas, and HCT-116 colon cancer cells (27). The mechanism of inhibition was associated with its direct binding to RSK2 to suppress RSK2 activity. We provided evidence showing that activation of the NTD of RSK2 is required for the activation of the ERKs-mediated CTD. Computational modeling predicted that kaempferol binds with the NTD, but not the CTD (27) to exert its activity. This prediction was confirmed experimentally, and we conducted mutagenesis experiments to clearly show that Val82 and Lys100 are critical amino acids for kaempferol binding and RSK2 activity (27).
Eriodictyol (Table 1) is a flavanone that is found in various fruits, and we discovered that it also binds to the NTD of RSK2 to inhibit RSK2 N-terminal kinase activity (79, 85, 98). In this same study, we found that ATF1 is a novel substrate of RSK2 and that RSK2-ATF1 signaling plays an important role in EGF-induced neoplastic cell transformation. RSK2 phosphorylated ATF1 at Ser63 and enhanced ATF1 transcriptional activity and tumor promoter-induced JB6 cell transformation. Eriodictyol had no effect on the phosphorylation of RSK, MEK1/2, ERK1/2, p38, or JNKs, indicating that this compound specifically suppresses RSK2 signaling. The phosphorylation of ATF1 by RSK2 and its susequent cellular effects, including Ras-mediated focus formation, were markedly attenuated with eriodictyol treatment (98). Besides the Ras/Raf/MEK/MAPK pathway, flavanoids also strongly interact with the Src and Fyn tryrosine kinases to interfere with their activity (Fig. 6).
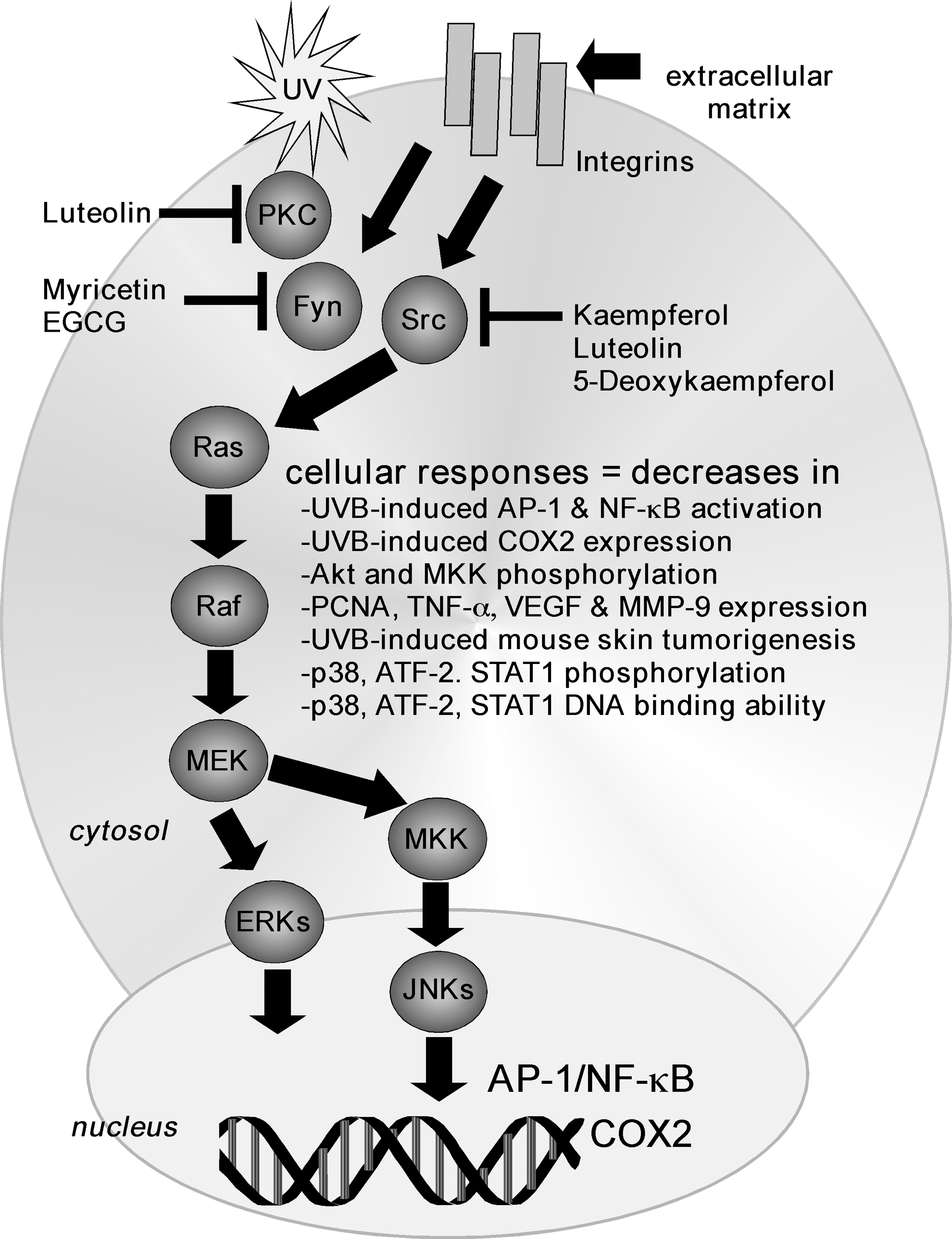
Fyn and Src inhibition by flavonoids
Src and Fyn are non-receptor tyrosine kinases that can phosphorylate tyrosine residues of numerous cytosolic, nuclear, and membrane proteins. The Src and Fyn proteins can also activate the Ras/Raf/MEK/MAPK pathways (Fig. 6). The Src protein has three major domains, Src homology 2 (SH2), SH3, and a kinase catalytic domain (or SH1). Both SH2 and SH3 participate in protein–protein interactions, whereas the kinase catalytic domain contains the kinase active site. Two major phosphorylation sites have been identifed in Src and include Tyr416 and Tyr527. Src is one of the nine members in a closely related family that often compensate for one another. Other Src family kinase members are Fyn and Yes, which are also widely expressed. The additional family members include Blk, Fgr, Hck, Lck, Lyn, and Yrk, which are expressed only in specific tissues. All have structurally similar SH2, SH3, and kinase domains and are capable of acting as signaling molecules in a similar manner to Src.
Fyn is involved in the UVB-induced phosphorylation of histone H3 at serine 10. UVB distinctly increases Fyn kinase activity and phosphorylation (55), and active Fyn phosphorylates histone H3 at serine 10. Recently, we also found that kaempferol suppressed UVB-induced COX2 protein expression in mouse skin epidermal JB6 P+ cells and attenuated the UVB-induced transcriptional activities of COX2 and AP-1 (90). In addition, kaempferol reduced UVB-induced phosphorylation of ERKs, p38, and JNKs and also inhibited the upstream Src kinase activity. Pull-down assays and computer docking data indicated that kaempferol competes with ATP for direct binding with Src in the ATP-binding site (90). Luteolin (Table 1) is another flavone that is present in various vegetables, including onion and broccoli, and it also has been reported to possess anticancer activities. We found that luteolin also suppressed UVB-induced COX2 expression and AP-1 and NF-κB activity in JB6 P+ cells. These effects were related to the direct binding of luteolin to protein kinase Cɛ (PKCɛ) and Src in an ATP-competitive manner (23). Other effects included an inhibition of UVB-induced Akt phosphorylation, tumor necrosis factor (TNF)-α, and proliferating cell nuclear antigen (PCNA). Notably, luteolin reduced tumor incidence, multiplicity, and overall tumor size in SKH-1 hairless mice exposed to chronic UVB, and a further analysis using skin lysates confirmed that luteolin inhibited PKCɛ and Src kinase activities in vivo (23). We further found that an analogue of kaempferol, 5-deoxykaempferol (5-DK; Table 1) also suppressed UVB-induced COX2 expression along with vascular endothelial growth factor (VEGF), PCNA, and MMP-9 expression (89). This compound had inhibitory effects on multiple proteins, including attenuation of phosphorylation of MKK3/6, MKK4, and Akt, but had no effect on phosphorylation of Src, ERKs, or RSK. Exposure to 5-DK significantly suppressed UVB-induced tumorigenesis in mouse skin in a dose-dependent manner. 5-DK directly bound to Src, PI3-K, and RSK2 competitively with ATP to exert its inhibitory effects, and our docking data suggested that 5-DK docks at the ATP-binding site of Src, PI3-K, and RSK2 (89).
Myricetin was shown to directly bind with Fyn and suppress Fyn kinase activity also in an ATP-competitive manner (67). Docking data revealed that myricetin docked to the ATP binding site of Fyn, which was located between the N and C lobes of the kinase domain. The binding of myricetin with Fyn corresponded with the reduction of UVB-induced COX2 expression and decreased the activation of AP-1 and NF-κB in JB6 P+ cells (67). Myricetin effectively suppressed Fyn kinase activity directly to reduce UVB-induced COX2 expression and UVB-induced skin tumor incidence in a dose-dependent manner (67). We also found that the tea polyphenol, (−)-epigallocatechin gallate (EGCG), could bind with Fyn at its SH2 domain but not at its SH3 domain (56). The binding was associated with decreased EGF-induced cell transformation that was mediated by Fyn kinase activity and phosphorylation. In addition, knockdown of Fyn or treatment with EGCG inhibited phosphorylation of p38, ATF-2, and STAT1 and decreased the DNA binding ability of AP-1, STAT1, and ATF-2 (56). In general, with the exception of EGCG, unlike their interaction with MEK, flavonoids compete with ATP for binding with Fyn or Src.
Inhibition of PI3-K/PTEN/Akt/mTOR signaling by flavonoids
The PI3-K/PTEN/Akt/mTOR pathway (Fig. 7) can become abnormally activated in many human cancers, and its activation can enhance cancer cell proliferation, tumor growth, angiogenesis, and survival (62, 106, 151). PI3-K, Akt, and mTOR are considered oncogenic proteins; whereas PTEN acts as a tumor suppressor that is involved in cell-cycle regulation, preventing cells from growing and dividing too quickly (30) and its loss or mutation is associated with increased tumor development in numerous organs. The development of inhibitors of this signaling axis is of great interest and importance. Delphinidin was also found to suppress UVB-induced COX2 expression, COX2 promoter activity, and prostaglandin E2 (PGE2) production in JB6 P+ mouse epidermal cells (85). AP-1 and NF-κB, crucial transcription factors involved in COX2 expression, were activated by UVB and delphinidin abolished this activation. UVB-induced phosphorylation of JNKs, p38, and Akt was inhibited by delphinidin. The activities of MKK4 and PI3-K were markedly reduced by delphinidin. A pull-down assay using delphinidin-conjugated Sepharose beads revealed that delphinidin binds directly with MKK4 or PI3-K in a manner that was competitive with ATP. Moreover, in vivo investigations using mouse skin revealed that the up-regulation of COX2 expression, MKK4 activity, and PI3-K activity induced by UVB was attenuated with delphinidin treatment. Collectively, these results demonstrated that delphinidin targets MKK4 and PI3-K directly to suppress COX-2 overexpression and UVB-induced skin carcinogenesis (85).
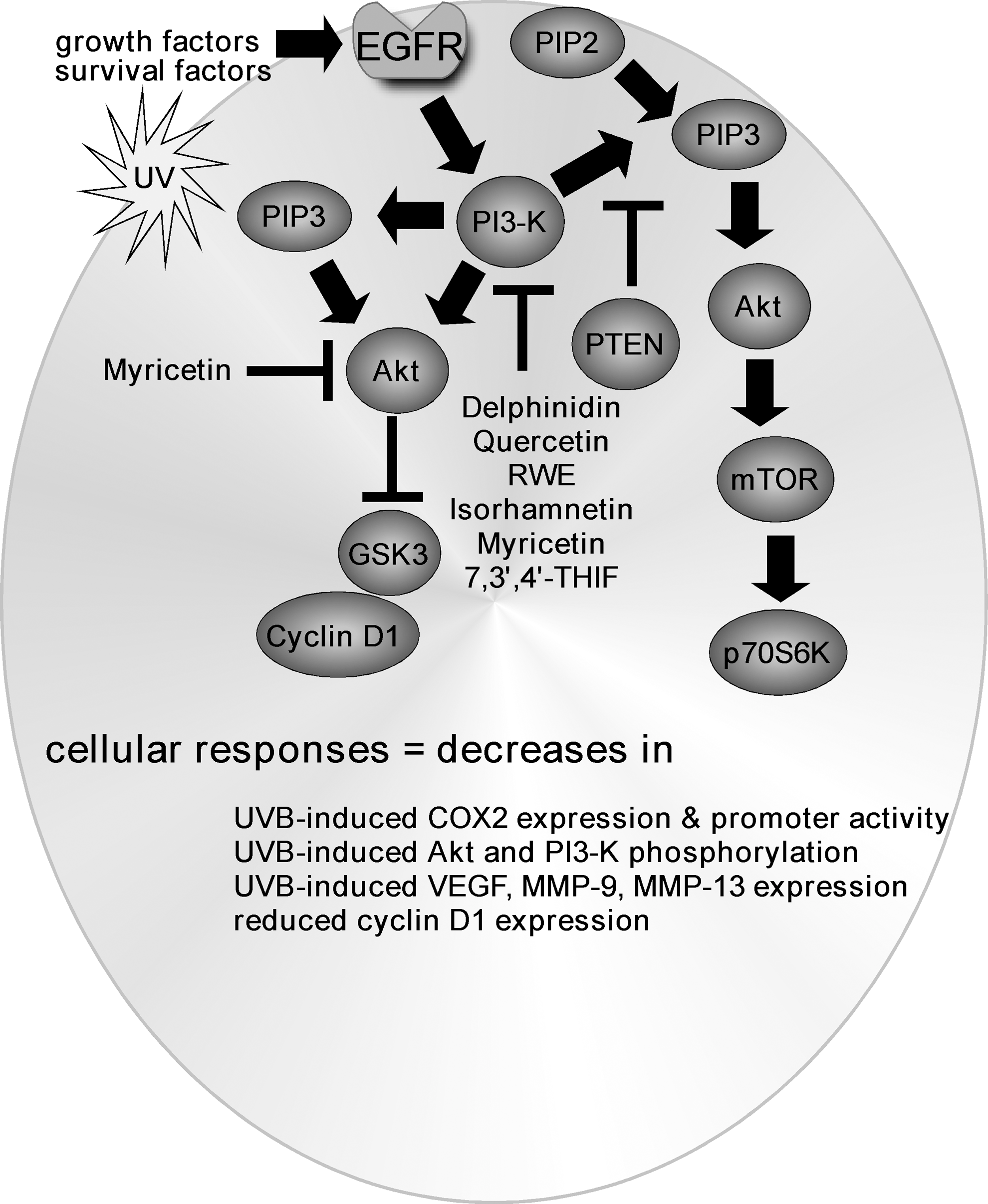
Furthermore, RWE and quercetin treatment also decreased TNF-α-induced up-regulation of MMP-9 and cell migration that were mediated by the suppression of Akt phosphorylation and AP-1 or NF-κB transactivation (61). In this case, besides binding with MEK, quercetin and RWE were also discovered to interact with and inhibit PI3-K activity (61). Additional studies showed that myricetin also suppressed TNF-α-induced VEGF expression by binding directly to MKK4 in an ATP-competitive manner to reduce its kinase activity, resulting in decreased JNKs and ERKs phosphorylation (74). Computer modeling suggested that myricetin docks onto the ATP-binding site in MKK4, which is located between the N- and C-lobes of the kinase domain (74). In addition, isorhamnetin also was found to bind to PI3-K in an ATP-competitive manner to inhibit the activity of that kinase (75).
Myricetin was also reported to bind to and inhibit Akt phosphorylation and activity in an ATP-competitive manner (83). We reported that topical treatment with myricetin reduced UVB-induced neovascularization in SKH-1 hairless mouse skin and suppressed UVB-induced VEGF, MMP-9, and MMP-13 expression and PI3-K activity (68). Myricetin directly bound to PI3-K, resulting in the subsequent attenuation of the UVB-induced phosphorylation of Akt/p70S6K in mouse skin (68). The daidzein metabolite, 7,3′,4′-THIF also binds to PI3-K, markedly inhibiting its kinase activity, thereby suppressing the Akt/GSK-3β/AP-1 pathway and resulting in a decrease in the expression of cyclin D1 (88). Overall, various flavonoids target and inhibit PI3-K and Akt activity by binding competively with ATP.
Other protein targets of flavonoids
In contrast to equol, the soy components, 7,3′,4′-THIF and 6,7,4′-THIF, do not target Raf or MEK activity (Fig. 8). Instead, 7,3′,4′-THIF prevented EGF-induced neoplastic transformation and proliferation of JB6 P+ mouse epidermal cells by blocking cell-cycle progression of EGF-stimulated cells at the G1 phase (88). It also suppressed the phosphorylation of retinoblastoma protein at Ser795 and Ser807/Ser811, which are the specific sites of phosphorylation by cyclin-dependent kinase (Cdk4). Furthermore, 7,3′,4′-THIF also attenuated the expression of G1 phase-regulatory proteins, including cyclin D1, Cdk4, cyclin E, and Cdk2. In addition to regulating the expression of cell-cycle-regulatory proteins, 7,3′,4′-THIF directly bound to Cdk4 and Cdk2 and strongly inhibited their kinase activities (88). Furthermore, similar to myricetin, 7,3′,4′-THIF also directly bound to and inhibited MKK4 activity and also suppressed Cot activity, resulting in the inhibition of UVB-induced COX2 expression (86). Cot is a serine/threonine kinase member of the MAPK kinase kinase family that phosphorylates histone H3, resulting in the activation of c-Fos and AP-1 (29). Topical application of 7,3′,4′-THIF clearly suppressed the incidence and multiplicity of UVB-induced tumors in hairless mouse skin by directly binding to and inhibiting Cot or MKK4 kinase activity. A docking study revealed that 7,3′,4′-THIF, but not daidzein, easily docked to the ATP binding sites of Cot and MKK4 located between the N- and C-lobes of the kinase domain of each (86).
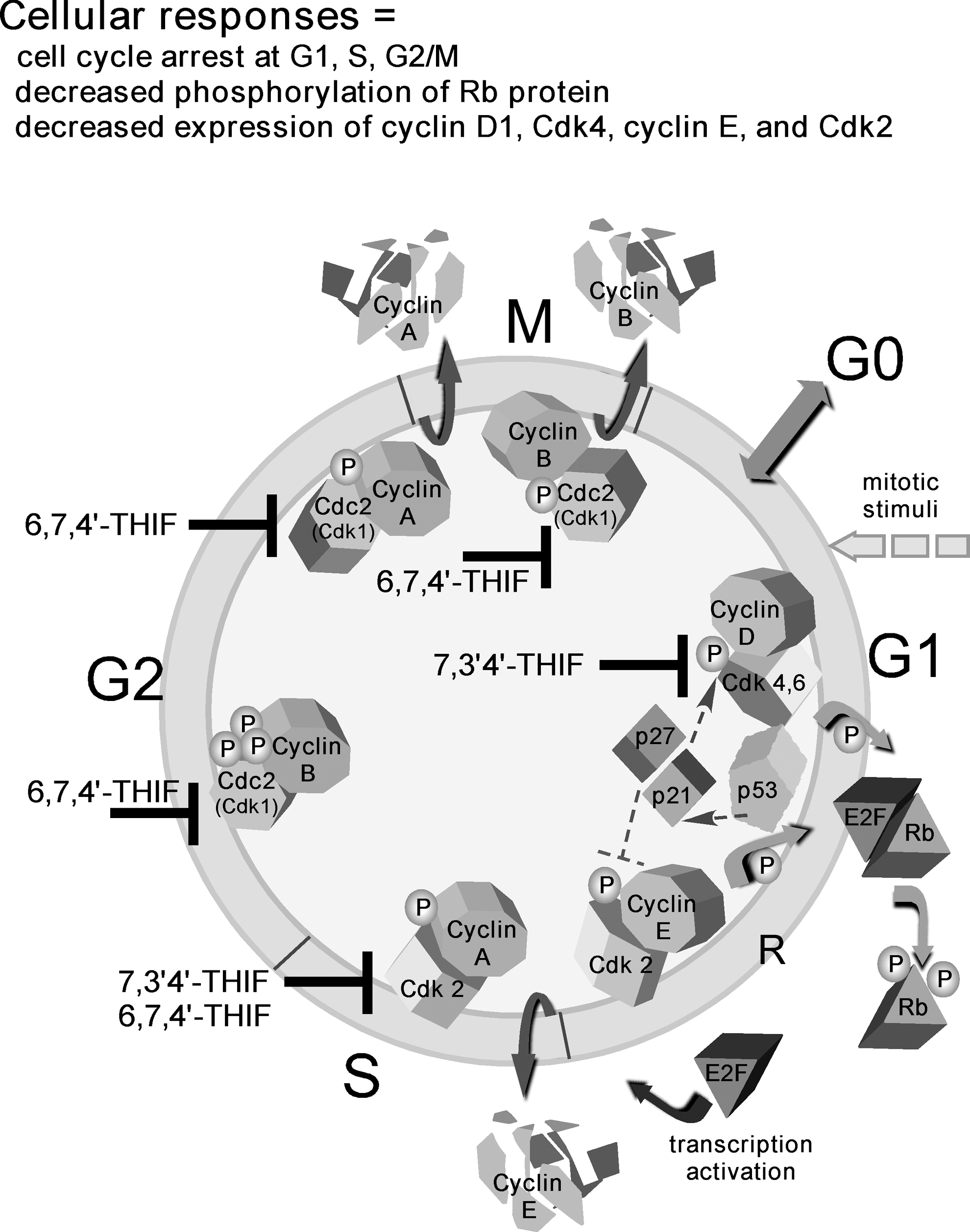
We investigated the biological activity of 6,7,4′-THIF in in vitro and in vivo models of human colon cancer. 6,7,4′-THIF suppressed anchorage-dependent and -independent growth of HCT116 and DLD1 human colon cancer cells more effectively than daidzein (87). In addition, 6,7,4′-THIF induced cell-cycle arrest at the S and G2/M phases in HCT116 human colon cancer cells and effectively suppressed the expression of Cdk2, but had no effect on other S- or G2/M-phase regulatory proteins such as cyclin A, cyclin B1, or Cdk1. 6,7,4′-THIF, but not daidzein, inhibited Cdk1 and Cdk2 activities in HCT116 cells by directly interacting with Cdk1 and Cdk2. Notably, 6,7,4′-THIF significantly decreased tumor growth, volume, and weight of HCT116 xenograft tumors and bound directly to Cdk1 and Cdk2 in vivo, resulting in the suppression of Cdk1 and Cdk2 activity in tumors corresponding with the in vitro results (87).
The JAK1/STAT3 pathway has been suggested to play a role in cell transformation and carcinogenesis, and myricetin was reported to directly bind to JAK1 and STAT3 to inhibit EGF-induced transformation of JB6 P+ cells (84). Data revealed that myricetin inhibited DNA binding and transcriptional activity of STAT3 and suppressed phosphorylation of JAK1 and STAT3 at Tyr705 and Ser727 (84).
One of the most well-studied flavanoids is the green tea catechin (−)-EGCG (Table 1) (18, 19, 136, 137). Unlike the flavonoid compounds discussed thus far, EGCG does not appear to target the major EGFR signaling pathways. This polyphenol reportedly prevents or decreases the risk of developing various cancers, obesity, diabetes, and cardiovascular disease. Over the last decade, several proteins that directly bind with EGCG have been identified and include plasma proteins, fibronectin, fibrinogen, and histidine-rich glycoprotein (127), apoptotic Fas (54), laminin and the 67-kDa laminin receptor (54, 137), fatty acid synthase (138), heat shock protein 90 (115, 150), and T-cell receptor CD4 (144). Even though these targets of tea polyphenols have been identified, the biologic and physiologic significance of their role in the anticancer effects of tea polyphenols is still not totally clear. A part of the challenge in these and other studies is whether EGCG or other flavonoid compounds can reach a level that is physiologically achievable and the extent to which these compounds can penetrate the cell membrane.
Identification of novel high-affinity EGCG-binding proteins could facilitate the design of new strategies to prevent cancer. We first used JB6 Cl41 cell lysates and EGCG-conjugated Sepharose 4B beads followed by two-dimensional electrophoresis and matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) analysis to identify the intermediate filament protein, vimentin (41), and glucose-regulated protein 78 (GRP78) (42) as novel EGCG-binding proteins. EGCG bound to and inhibited phosphorylation of vimentin (Ser50/55), and the binding was associated with decreased breast cancer cell proliferation. Notably, the binding affinity of vimentin and [3H]-labeled EGCG exhibited a Kd value of 3.3 nM, which is clearly achievable physiologically. GRP78 is associated with the multidrug resistance phenotype of many types of cancer cells, and EGCG binding caused the conversion of GRP78 from its active monomer to the inactive dimer and oligomer forms (42). The binding of EGCG interfered with the formation of the anti-apoptotic GRP78-caspase-7 complex, which resulted in an increased etoposide-induced apoptosis in cancer cells (42).
We also used EGCG-conjugated Sepharose 4B affinity beads and found that EGCG directly binds with the insulin-like growth factor-I receptor (IGF-IR) and acts as a small-molecule inhibitor of IGF-IR activity (IC50 of 14 μM) competitive with ATP (96). IGF-IR has been implicated in cancer pathophysiology, and suppressing IGF-IR signaling in various cancer cell lines causes inhibition of the transformed phenotype as shown by the attenuation of colony formation in soft agar and the reduction of tumor formation in athymic nude mice.
The zeta chain associated protein of 70 kDa (ZAP-70) tyrosine kinase plays a critical role in T-cell-receptor-mediated signal transduction, and the immune response and a high level of ZAP-70 expression are observed in leukemia. Our results showed that EGCG could form numerous intermolecular hydrogen bonds and hydrophobic interactions within the ATP binding domain of ZAP-70 with a high affinity (Kd=0.6207 μM) (130). The binding was associated with decreases in ZAP-70 activity and AP-1 activation along with an induction of caspase-mediated apoptosis in leukemia cells expressing ZAP-70 but not in ZAP-70 deficient cells (130). We also found that EGCG interacted with the Ras-GTPase-activating protein SH3 domain-binding protein 1 (G3BP1) with good binding affinity (Kd=0.4 μM). H1299 and CL13 lung cancer cells highly express G3BP1, and EGCG effectively suppresses anchorage-independent growth of these cells but was less effective in cells with a low expression of G3BP1 (131). Finally, we (113, 119, 139) found that EGCG binds with the human peptidyl prolyl cis/trans isomerase [Pin1 (139)], which plays a critical role in oncogenic signaling (40, 128). In this case, we were able to combine crystallography, computational biology and cell- and animal-based studies to identify Pin1 as a novel binding protein of EGCG. Pin 1 comprises two distinct functional domains, the protein–protein interaction N-terminal WW domain (1–39 aa) and the isomerization catalysis PPIase domain (45–168 aa) connected by a flexible linker region (40–44 aa) (119). This protein is very likely required for the full activation of mutiple signal transduction pathways, including NF-κB, AP-1, β-cateinin, and NFAT (124, 145, 146). We solved the X-ray crystal structure of the Pin1/EGCG complex resolved at 1.9 Å resolution, and the structure revealed the presence of EGCG in both the WW and PPIase domains of Pin1. Direct binding between Pin1 and EGCG was confirmed and resulted in the inhibition of Pin1 PPIase activity, growth of Pin1-expressing cells, and xenograft tumor growth (139). Importantly, the binding of EGCG with Arg17 in the WW domain prevented the binding of c-Jun, a well-known Pin1 substrate (139).
All of these results suggest that EGCG is quite promiscuous, because it interacts with many proteins to suppress their activities. However, we also determined that EGCG did not bind to other tyrosine kinases, including Abl, platelet-derived growth factor receptor (PDGFR), c-Src, Bmx, or Yes (96).
Summary and Conclusions
Multiple signaling pathways appear to be major targets of many flavonoid compounds. These pathways are known to play important roles in cancer development, and a great deal of effort has focused on developing inhibitors against specific components in the pathway. One might argue that food factors or other natural compounds targeting multiple molecules could be the most highly efficacious chemopreventive agents, because theoretically they could be combined with traditional chemotherapeutic agents to overcome drug resistance by targeting multiple upstream or downstream molecules. They might also be used at lower concentrations in combination with each other or other anticancer agents. One could also argue that eating whole food or a complete diet containing many of the flavonoid compounds would be the wisest and safest approach to preventing cancer. The apparent promiscuity of compounds such as the flavonoids actually might provide at least a partial explanation in support of the epidemiological evidence, suggesting that consumption of fruits and vegetables reduces cancer risk. On the other hand, many food components, including many flavonoid compounds, are sold as supplements. Notably, the beneficial effects of supplements have not been supported in controlled human studies. In spite of the interest in food-derived compounds such as supplements, research on the effectiveness of supplementation is still in its infancy and the effects of long-term use of supplements in humans are not known.
With the possible exception of EGCG, much of the published work surrounding protein targets of flavonoids has focused on skin cancer. Obviously, the effects of flavonoids and identification of their cellular targets need to be assessed in other tissue and organ types. Clearly, the future of flavonoid research requires not only the identification of specific molecular targets but also a more complete understanding of their intake, bioavailability, metabolism, and excretion. Challenges include recognizing appropriate in vitro and in vivo models, developing standard measurements of analysis, and identification of suitable clinical biomarkers. Until these challenges are met, the clinical use of flavonoids in the prevention or treatment of cancer will be limited.
Footnotes
Acknowledgments
This work is supported by The Hormel Foundation and NIH grants CA077646, CA027502, CA120388, R37 CA081064, and ES016548.