
Abstract
Introduction
T
SODs and Early Stage Cancer Prevention
Oxidative stress contributes to the development of human cancers, which can result from decreased activities of antioxidant enzymes. SODs, providing the first defending line against superoxide radicals, have been studied in human cancers (Fig. 1). In human patient samples, Cu-ZnSOD activity is decreased in breast carcinoma (61). In gastric samples (adenocarcinoma) and healthy tissues, Cu-ZnSOD activity was significantly lower in cancer samples with respect to normal mucosa (86).
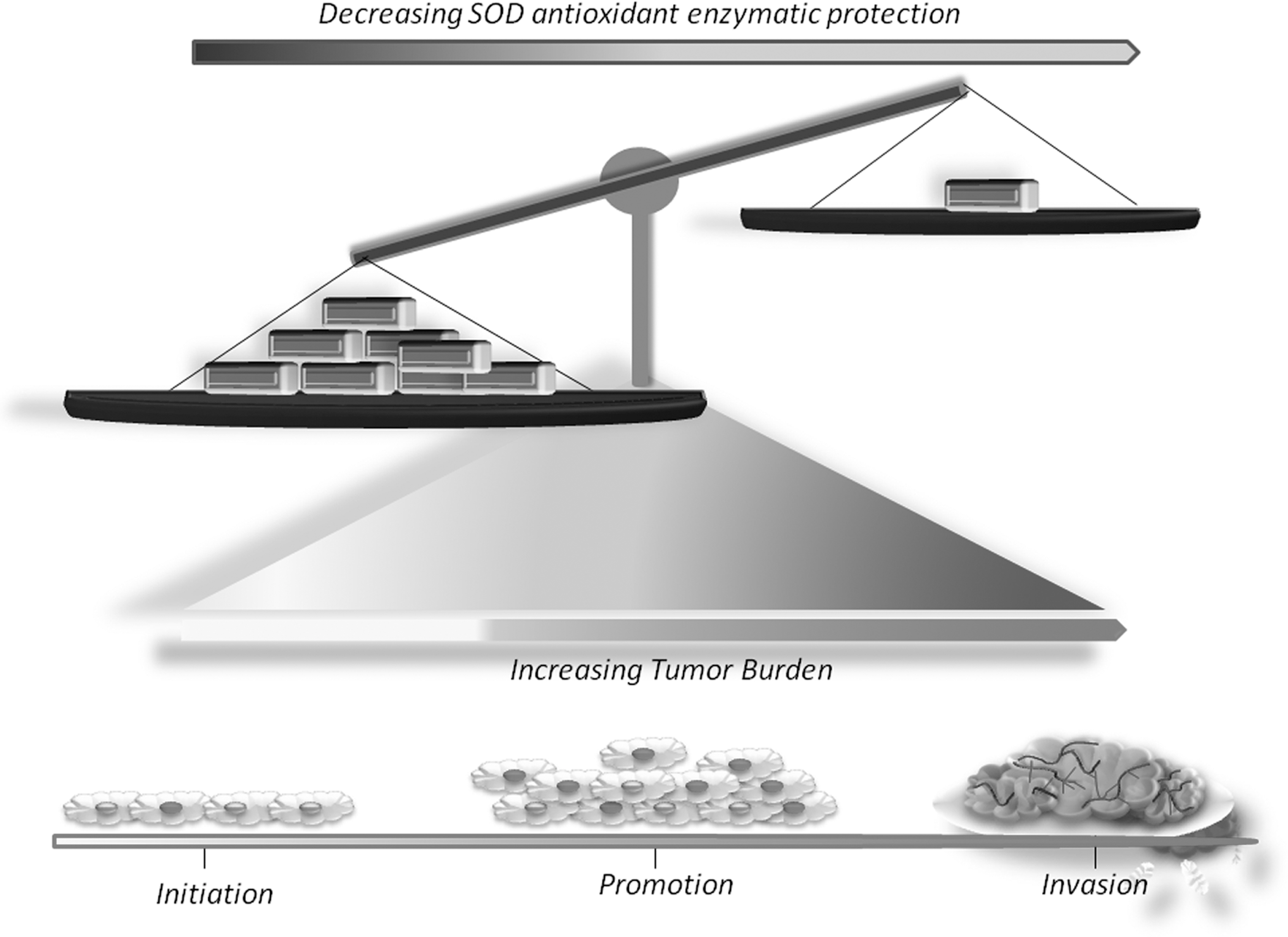
MnSOD is often dysregulated during cancer development. In human breast cancer, patients' fixed tissue samples (including ductal carcinoma in situ [DCIS] tissues, invasive ductal carcinoma (IDC) tissues, and adjacent cancer-free tissues), expression levels of MnSOD are lower in DCIS and IDC (140). In human oral cancers, a high expression level of MnSOD was associated with better disease-specific survival, especially for patients with moderate or poor cell differentiation, and early stage buccal mucosal squamous cell carcinomas (39). In human esophageal cancers, studies have shown that decreased MnSOD levels are associated with increased incidences of esophageal adenocarcinoma (EAC) (82).
Current studies suggest that extracellular superoxide dismutase (EcSOD) may play an important role in human lung cancer. EcSOD expression is down regulated in lung carcinomas, which is due to aberrant promoter hypermethylation (136).
In animal cancer models, aged Cu-ZnSOD knockout mice developed hepatocellular carcinoma (32, 33). Moreover, aged MnSOD knockout mice showed an increased incidence of cancers which was dominant in lymphomas (142). In a mouse model of T cell lymphoma (Lck-Bax38/1 transgenic mice), tumor formation was decreased when the mice were crossed with mice overexpressing MnSOD (141). In a skin carcinogenesis mouse model, MnSOD transgenic mice exhibited reduced oxidative stress and skin tumor formation (160); similar results have also been obtained when a SOD mimetic was applied topically to mouse skin (158). However, clinical observations have shown that SODs were higher in tumor tissues (51). For instance, increased activity of MnSOD was detected in advanced stages of head and neck squamous cell carcinoma (121). In a human tissue microarray analysis, MnSOD expression was higher in malignant tissues compared with normal tissues (49).
How do these controversies occur? There are several opposing views on how MnSOD expression may or may not contribute to cancer development (139). Some suggest that MnSOD is pro-oxidant, promoting the accumulation of H2O2 which can further lead to activation of various oncogenic pathways (155). Other investigators suggest that only the protein levels are increased during tumorigenesis, while enzymatic activity levels are decreased. For instance, MnSOD has been well-established as an enzyme that can dismutate superoxides; however, its ability to produce H2O2 has received conflicting interest concerning its role in the later stages of cancer development. H2O2 is considered as a metabolic by-product of oxidative metabolism; however, its ability to oxidatively inactivate regulatory proteins shifts the paradigm of H2O2 from being a simple by-product to an important oncogenic regulator in carcinogenesis. Melendez's group has reported that modulating antioxidant expression can promote PI3K activation via altered levels of H2O2 and oxidative inactivation of the pathway's inhibitory regulator phosphatase and tensin homolog (PTEN) (20). Furthermore, they suggest that mitochondrial origins of H2O2 can promote angiogenesis via increased Akt signaling and vascular endothelial growth factor (VEGF) expression; hence, the significance of MnSOD upregulation reported during the later phases of cancer development. Thus, this report further confirms the contributing role of MnSOD-mediated reactive oxygen species (ROS) during angiogenesis and metastatic spread.
Other investigators have reported an interesting correlation between SOD expression and H2O2 contributing to angiogenic induction of lymphocytes when other H2O2-detoxifying enzymes are low. Monte et al. reported that the addition of SOD to normal lymphocytes in vitro stimulated vascular density and angiogenesis (87). Moreover, the addition of H2O2-detoxifying enzyme, catalase (CAT), blocked the angiogenic response suggesting that SOD-mediated production of H2O2 was the main angiogenic factor in stimulating lymphocyte activation and their intermediate role in the induction of neovascularization.
The following two studies provide an additional perspective of protein expression versus enzymatic activity during cancer development. In patients' samples, the activities of Cu-ZnSOD and MnSOD were decreased in liver cirrhosis. However, higher protein level and activity of SOD isoforms were detected in liver malignant tumors when compared to levels in benign tumors (125). In a chemically induced mouse skin carcinogenesis study, MnSOD expression was suppressed in the early stage but increased at late stages of skin carcinogenesis (28).
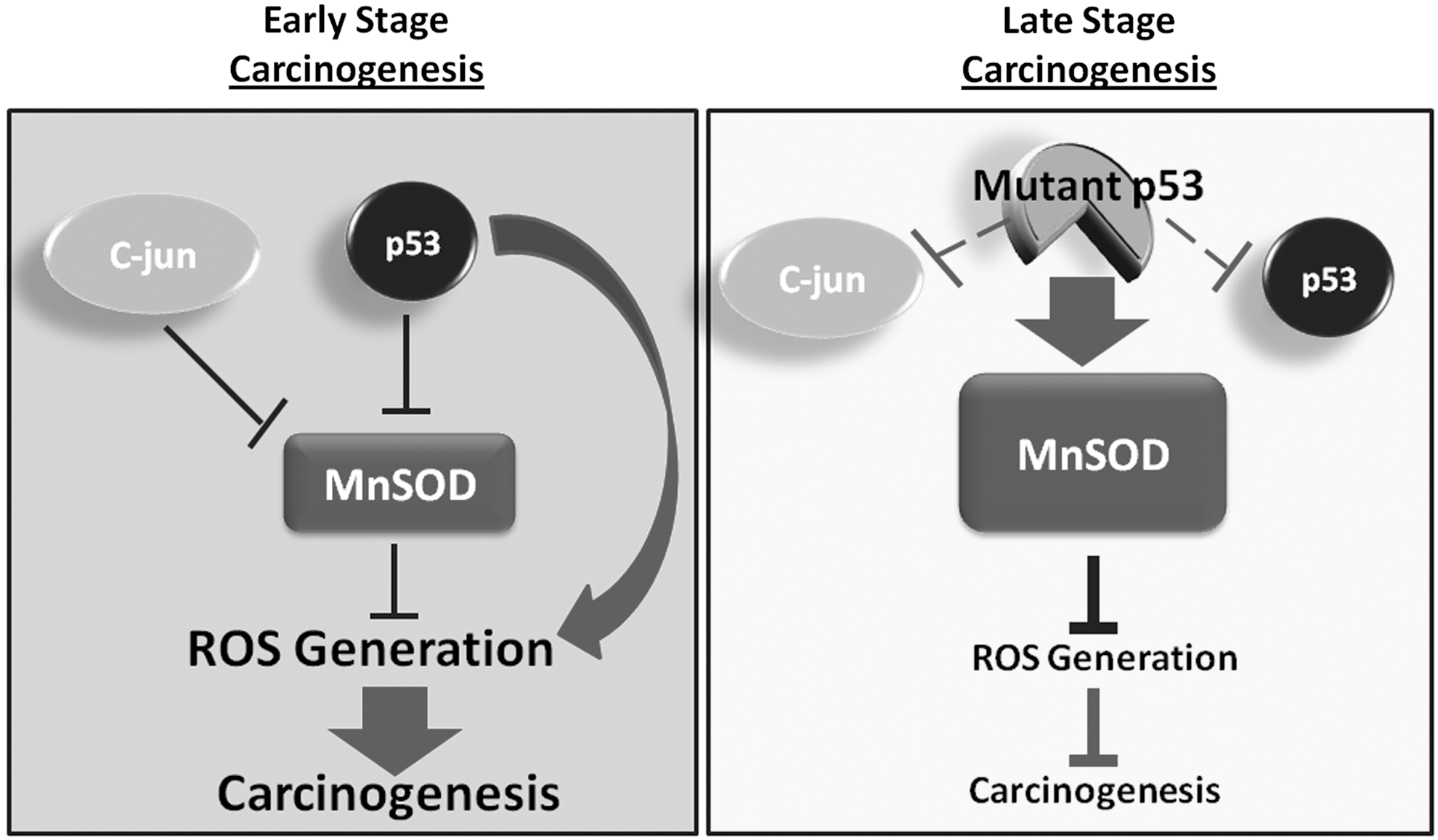
How could this shift in MnSOD expression occur? It is speculated that during carcinogenesis, SODs can be inactivated by oncogenes. For instance, c-jun, which is overexpressed in a number of human cancers, encodes a critical component of the activator protein-1 (AP-1) complex. c-jun mediates repression of MnSOD, resulting in expression of key regulators of ROS production and promoting cell survival (60). The suppression of MnSOD expression during early skin carcinogenesis is also mediated by the tumor suppressor p53. At late stages of carcinogenesis, p53 is often mutated, and therefore, loses this inhibitory effect (28) (Fig. 2). This SOD shift in expression also suggests that increasing SOD activity is a suitable strategy for cancer prevention. Such studies have been tested in human and animal models (Table 1).
Cu-ZnSOD, copper-zinc superoxide dismutase; CEBPD, CCAAT/enhancer binding protein delta; CML, chronic myelogenous leukemia; DMBA, 7,12-dimethylbenz(a)anthracene; EcSOD, extracellular superoxide dismutase; HIF-1α, hypoxia-inducing factor-1 alpha; MnSOD, manganese superoxide dismutase; MnSOD PL, manganese superoxide dismutase plasmid liposome; MnTE-2-Pyp(5+), Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin; MnTnHex-2-Pyp(5+), ortho Mn(III) meso-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin; SOD, superoxide dismutase; SOD1, superoxide dismutase 1; TPA, 12-O-tetradecanoylphorbol 13-acetate.
MnSOD overexpression has previously been shown to confer protection against radiation, as well as an effective chemopreventive approach to antioxidant based therapeutics. Tarhini et al. utilized a therapeutic DNA/liposome containing the human MnSOD transgene (133). Specifically, the MnSOD plasmid liposome consists of a double-stranded DNA plasmid, which contains the human MnSOD cDNA combined with two lipids {cholesterol and DOTIM (1-[2-[9-(Z)-hepta-decenyl]-3-[hydroxyethyl]imidazolinium chloride)} (133). A phase I study of MnSOD plasmid liposome in combination with traditional treatment modalities (carboplatin and paclitaxel) and radiation therapy in nonsmall-cell lung cancer was performed. Overall response rate for the standard chemoradiation regimen was satisfying (70%), and the oral administration of MnSOD plasmid liposome was determined as feasible, safe and protected patients from irradiation damage (133).
SOD mimetics are Mn-based metalloporphyrin complexes that scavenge superoxide, hydrogen peroxide, peroxynitrite and lipid-peroxyl free radicals (4, 24 –26, 35, 62, 104, 115). It has been demonstrated that metalloporphyrins with higher SOD activity have higher CAT activity suggesting the utility of SOD mimetics in ROS-mediated in vitro and in vivo models of disease (104). In an EAC rat model, the SOD mimetic Mn(III)tetrakis(4-benzoic acid) porphyrin (MnTBAP) decreased the incidence of EAC, which is correlated with a 25% increase in SOD enzymatic activities, suggesting MnTBAP may be able to serve as a chemopreventive agent for EAC (82). Another SOD mimetic (MnTE-2-PyP5+) has been tested in the chemically-induced skin carcinogenesis mouse model (158). The results showed that the SOD mimetic reduced tumor incidence by 40% when applied before tumor promoter treatment. Interestingly, when the mimetic was applied after tumor promoter treatment, tumor incidence was reduced further to 80%. This study also suggests that the timing of SOD intervention can be critical for its cancer preventive activity.
SOD Chemoprevention via Supplementation
Overexpression of antioxidant enzymes has been shown to suppress carcinogenesis both in vitro and in vivo, suggesting that the induction of endogenous antioxidant enzymes may be a potential target for cancer prevention. In an attempt to validate antioxidant enzyme induction as a potential novel approach to chemoprevention, SOD chemoprevention via supplementation has been tested. These supplements are often natural products with antioxidant activities.
Curcumin, derived from the Curcuma longa plant, is a natural product utilized in traditional Indian medicine and has been shown to mediate antioxidant, anti-inflammatory and antiangiogenic effects in several in vitro and in vivo carcinogenesis models. A recent study demonstrated that pretreatment with curcumin oil preserved MnSOD expression/activity and delayed bile acid-induced esophageal injury and potential cancer progression in vitro (123). Moreover, Curcuma aromatica, an herbal medicine, has been used for the prevention of EAC, possibly through its ability to induce MnSOD functional preservation. The results demonstrated that C. aromatica oil induced MnSOD expression and activity, which is associated with decreased incidence of EAC (74). In a human breast cancer xenograft model, curcumin reduced breast metastasis to lung tissues by ∼40% via dietary administration compared to control; and was further reduced when combined with paclitaxel (2). Furthermore, there are several clinical trials that are utilizing curcumin supplementation as a potential therapeutic for various diseases, such as colon/colorectal cancers, multiple myeloma, advanced cancers, lymphoma, breast cancer, as well as Type II diabetes (Table 2). On the contrary, curcumin has failed in clinical trials with Alzheimer's disease patients due to its inability to cross the blood-brain barrier. Moreover, it has been reported that curcumin has poor bioavailability and absorption, rapid metabolism, and systemic elimination, which contributes to a reduction in its redox-potency (Table 3). Recent studies suggest that curcumin derivatives may cross the blood-brain barrier potentially increasing the efficacy of these compounds in brain-specific diseases (15, 71). These results suggest the translational potential of natural product SOD-inducers may be questionable and tissue-specific; however, for various disease types SOD-inducers and their derivatives potentially have broad applicability in disease prevention.
CAT, catalase; COX-2, cyclooxygenase 2; CYP2E1, cytochrome P450 2E1; CYP3A4, cytochrome P450 3A4; GPx, glutathione peroxidase; GSH, glutathione; LPO, lipid peroxides.
In a colon cancer prevention study, rats were given hesperetin supplemented diet, and colon carcinogenesis was induced by 1,2-dimethylhydrazine (DMH). In DMH-treated rats, increased tumor incidence was accompanied by decreased activities of antioxidant enzyme glutathione S-transferase, glutathione peroxidase (GPx), SOD, and CAT. Administration of hesperetin to DMH-treated rats significantly decreased tumor incidence, and enhanced the activities of these antioxidant enzymes (3) (Table 4).
ATRA, all-trans retinoic acid; MnTBAP, Mn(III)tetrakis(4-benzoic acid) porphyrin; IL, interleukin; TBARS, thiobarbituric acid reactive substances.
Lycopene, a carotenoid responsible for the red pigment in ripe tomato and tomato products, has been inversely associated with cancer development. Moreover, in a 7,12-dimethylbenz(a)anthracene (DMBA)-induced mammary carcinogenesis rat model, lycopene alone or combined with melatonin was administered immediately after DMBA treatment. The results showed an elevation in the levels of malondialdehyde and nitric oxide in serum and breast tissues of DMBA injected rats. The activities of SOD, CAT, and GPx were increased in lycopene treated rats. Thus, the combined treatment suppressed mammary carcinogenesis (88).
Astaxanthin (ASTA), a marine, fat soluble xanthophyll, has been shown to exert antioxidant, anti-inflammatory, and anticancer functions. Yasui et al. demonstrated the chemopreventive effects of ASTA in a mouse model of colon carcinogenesis (152). Mice that were treated with ASTA exhibited a significant reduction in the occurrence of colonic muscosal ulcers, dysplastic crypts, and colonic adenocarcinoma (152). The anti-inflammatory activity of ASTA via ROS/reactive nitrogen species scavenging was also demonstrated by the suppression of inflammatory cytokines, including redox-sensitive nuclear factor–kappaB (NF-κB), tumor necrosis factor (TNF)-alpha, and interleukin-8 (152) (Table 4). In addition, ASTA inhibited proliferation, and induced apoptosis in the colonic adenocarcinomas. (152). However, Ohno et al. reported that ASTA can significantly induce cytochrome P450 CYP1A expression enhancing the mutagenicity of benzo[a]pyrene in male Wister rats (100).
On the other hand, naturally occurring and synthethic selenium has also shown promising chemopreventive results. Interestingly, historical events rendered selenium as a highly toxic element due to several reports of illnesses in humans via occupational exposure or environmental exposure in mammals (7, 38, 96, 110) Nonetheless, in 1973, Rotruck et al. demonstrated the chemopreventive effects of dietary selenium via GPx in protecting rat erythrocytes from oxidative injury in vitro (116). These same chemopreventive effects were echoed in clinical trials as well. However, it is known that the biological activity of selenium is determined by the chemical form administered. Moreover, selenium is a component of GPx localized within the active site of the enzyme; therefore, synthethic selenium compounds are potential donors of selenium for GPx enzymatic activity. Synthetic organoselenocyanates are receiving considerable attention as effective chemopreventive agents due to lower toxicity when compared to inorganic selenium (Table 4). Moreover, organoselenocyanates have shown chemopreventive activity during carcinogenesis at all stages; initiation, promotion, and progression, via the antioxidant properties of these synthetic compounds (7, 22, 23, 55, 138) (Table 4). A few examples of organoselenocyanates include benzyl selenocyanate (37), diphenylmethyl selenocyanate (18, 19), as well as ortho and meta isomers of phenylenebis(methylene)selenocynate (102, 124). Das et al. found that diphenylmethyl selenocyanate exerted chemopreventive activity in the pre- and postinitiation phases of the two stage skin and colon carcinogenesis mouse models. Their results demonstrated a significant reduction in skin papilloma incidence and multiplicity via the induction of phase II detoxifying enzymes and SOD (22, 23).
Ginger is a natural dietary component, which has antioxidant and anticarcinogenic properties (Table 4). Ginger consists of various phenols with antioxidant, as well as physiological and pharmacological activities which include gingerol, shogaol and zingerol (93). When tested in a DMH-induced colon carcinogenesis rat model, ginger supplementation increased the activities of SOD, CAT, GPx, glutathione-S-transferase, and glutathione reductase leading to decreased oxidative stress, tumor incidence, and tumor multiplicity (81). As previously mentioned, ginger consists of several components that possess chemopreventive potential. The [6]-gingerol chemical family of the ginger rhizome (Zingiber officinale Roscoe, Zingiberaceae) has been shown to have chemopreventive effects against benzo[a]pyrene induced skin carcinogenesis in mouse models (93). These results suggest that topical application of [6]-gingerol modulates p53 cellular localization inducing apoptosis, delaying carcinogenesis as a mechanism of cancer prevention.
A comprehensive antioxidant supplement study resulted from the dietary combination Protandim (92). Protandim consists of five well-established medicinal plants that all have been shown to induce endogenous antioxidant enzymes. The components include Bacopa monniera, Silybum marianum (milk thistle), Withania somnifera (Ashwagandha), Camellia sinensis (green tea), and Curcuma longa (turmeric) (92). An early human study showed that after 120 days of supplementation, the activity of erythrocyte SOD was increased by 30% and CAT by 54%. The chemopreventive activity of Protandim was later tested in a chemically-induced mouse skin carcinogenesis study (77). Mice were given the Protandim diet 2 weeks before chemical carcinogen treatment, and remained on the diet throughout the study. After 2 weeks of Protandim supplementation, both the activities of SOD and CAT were increased by 30%. Finally, Protandim suppressed tumor incidence by 33% and tumor multiplicity by 57% (77).
Mechanisms of Action of SODs in Cancer Prevention
The unique multi-modal advantages of targeting oxidative stress in chemoprevention have been well-studied and exploited in both basic science research and clinical investigation. In summary, cell proliferation, angiogenesis, inflammation, and cell death pathways are often involved in the actions of SOD-mediated cancer prevention.
In a chemically-induced skin carcinogenesis mouse model, chemical carcinogens activated the oncogenic Ras-Raf-mitogen-activated protein kinase (MAPK)-AP1 pathway, partially due to oxidative stress. However, MnSOD overexpression or knockout did not affect Ras mutation, instead, decreased (160) or increased (161) AP-1 activity. In addition, MnSOD overexpression also suppressed the activation of protein kinase Cɛ (PKCɛ), a PKC isoform activated by tumor promoter TPA (160). The SOD mimetic MnTE-2-PyP5+ showed similar anti-AP-1 activity in the skin carcinogenesis mouse model. Moreover, in a mouse model of breast cancer, the SOD mimetic suppressed angiogenesis via inhibiting the HIF-VEGF pathway (108). Furthermore, derivatives of SOD mimetics may also act as a pro-oxidant inducing cell death suggesting chemotherapeutic potential (131). Nevertheless, in a mouse model of T-cell lymphoma, suppressing chromosomal instability by MnSOD overexpression may potentially contribute to its antitumor effect (141). In the previously mentioned Cu-ZnSOD deficient mice study (32), there was increased cell proliferation due to progressive increases in the cell cycle control proteins cyclin D1 and D3, and the hepatocyte growth factor receptor Met. In the prostate tissue of Cu-ZnSOD deficient mice, there was decreased DNA methylation, which may cause aberrant expression of oncogenes (8). These results demonstrated that Cu-ZnSOD deficiency had a significant impact on the expression of NF-κB related genes in both kidney and liver. The differences in gene expression reported in their work may contribute to understanding of the molecular mechanisms underlying phenotypic abnormalities in Cu-ZnSOD-deficient mice (e.g., increase in the incidence of liver cancer) (9).
Stabilization of p53 has become an important target in cancer drug development. Recent studies demonstrated a novel role of mitochondrial p53 (99). It has been suggested that p53 may play a direct role in oxidative stress propagation (159). Upon p53 activation, the tumor suppressor translocated to mitochondria. Once localized in mitochondria, p53 physically interacted with MnSOD. As a result, this interaction inhibited the free radical scavenging abilities of MnSOD; therefore, providing enhanced ROS generation (159).
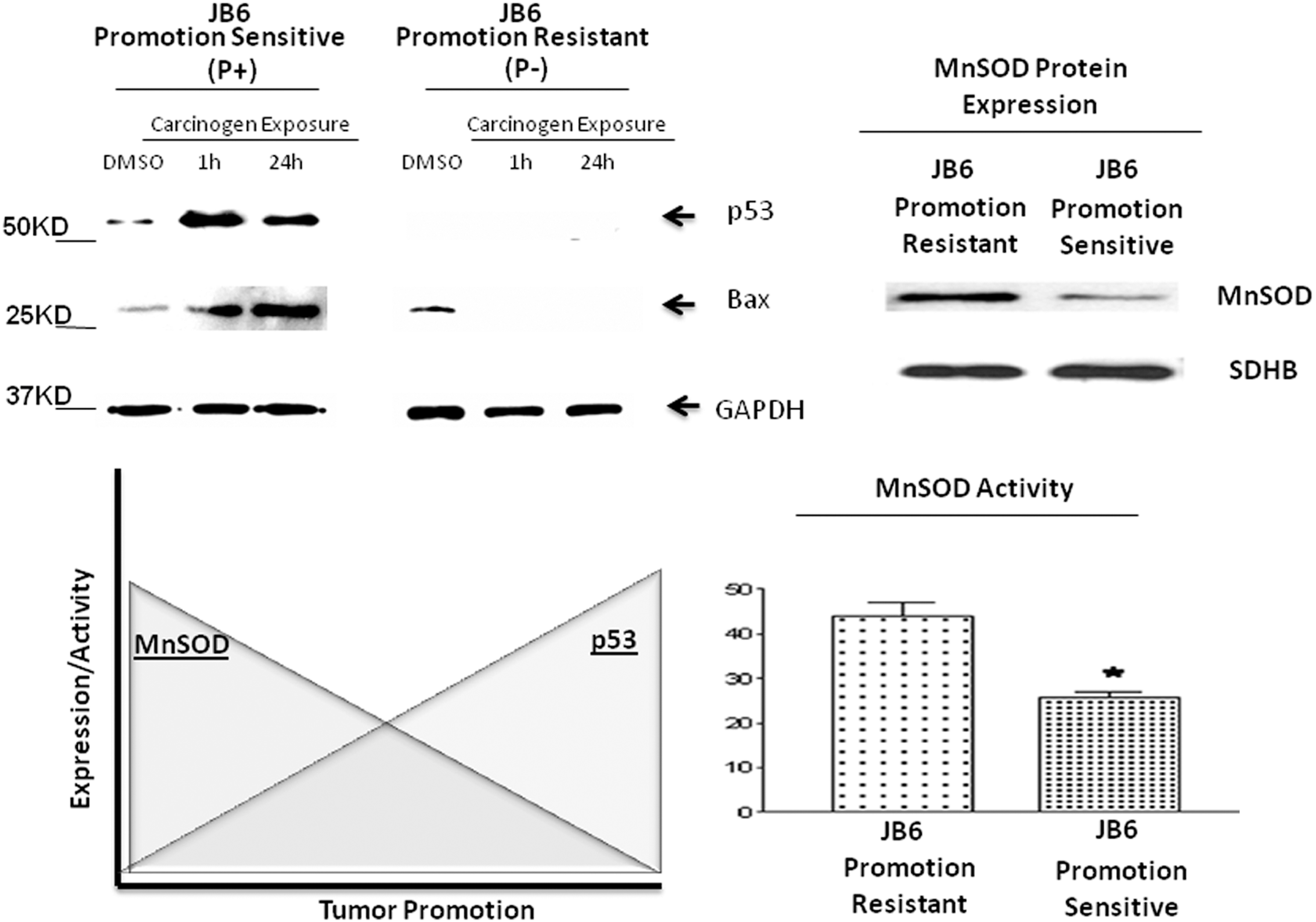
As we know, oxidative stress is a key player in the pathogenesis of numerous diseases. Furthermore, the inactivation of the p53 pathway is observed in most human cancers, with mutations in p53 occurring in at least 50% of all tumors (128). In addition to the lack of tumor suppressive functions, p53 mutants gain oncogenic activities contributing to carcinogenesis and drug resistance (27, 85, 144). The significance of p53-mediated ROS generation in early stage tumor promotion has been studied in mouse skin epidermal JB6 promotable (P+) and nonpromotable (P−) cells via a gene transfection approach. The results reveal that the tumor promotable phorbol ester 12-O-tetradecanoylphorbol 13-acetate (TPA) is capable of inducing p53 expression in JB6 (P+) cells, but not in JBP (P−) cells (112). More importantly, the results from promotion-sensitive p53-expressing JB6 (P+) cells transfected with short interfering RNA (siRNA) targeting p53 and promotion-resistant p53-deficient (P−) cells transfected with wild-type p53 and mutant p53 demonstrated that the presence of wild-type p53 expression during the early stage of tumor promotion was necessary to promote ROS-mediated anchorage-independent growth and skin cell transformation (Fig. 3). Furthermore, this phenomenon is believed to exist via the p53-MnSOD mitochondrial interaction as reported by Zhao et al. (159).
Another transcription factor and key modulator of tumorigenesis, NF-κB, plays a fundamental role in the formation and development of malignant changes caused by inflammation (106). Continued activation of NF-κB leads to increased expression of genes that encode proinflammatory cytokines, adhesion molecules, cyclooxygenase 2 (COX-2), and antiapoptotic genes, such as B-cell lymphoma 2 (BCL2), which can promote NF-κB-mediated cell survival (143). NF-κB is also redox sensitive. Oxidants can act as signaling molecules changing the redox status of transcription factors, and altering their capacity of DNA binding and transactivating genes involved in the regulation of cell growth (1). Moreover, ROS have been implicated in the pathogenesis of various hyperproliferative and inflammatory diseases (79).
In the two-stage skin carcinogenesis mouse model, chemical carcinogen treatment induced the markers of inflammation which includes skin epidermal hyperplasia, macrophage infiltration, intercellular adhesion molecule-1 (ICAM-1)/vascular cell adhesion molecule-1 (VCAM-1) expression, and increased NF-κB DNA binding activity (77). These changes were accompanied by increases in cell proliferative signaling pathways via increased DNA binding activity of AP-1 subunits, c-jun and Jun-D, and activation of phosphorylated c-Jun N-terminal kinase (JNK)—all important oncoproteins that contribute to tumor promotion. These results corroborate a growing body of literature that has shown direct association of inflammation and carcinogenesis (18). We have demonstrated that the subsequent induction of various antioxidant enzymes can reduce the inflammatory response through the suppression of oxidative damage and adhesion molecule expression. In addition, antioxidant induction can modulate DNA binding evidenced by reduced NF-κB DNA binding activity (77). Therefore, the induction of endogenous antioxidant enzymes can suppress inflammation as an additional mechanism of chemoprevention
Mitochondrial Involvement in Cancer
Mitochondria serve as scaffolds for a diverse array of cellular signaling pathways, such as adenosine triphosphate (ATP) and fatty acid synthesis, ROS generation, β-oxidation, calcium (Ca2+) homeostasis and apoptosis. In the 1920's, Warburg not only observed higher rates of glycolysis in cancer cells, but also hypothesized that mitochondrial dysregulation contributed to the pathogenesis of cancer (147). Interestingly, several decades later, the role of oxidative mitochondrial metabolism and overall mitochondrial physiology has received considerable attention in effectively targeting cancer pathophysiology. Several have observed mitochondrial metabolic adaptations in cancer cells which include modulation of mitochondrial respiration. A previous study found that defects in mitochondrial respiration promoted a survival advantage (105). It was demonstrated that deficiencies in mitochondrial respiration lead to reduced nicotinamide adenine dinucleotide (NADH) accumulation resulting in inactivation of PTEN and activation of the Akt survival pathway. Our previous studies have revealed that tumor promoters can decrease mitochondrial respiration which coincides with early metabolic changes as a mechanism to promote carcinogenesis (111, 146, 150).
Although decreased mitochondrial respiration has been shown in various cancer cell lines, this is not true for all cancer cells. Rodriguez-Enriquez et al. found that AS-30D hepatoma cells exhibit high glycolytic activity along with active oxidative phosphorylation (113). In addition, several cancer cell lines have demonstrated active oxidative mitochondrial metabolism, such as the PC-3 prostate cancer cell line (83), HeLa cells (117), MCF7 breast cancer cells (44) and D-54MG, GL261 glioma cell lines (43), suggesting the heterogeneity of mitochondrial metabolism among cancer cells and the need for more target specific therapeutics.
Several studies have suggested that targeting both glycolytic and mitochondrial metabolism may serve as an effective mechanism for anticancer drug development (36, 57, 67). It has been demonstrated that delocalized lipophilic cations accumulate within the mitochondria of transformed cell lines due to the constitutively higher mitochondrial membrane potential compared to their normal counterparts (6, 89). In screening for small molecules that selectively inhibit cell proliferation via a mitochondrial mediated pathway, Fantin et al. found that the small molecule F16, characterized as a delocalized lipophilic cation, selectively accumulates in the mitochondria to induce cell cycle arrest and increased apoptosis in various transformed cell lines (34). Moreover, inhibition of oxidative phosphorylation can induce mitochondrial depolarization and apoptosis which can be used as a therapeutic mechanism for anticancer drug development. Cariati et al. demonstrated that the arotinoid motarotene (Ro-408757) is a novel therapeutic approach against Burkitt's lymphoma (13). Ro-408757 induced apoptosis by downregulating the NADH dehydrogenase subunit 1 of the mitochondrial respiratory chain leading to caspase activation and enhanced ROS generation (13). Similar results were found utilizing mitochondrial respiration inhibition as a therapeutic target in osteosarcoma cells. Hagen et al. reported that the estradiol metabolite, 2-methoxyestradiol, inhibited mitochondrial respiration, promoting protein degradation of hypoxia-inducing factor-1α (HIF-1α) and apoptosis as a potential therapeutic approach (45). Interestingly, Cariati also demonstrated that inhibition of SOD and free radical scavengers has therapeutic potential (13). Although enhanced ROS generation is associated with inhibition of mitochondrial respiration and apoptosis, SODs can be involved in protecting the cell from oxidative injury and survival (10, 40, 107, 126). Consistent with these results, several studies have reported the chemoresistant potential of MnSOD expression and activity (16, 41, 53, 103, 134, 154). Thus, the presence of SODs can decrease the efficacy of anticancer drugs with a ROS-dependent mechanism of action, such as mofarotene, adriamycin and mitomycin C (13, 50, 53).
SOD and Cell Metabolism in Cancer Prevention
Warburg observed that cancer cells have a preference to metabolize glucose via glycolysis, even though this method of ATP production is less efficient when compared to oxidative phosphorylation. Interestingly, cancer cells utilized glycolysis in presence of oxygen. This altered metabolism provides a selective advantage for survival and supports rapid cancer cell proliferation in a unique tumor microenvironment. Some key components of the Warburg effect include increased glucose consumption, decreased oxidative phosphorylation, and lactate production, which are similar characteristic of oncogene activation.
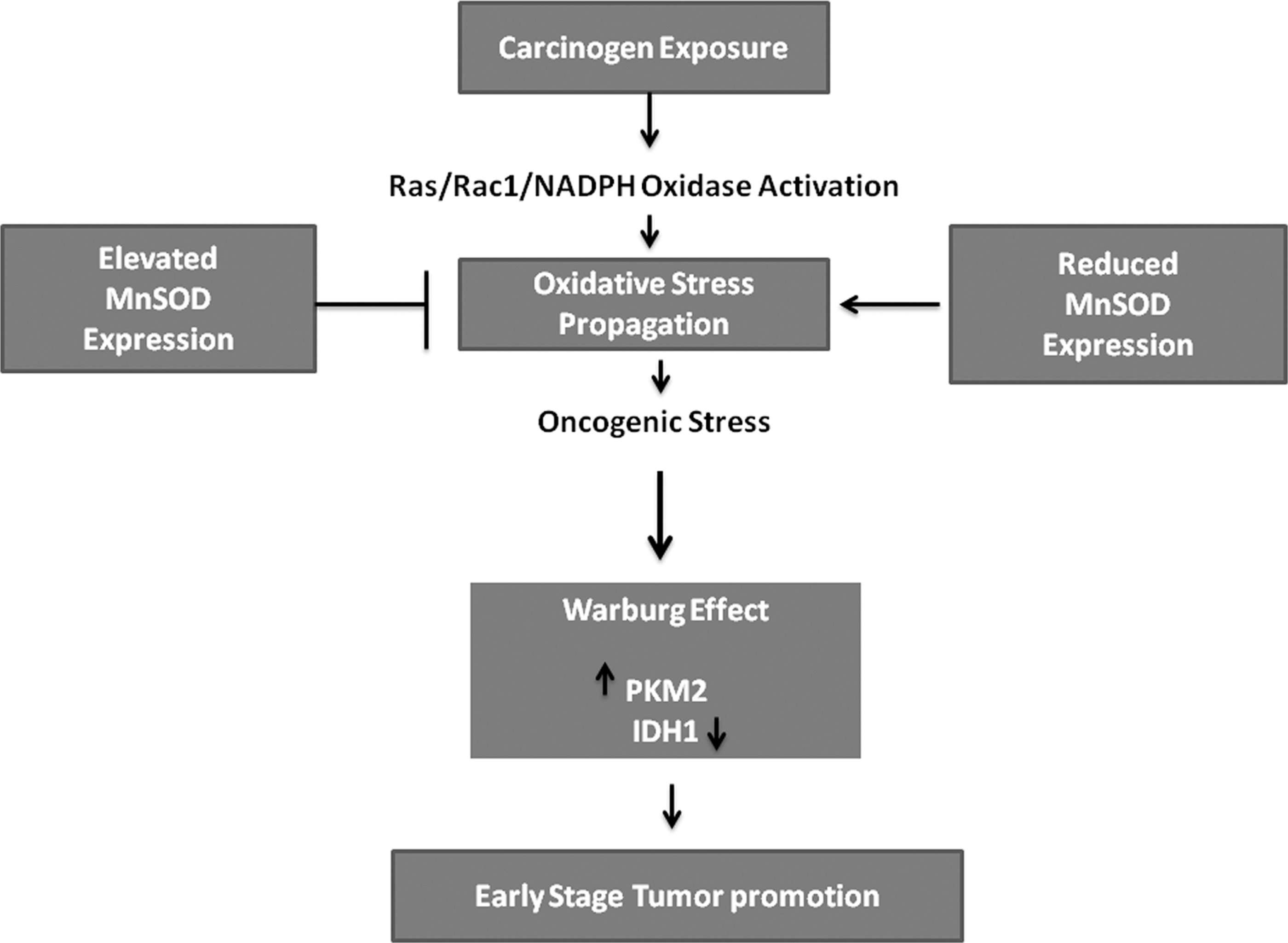
Can cell metabolism be targeted for cancer prevention? What role can SOD play in this approach? Fantin et al. (34a) described a link between altered cancer cell glucose metabolism and mitochondrial physiology. The glycolytic switch that occurs in cancer cells results in decreased dependence on mitochondrial respiration. Several studies have shown alterations in mitochondrial physiology and decreasing oxidative phosphorylation capacity is advantageous to the malignant phenotype (12, 31). However, is decreased mitochondrial respiration an essential tumor phenotype, and is there a link between altered mitochondrial metabolism and cell transformation? A study utilizing the tumor promoter sensitive mouse JB6 skin epidermal cell line and mitochondrial respiration substrates malate/pyruvate and succinate demonstrated that enhanced mitochondrial respiration had significant effects on suppressing skin cell transformation, suggesting a functional link between altered mitochondrial physiology and cell transformation during the early stage of skin carcinogenesis. Which metabolic enzymes may mediate this effect? A more specific marker of the glycolytic phenotype, pyruvate kinase, draws attention. Specifically, we look to the M2 isoform of pyruvate kinase. The embryonic M2 isoform has been shown to be exclusively expressed in tumor cells. Christofk et al. observed that stable knockdown of pyruvate kinase M2 isoform (PKM2) in the human lung cancer cell line H1299 resulted in decreased rates of glucose metabolism and reduced cell proliferation (17). These results were echoed in clinical studies reported by Roigas et al., who observed that PKM2 levels in patients with malignant renal tumors were significantly higher than in healthy donors (114). In addition, PKM2 has been shown to be capable of predicting disease recurrence in renal cell carcinoma patients after nephrectomy (95). These studies suggest a direct correlation of PKM2 upregulation and cancer progression (Fig. 4). To provide evidence that alterations in mitochondrial respiration have a direct impact on the glycolytic phenotype, PKM2 protein expression and activity in skin epidermal JB6 P+ cells following mitochondrial respiration substrate pretreatment were measured and the results showed that PKM2 expression was significantly decreased (150). Although the expression of pyruvate kinase M1 isoform (PKM1) occurs in normal adult cells, tumor cells express the PKM2 isoform which has been reported as an alternative splice variant of the M1 isoform. However, at what stage this metabolic switch occurs is only partially understood. In a two-stage skin carcinogenesis mouse model, the results showed increased PKM2 activity and decreased PKM1 activity, demonstrating a potential role of the PKM1/PKM2 switch in early stage skin carcinogenesis (150). It is also important to emphasize that upregulation of PKM2 expression and activity is only seen in wild-type mice, not in the MnSOD transgenic mice, suggesting MnSOD may also inhibit glycolysis as a novel mechanism of cancer prevention (150).
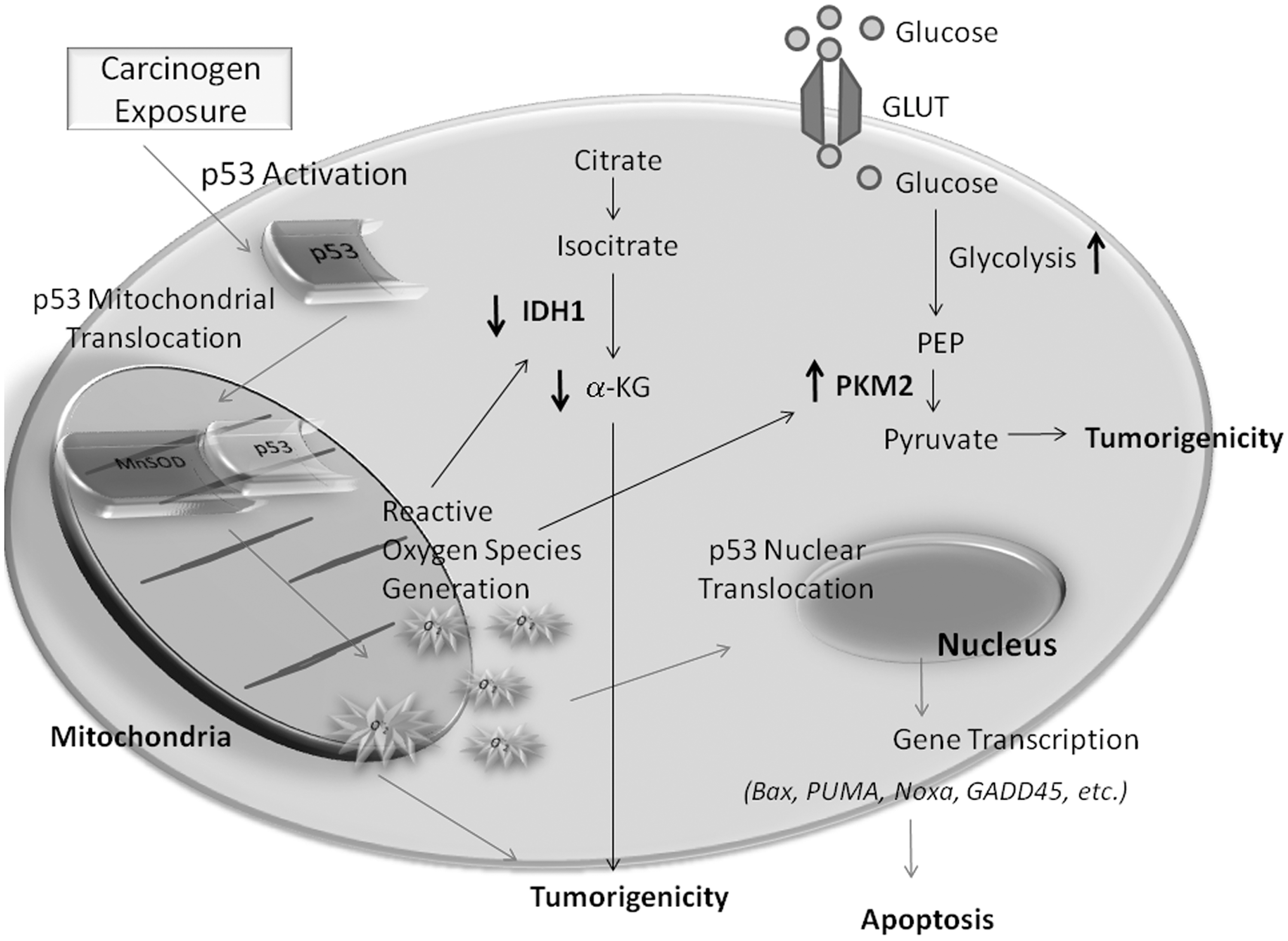
Another metabolic enzyme which is involved in early carcinogenesis is isocitrate dehydrogenase 1 (IDH1), which is decreased in skin tissues and skin cells following carcinogen exposure via the phorbol ester TPA or the environmental carcinogen ultraviolet electromagnetic radiation subtype C (UVC) (111). Nakamura suggests that IDH enzymes confer cellular protection by producing nicotinamide adenine dinucleotide phosphate (NADPH), which is essential for the reduction of glutathione reductases and for the activity of thioredoxin (90). Park and colleagues showed that IDH1 deficiency leads to increased markers of oxidative damage including lipid peroxidation, oxidative DNA damage, and decreased cell survival, suggesting that IDH1 can modulate the response to oxidative damage in cells. On the other hand, IDH enzymes can be inactivated via oxidation. Using MnSOD transgenic mice and their nontransgenic counterparts, our study demonstrates that IDH1 expression is significantly decreased during skin carcinogenesis only in nontransgenic mice, not in the MnSOD transgenic mice, suggesting that redox signaling may play a role in regulating IDH1 expression (111) (Fig. 5). Xu et al. demonstrated that a cell permeable α-ketoglutarate derivative can reverse the effects of inhibitors of α-ketoglutarate dependent dioxygenases (151). Therefore, if α-ketoglutarate levels are low, which has been reported in certain gliomas, or if IDH1 is mutated producing the oncometabolite, 2-hydroxyglutarate, α-ketoglutarate derivative can be used to reverse inhibition of α-ketoglutarate-dependent dioxygenases, as well as replenish intracellular levels of α-ketoglutarate to maintain cellular homeostasis and prevent cancer development (Fig. 5). Moreover, drugs that mimic α-ketoglutarate may be effective in combining with current treatment modalities to reduce tumor growth and angiogenesis. Furthermore, we have previously suggested that IDH1 downregulation may be due to ROS oxidative damage. In addition, we have shown that elevated levels of MnSOD prevented carcinogen-induced downregulation of IDH1 (111). Thus, regulating the intracellular redox status may be a novel effective approach to regulate IDH1 expression and activity.
Clinical Implications: SODs and Defining Mechanisms and Potential Targets for Cancer Prevention
Previous studies have demonstrated a significant decrease in SOD enzymatic activity in a variety of human cancers (65). As a result, increased levels of oxidative stress enhance the progression of tumor formation and cancer incidence. Although aberrant signaling and epigenetic regulation have been implicated in cancer progression, the lack of protection against oxidative injury seems to be a potent causative factor when SOD activity is diminished. With the SOD family being a first line of defense against tumor-associated ROS, animal models have suggested the utilization of dietary MnSOD inducers and SOD mimetics as effective modes of cancer prevention. Therefore, cancer prevention via dietary supplementation has received increasing attention due to the practicality of administration and long term low toxicity. However, clinical trials utilizing dietary supplements and combinations have failed, showing no significant effects on cancer incidence or cancer death. In addition, several studies have suggested that many of the natural products aforementioned [i.e., curcumin (11, 72, 109); hesperetin (14); lycopene (153); and ginger (70, 98)] are redox-active resulting in a pro-oxidative mechanism of action triggering potential adverse responses in vivo and in patients. Thus, these results suggest the need for more intense mechanistic clarity of dietary supplements in translational medicine. The focus of this review has been the chemopreventive aspects of SOD enzymes and the potential mechanistic targets utilized to mediate suppression and elimination of preneoplastic cells from progressing to malignancy. The multiple targets within tumor cells and tumor microenvironments discourage the utilization of single agents for effective chempreventive approaches. Therefore, multiple agents must be used to establish an optimal effect in preventing oncogenic activity and the overall opportunity to transform into invasive cancers. The advantages of the multi-modal approach of SOD-based chemoprevention allows investigators to broaden their therapeutic targets to encompass tumor-associated stromal and endothetial cells, as well as infiltrating macrophages that can be recruited to carcinogenic microenvrionments. As aforementioned, these dietary agents have been shown to have antitumorigenic, anti-inflammatory and antiangiogenic effects; therefore, strategies to combine dietary agents with chemopreventive properties warrants further testing. Currently, the dietary combination supplement Protandim has been effective in suppressing tumor promotion. As aggressive tumors progress, ROS increase lipid peroxidation and promote higher levels of reactive metabolites to maintain a mileu of persistent oncogenic stress. The dietary combination, Protandim, has been shown to reduce lipid peroxidation products in humans as evidenced by decreased thiobarbituric acid-reacting substances (92). This same theory was echoed in the two-stage skin carcinogenesis mouse model as a mechanism of chemoprevention (77). Nevertheless, these results are conclusive to suggest that targeting ROS generation via an antioxidant approach has broad applications to chronic diseases mediated by oxidative stress. However, for dietary supplements to have preventive efficacy, several factors must be considered (i) mechanistic verification; (ii) multi-component synergy for optimal effects; and (iii) compound stability. ROS are known to act as signaling molecules that modulate key signal transduction pathways during cancer development. In previous studies, proliferative pathways have been targeted and suppressed following dietary supplementation, but failed to translate to clinical trials. Gaining a clear understanding of the mechanisms involved and targeted by certain treatment strategies are key in preventive efficacy. The combination of multiple dietary supplements with widely-studied and well-established mechanisms of action provides for a smooth translational process from bench to bedside.
Gaining mechanistic clarity and targeting hallmark pathways in carcinogenesis are key in evaluating the therapeutic efficacy of dietary supplementation in cancer prevention. In using Ingenuity Pathway Analysis, many of the genes associated with oncogenic signaling and cell cycle were significantly decreased 2-fold on average by the dietary supplement Protandim (30). The effects were due to the potent synergy of the five active components, which were not seen in single compounds alone (30). As previously noted, proliferative, as well as proinflammatory signaling pathways are targeted in the utilization of multi-component dietary supplements. In addition, use of dietary supplementation not only reduced tumor multiplicity and incidence, but modulated the tumor microenvironment by reducing tumor associated macrophage infiltration and subsequent ROS generation (77). In targeting ROS generation, it is known that oxidative stress is associated with mitochondrial oxidative pathways and energy homeostasis. On the molecular level, decreases in ROS generation via SOD expression/activity reversed the glycolytic switch from aerobic glycolysis to oxidative phosphorylation via the mitochondria. Moreover, enhanced MnSOD activity suppressed IDH1 downregulation during the early stage of skin carcinogenesis (150). However, Xu et al. demonstrated that mutations within IDH1/2 can produce oncometabolites that affect downstream regulators of epigenetics (151). Therefore, evaluating genome-wide alterations following antioxidant induction warrants further investigation.
With a surge in the development of drugs that target ROS signaling, compound stability and bioavailability are important considerations in translational medicine. With SOD being a protein, effective delivery and stability are factors that can decrease preventive efficacy. SOD mimetics are potent catalytic scavengers of ROS that mimic the activity of endogenous SOD enzymes. Previous studies have suggested that MnSOD can be targeted specifically. Hodge et al. found that the epigenetic drug, zebularine, reverses MnSOD promoter methylation, increasing MnSOD protein and enzymatic activity in KAS 6/1 human multiple myeloma cells (47) Other epigenetic modulators, such as the histone deacetylase inhibitors trichostatin A and sodium butyrate also increase the expression of MnSOD in breast cancer cells by modulating histone methylation and acetylation resulting in altered expression of MnSOD (46). These results were also echoed in mouse myoblast cells. Maehara et al. demonstrated that trichostatin A induces dissociation of the Sp1-HDAC1 inhibitor complex from the promoter of MnSOD, suggesting the importance of MnSOD silencing via epigenetic mechanism in cancer development and drug targeting (80).
Interestingly, the utilization of adenoviral vectors to overexpress SODs has shown significant effects in reducing cancer growth when combined with traditional chemotherapeutics. Weydert et al. directly injected Cu-ZnSOD or MnSOD adenoviral vectors concomitantly with 1,3-bis(2-chloroenthyl)-1-nitrosurea into breast tumor xenografts which significantly decreased breast cancer cell growth and increased nude mice survival compared to treatment alone (149) Nonetheless, many have demonstrated the chemopreventive effects of MnSOD with the premise of cancer cells expressing lower MnSOD expression. However, other investigators have reported variable expression of MnSOD in cancerous versus noncancerous tissues, with higher expression of MnSOD in thyroid (94), brain (68), esophageal, and gastric (52) and colorectal cancers (56). Therefore, targeting cancers with higher MnSOD may be counterintuitive. Interestingly, Li et al. found that increasing MnSOD-mediated ROS production reduced cancer cell growth and sensitized cells to chemotherapeutic ROS generating agents (75). Thus, MnSOD overexpression in cancer cells with high levels of MnSOD is an effective approach if intracellular ROS/peroxide removal enzymatic systems remain at lower levels. Moreover, Zhang et al. reported similar results utilizing dual gene virotherapy consisting of an adenovirus that contained both human MnSOD and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). It was reported that the production of MnSOD-mediated hydrogen peroxide production, along with TRAIL overexpression, significantly eliminated colorectal tumor xenografts via apoptosis (157).
As previously mentioned, SOD mimetics have been shown to suppress oxidative injury and biomarkers of cell proliferation. Moreover, synthetic SOD mimetics have been shown to cross the blood brain barrier possessing preventive efficacy in providing protection against neurodegenerative diseases (54, 59, 104, 115, 120). Ultimately, these results suggest that the chemopreventive properties of MnSOD inducers and SOD mimetics extrapolate therapeutically to a myriad of chronic diseases mediated by ROS generation, signaling, and–mediated injury (119).
Concluding Remarks
The concept of targeting ROS generation in chemoprevention is a broad therapeutic approach that involves the activation of multiple signaling pathways, alterations in epigenetic regulation and various extracellular components that modulate the tumor microenvironment. Therefore, future therapeutic approaches involving dietary supplementation must be multicomponent cocktails that synergistically target a diverse array of signaling pathways, as well as proapoptotic mechanisms. In addition, clinical trials must include long term evaluations of potential antioxidant based therapy-induced resistance and toxicity in human populations. Ultimately, antioxidant-based mimetics may potentially be the future of oxidative stress targeted therapies in chemoprevention.