
Abstract
Introduction
T
Previous studies have demonstrated that reactive oxygen species (ROS), which are produced after glycoprotein VI (GPVI) stimulation, are responsible for a series of platelet-activating events. However, the mechanism by which ROS positively regulate the GPVI signaling cascade remains to be established.
The present study is the first to demonstrate that SH2 domain-containing PTP-2 (SHP-2) is targeted by ROS produced in collagen-stimulated platelets and suggests that a novel mechanism for the regulation of platelet activation by ROS is due to oxidative inactivation of SHP-2, which promotes tyrosine phosphorylation-based signal transduction.
Previous studies have demonstrated that reactive oxygen species (ROS), which are produced after GPVI stimulation (4, 7, 30, 49, 59), are responsible for a series of platelet-activating events, including PLCγ2 activation (49), cytosolic calcium elevation (49), αIIbβ3-integrin activation (7), granule release (4, 30), aggregate formation (7, 49), and thrombus formation (7). However, the mechanism by which ROS positively regulate the GPVI signaling cascade remains to be established.
One redox-dependent regulatory mechanism is via PTPs, which have a cysteine in the catalytic site that can be reversibly oxidized by ROS (8, 40, 50, 51). ROS are actively produced by cells stimulated with growth factors (34, 38), insulin (36), and B-cell or T-cell ligands (32, 52), and their production is considered to be important for efficiently propagating the tyrosine phosphorylation-dependent signal transduction. The role of PTPs in regulating platelet activation is not as well understood as the role of protein–tyrosine kinases. Accumulating data indicate that several PTPs, including LMW-PTP, SHIP-1, PTEN, and SH2 domain-containing phosphatase-2 (SHP-2), negatively affect GPVI-mediated platelet activation (10, 37, 58). It has been suggested that SHP-2 is activated by an interaction with platelet–endothelial cell adhesion molecule-1 (PECAM-1) and then plays a negative regulatory role in GPVI-mediated platelet activation by dephosphorylating important signaling effectors and adaptor molecules (28, 39, 46). Hyperglycemia-induced ROS production causes oxidative inactivation of PTPs and enhanced tyrosine phosphorylation of Syk in platelets exposed to collagen (59), but currently, there is no evidence that a specific PTP is regulated by ROS in collagen-stimulated platelets.
The current study sought to elucidate the redox-dependent control of the GPVI signaling cascade by intracellular ROS production in platelets. Employing biochemical and genetic approaches, we show that ROS produced upon GPVI stimulation lead to the oxidative inactivation of SHP-2, which potentially promotes the phosphorylation of tyrosine residues on signaling molecules, culminating in PLCγ2 activation and platelet aggregation. Our results suggest that collagen-induced ROS generation initiates a positive feedback regulatory loop involving SHP-2 that, at least in part, controls the rate and extent of platelet activation.
Results
Collagen-induced ROS generation enhances a cascade of tyrosine phosphorylation events
To determine whether collagen-induced ROS affect platelet activation, we stimulated platelets with collagen after pretreatment with N-acetyl-
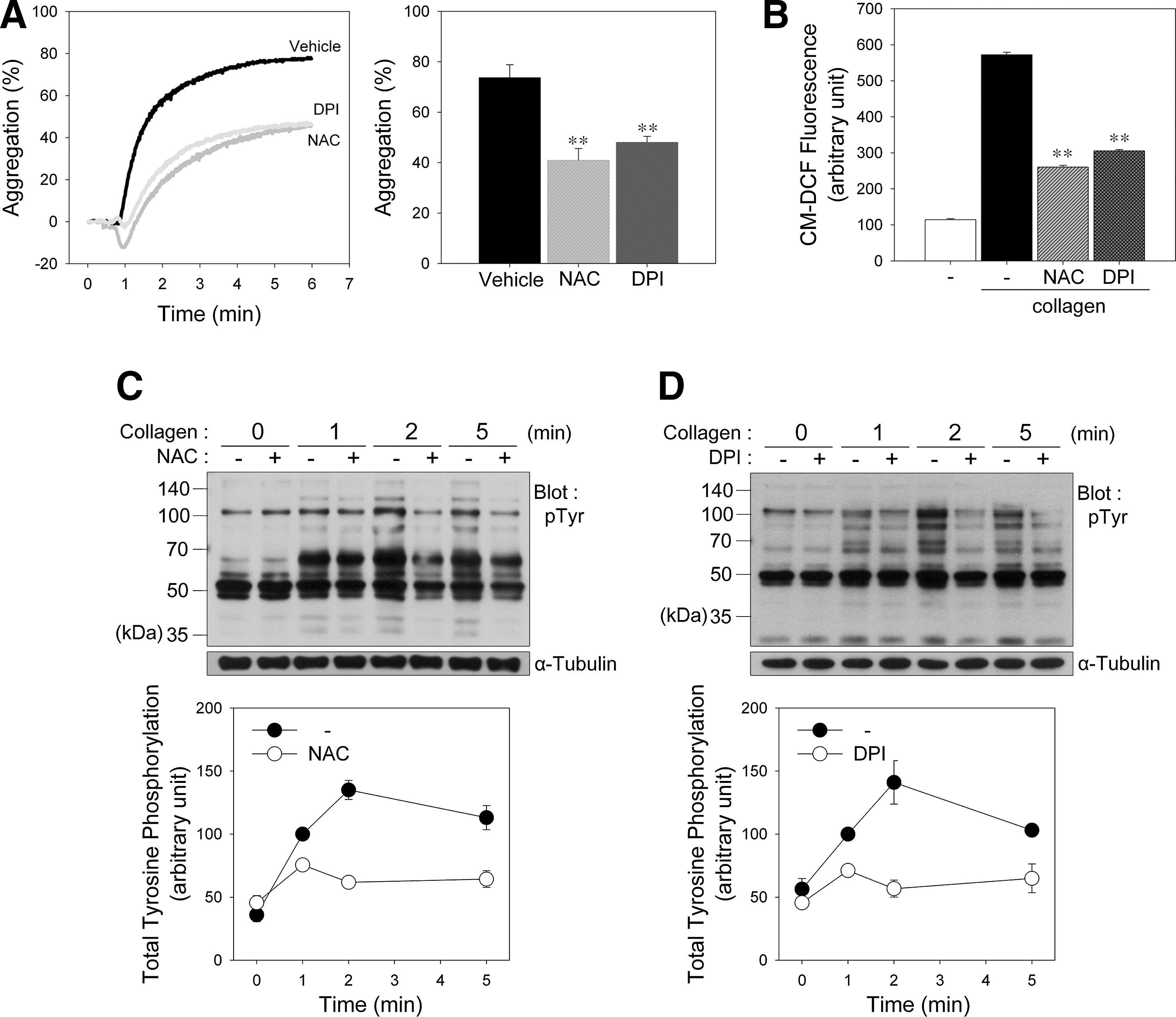
To compare the effects of platelet agonists on aggregation and ROS production, platelets were also stimulated with collagen, thrombin, TXA2 analogue U46619, or adenosine diphosphate (ADP) (Supplementary Fig. S1A; Supplementary Data are available online at
We next examined the effect of ROS on collagen-induced tyrosine phosphorylation in platelets. Incubation of platelets with collagen caused increased tyrosine phosphorylation of numerous proteins. In contrast, platelets pretreated with NAC (Fig. 1C) or DPI (Fig. 1D) showed a marked reduction in tyrosine phosphorylation, indicating that ROS generated after collagen stimulation are needed to induce an optimal tyrosine phosphorylation response for platelet activation through PTP oxidation.
Collagen-induced ROS generation leads to SHP-2 oxidation
Because SHP-2 is recruited to the signaling complex containing a GPVI/FcRγ chain and LAT and plays a negative regulatory role in GPVI-mediated platelet activation (33, 39, 41), we assessed SHP-2 oxidation in platelets after collagen stimulation. To specifically detect the oxidation of the catalytic cysteine on SHP-2, we used a monoclonal anti-oxPTP antibody that was specific for the conserved active site of classical PTPs oxidized to sulfonic acid (VHCSO3HSAG) (48). The signature motif is conserved in SHP-2; therefore, the anti-oxPTP antibody has previously been used to monitor SHP-2 oxidation (29, 48, 57). Oxidized SHP-2 had an increased signal after cysteine oxidation in collagen-stimulated platelets (Fig. 2A). However, the oxidation was transient, indicating that collagen-stimulated SHP-2 oxidation is reversible.
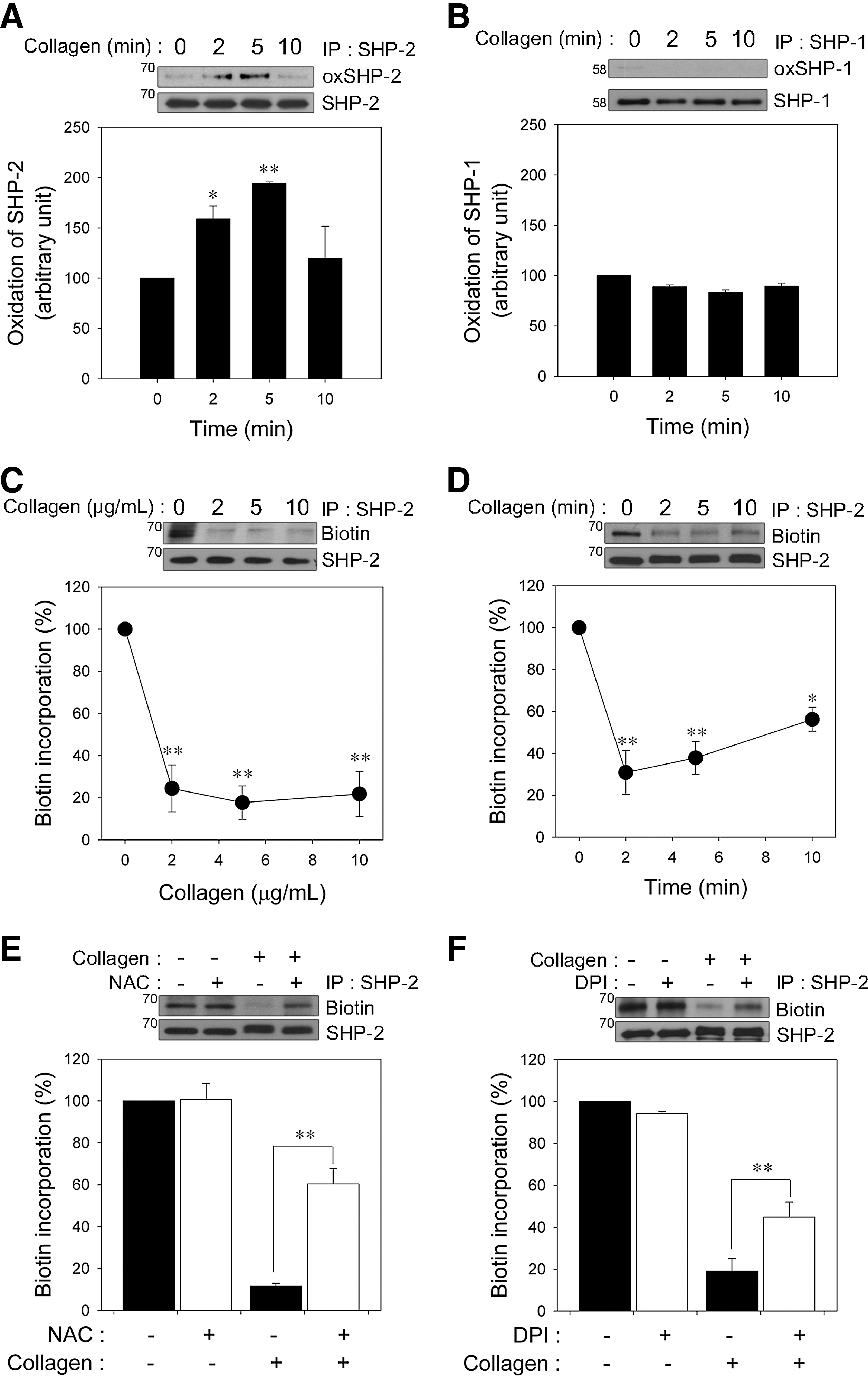
SHP-1 and SHP-2 are structurally homologous PTPs that possess tandem SH2 domains and have been thought to inhibit GPVI-regulated signal transduction (45). Thus, we examined whether SHP-1 is also oxidized. Under the same conditions in which reversible oxidation of SHP-2 was observed, SHP-1 oxidation was barely detected (Fig. 2B), although SHP-1 was oxidized by pervanadate as positive control (Supplementary Fig. S3). Similarly to previous studies in other cells (29, 32), our results also suggested that SHP-2 might be more redox sensitive than SHP-1 in collagen-stimulated platelets.
Next, we used the (+)-biotinyl-iodoacetamidyl-3,6-dioxaoctanediamine (PEO-iodoacetyl-biotin) reagent, a biotinylated thiol reactive probe, to confirm SHP-2 oxidation by selective direct labeling of the reduced thiols on the catalytic cysteine of SHP-2. Biotin incorporation into immunoprecipitated SHP-2 readily detected in unstimulated platelets was rapidly decreased in a dose-dependent manner (Fig. 2C), and the loss of biotin incorporation was recovered slowly during the follow-up period (Fig. 2D), indicating that the majority of the iodoacetamide labeling of SHP-2 was at the active cysteine site, and this oxidation was reversible. Consistent with a previous study (57), we found that SHP-2 oxidation detection using the PEO-iodoacetyl-biotin method, in comparison, was more sensitive than the anti-oxPTP antibody protocol. Thus, the former method was subsequently used to analyze SHP-2 oxidation in this study. To show that ROS generation leads to SHP-2 oxidation after specific stimulation of GPVI, platelets were stimulated with collagen-related peptide (CRP). As shown in the Supplementary Figure S4, the GPVI-specific agonist CRP also induced SHP-2 oxidation.
To further confirm the causal role of ROS in SHP-2 oxidation, the platelets were pretreated with NAC (Fig. 2E), Nox inhibitors including DPI, apocynin, 2-acetylphenothiazine, and VAS2870 (Fig. 2F and Supplementary Fig. S5) before the collagen stimulation. As expected, the loss of SHP-2 labeling was inhibited by ROS scavengers or Nox inhibitors, implying that ROS produced upon collagen stimulation are responsible for SHP-2 oxidation and inactivation in platelets.
SHP-2 associates with multiple proteins in the LAT signaling complex upon stimulation
After stimulation, SHP-2 directly or indirectly associates with signaling molecules, such as PECAM-1, Syk, LAT, Vav1, and SH2 domain-containing leukocyte protein of 76 kDa (SLP-76), in platelets and other cell systems (17, 20, 32, 39, 41); therefore, we examined the association between SHP-2 and other proteins using co-immunoprecipitation studies. SHP-2 immunoprecipitation from collagen-stimulated platelets showed an increased association of multiple proteins (Fig. 3A). As previously demonstrated (39), the association between PECAM-1 and SHP-2 was increased upon GPVI stimulation. Similar to previous results for the association of Syk with SHP-1 in GPVI-stimulated platelets (45), the association between Syk and SHP-2 also distinctly increased following collagen stimulation. Upon GPVI stimulation, LAT forms a platform for the assembly of a signaling complex that includes SLP-76, Grb2-like adapter downstream of Shc (Gads), Vav1, Bruton's tyrosine kinase (Btk), and PLCγ2, which culminates in PLCγ2 activation (56). In addition, LAT deficiency reduces the phosphorylation of Syk and PLCγ in GPVI-mediated platelet activation (24). LAT, SLP-76, Vav1, Btk, Gads, and PLCγ2 were detected in SHP-2 immunoprecipitates from stimulated platelets but were barely detected in resting platelets. However, there were no apparent changes in the level of the adhesion- and degranulation-promoting adapter protein (ADAP) or Grb2-associated binder 2 co-immunoprecipitated with SHP-2, implying a selective effect. Next, to confirm the recruitment of SHP-2 to the LAT signaling complex, LAT immunoprecipitates were examined (Fig. 3B). A low level of SHP-2 was present in the LAT immunoprecipitates from unstimulated platelets, and this association increased notably following collagen stimulation. Increased binding of SLP-76, Gads, Vav1, Btk, and PLCγ2 to immunoprecipitated LAT in stimulated platelets was also observed. These results and previous reports support the direct or indirect participation of SHP-2 in the LAT signaling complex in collagen-stimulated platelets.
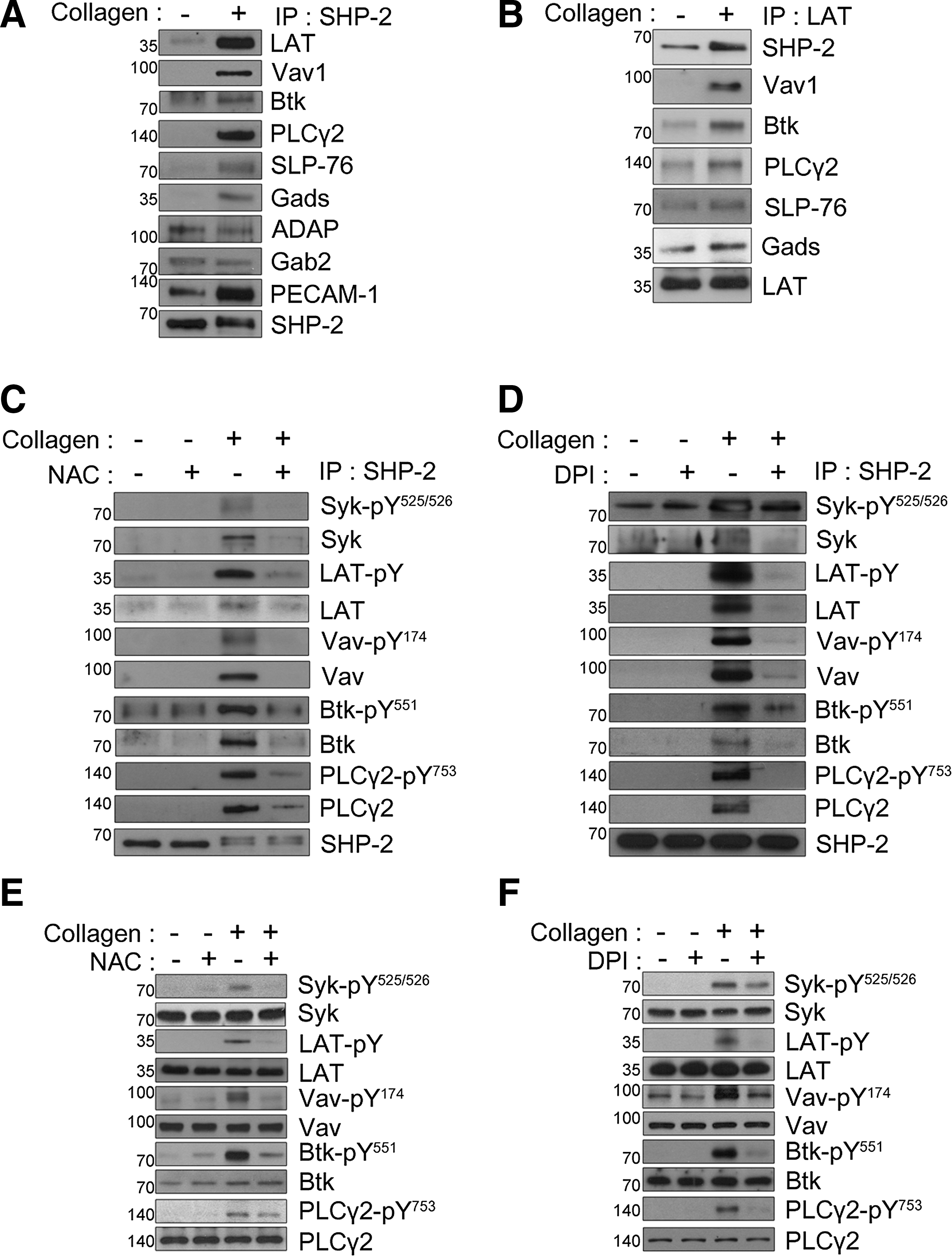
Effects of collagen-induced ROS generation on LAT-mediated PLCγ2 activation
The loss of the PTP activity caused by the oxidation or mutation of the catalytic cysteine leads to increased binding affinity for phosphorylated substrates (8, 16). Among the proteins present in the SHP-2 immunoprecipitates from the collagen-stimulated platelets (Fig. 3A), Syk, Btk, and Vav1 have already been proposed as potential substrates for SHP-2 or SHP-1 (14, 32, 35, 45, 59), suggesting a possible association between SHP-2 and these phosphorylated proteins that changes depending on ROS-mediated SHP-2 oxidation. To investigate this possibility, we analyzed SHP-2 immunoprecipitates with phospho-specific antibodies for Tyr525/Tyr526 on Syk, Tyr174 on Vav1, and Tyr551 on Btk. These tyrosine phosphorylations are necessary for the activation of these signaling molecules and can be used as markers of the activity of these molecules. The collagen-induced association of SHP-2 with phosphorylated Syk, Vav1, or Btk was apparently decreased by NAC or DPI (Fig. 3C, D). Upon collagen stimulation, Syk phosphorylates multiple tyrosine residues on LAT (44), and Btk seems to be responsible for phosphorylation of Tyr753 and Tyr759 at PLCγ2 (3, 54). Concomitant with the major changes in phosphorylated Syk and Btk, the amount of phosphorylated LAT and PLCγ2 present in SHP-2 immunoprecipitates was decreased in the NAC- or DPI-treated platelets. These results suggest that Syk, Btk, and Vav1 are potential substrates of SHP-2, and their activity can be regulated by SHP-2 in a redox-dependent manner in collagen-stimulated platelets.
Because SHP-2 oxidation was inhibited by ROS quenching, an antioxidant should decrease tyrosine phosphorylation in SHP-2 substrates. Thus, the effects of NAC or DPI on specific tyrosine phosphorylation on Syk, Vav1, and Btk were analyzed in total cell lysates (Fig. 3E, F). NAC or DPI inhibited collagen-stimulated Syk phosphorylation at Tyr525/Tyr526, which is critical for activating kinase function (18, 19). The inhibitory effects of NAC or DPI on the tyrosine phosphorylation of LAT might have resulted from the downregulation of the Syk activity. The data support a model in which SHP-2 is brought in proximity to the LAT signaling complex and downregulates Syk phosphorylation, which inhibits the ability of Syk to phosphorylate LAT and to induce assembly of the LAT signaling complex. Phosphorylation of Tyr174 on Vav1 and Tyr551 on Btk was also inhibited by a ROS scavenger, implying that a decrease in their activation can diminish PLCγ2 activity (3, 47, 54). Collagen-stimulated tyrosine phosphorylation of PLCγ2 at Tyr753, a phospho-site associated with an increased activity, was also inhibited by NAC or DPI treatment. The data suggest that ROS control tyrosine phosphorylation of Vav1 and Btk in the LAT signaling complex and subsequent PLCγ2 activation.
Glutathione peroxidase 1/catalase double deficiency leads to enhanced activation of PLCγ2 via oxidative inactivation of SHP-2 in collagen-stimulated platelets
Among ROS, H2O2 is readily produced and removed following a physiological stimulus and can freely diffuse across cellular membranes; thus, H2O2 exhibits properties that are ideal for regulating signaling (12, 53). Glutathione peroxidase 1 (GPx1) and catalase are the principle enzymes responsible for eliminating H2O2. To establish a role for H2O2 in redox regulation of SHP-2, platelets from GPx1−/−Cat−/− and wild-type (WT) mice were studied (Fig. 4A). To exclude a possible change in GPVI expression level in GPx1−/−Cat−/− platelets, we measure the expression level. GPx1−/−Cat−/− platelets expressed similar level of GPVI as WT platelets (Supplementary Fig. S6).
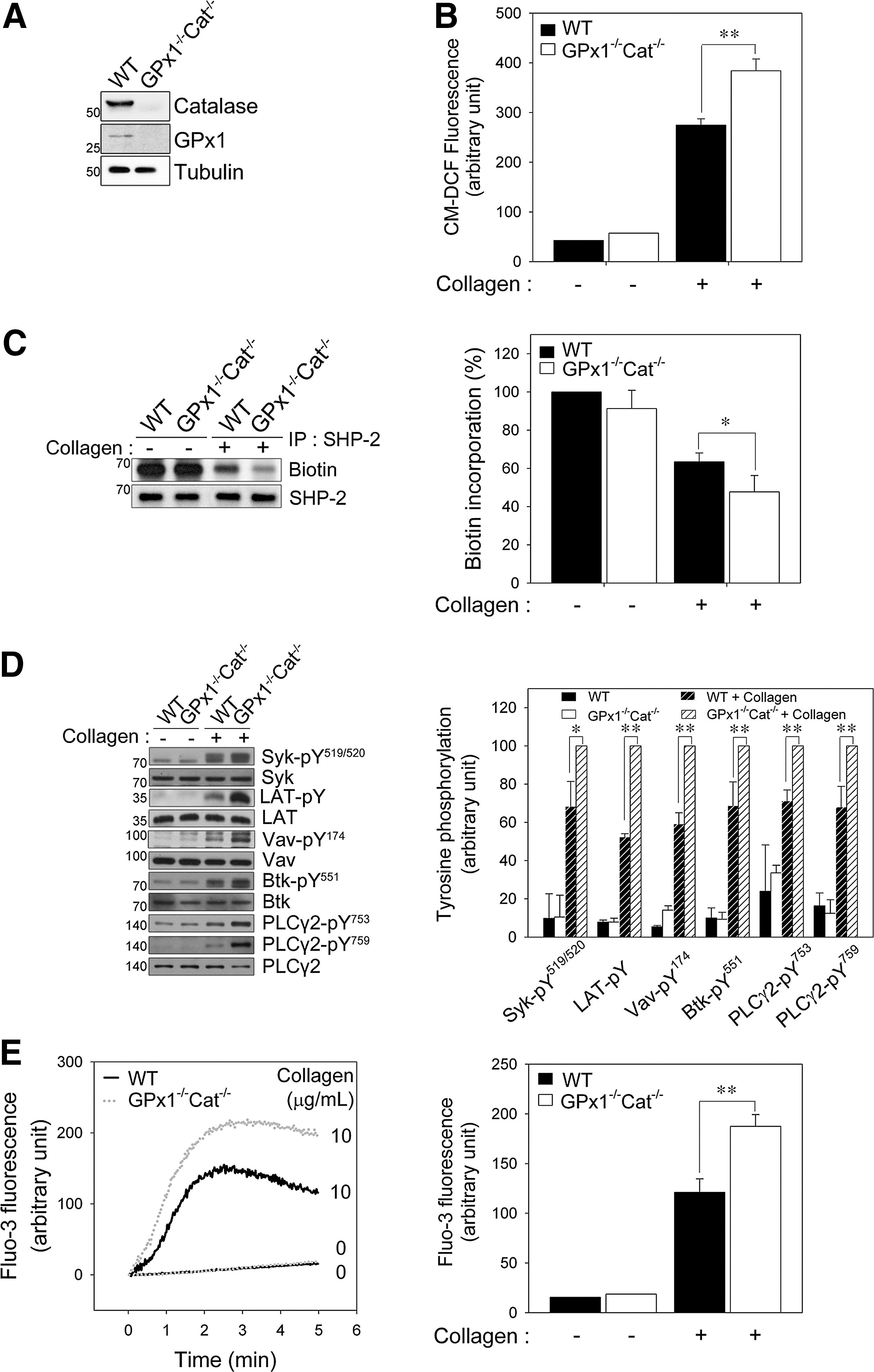
The intracellular ROS level of GPx1 or catalase single knockout platelets was higher than that of the WT platelets but is not increased as much as that of double knockout platelets (Supplementary Fig. S7). To more effectively regulate the level of ROS, we used GPx1/catalase double-deficient mice in this study. The intracellular ROS level was significantly higher in the GPx1−/−Cat−/− platelets after collagen stimulation than the WT platelets (Fig. 4B), indicating that the increase in intracellular ROS is limited by H2O2-eliminating enzymes. To more precisely define the impact of GPx1/catalase double deficiency on collagen-dependent signaling, we first examined SHP-2 oxidation. Biotin incorporation demonstrated that the SHP-2 oxidation level was greater in the GPx1−/−Cat−/− platelets than the WT platelets (Fig. 4C), indicating that the H2O2 produced after collagen stimulation is responsible for SHP-2 oxidation and inactivation in platelets.
We have demonstrated that SHP-2 can regulate GPVI signaling pathways in a redox-dependent manner, and Syk, Btk, and Vav1 are potential SHP-2 substrates. Therefore, we analyzed the activation of downstream signaling molecules using phospho-specific antibodies (Fig. 4D). The phosphorylation of Syk at Tyr519/520 (Tyr525/Tyr526 in the human protein), of Vav1 at Tyr174, and of Btk at Tyr551 was elevated in the GPx1−/−Cat−/− platelets compared to the WT platelets after collagen stimulation. Subsequently, collagen-stimulated PLCγ2 phosphorylation at Tyr753/759 was also increased in the GPx1−/−Cat−/− platelets compared to the WT platelets. Given that the release of calcium from intracellular stores was controlled by PLCγ2-mediated inositol 1,4,5-triphosphate production, we examined the effect of GPx1/catalase double deficiency on the rise in cytosolic calcium. Concomitant with the change in ROS-dependent PLCγ2 activation, the collagen-induced increase in cytosolic calcium was significantly greater in the GPx1−/−Cat−/− platelets than the WT platelets (Fig. 4E). These results support a model in which the H2O2 produced upon collagen stimulation leads to SHP-2 oxidation and promotes tyrosine phosphorylation of its substrates, which causes the eventual phosphorylation and activation of PLCγ2 and release of calcium from intracellular stores.
GPx1/catalase double deficiency enhances platelet activation and aggregation in a redox-dependent manner
To further examine the redox-dependent platelet activation events, we monitored the surface expression of P-selectin, a marker of degranulation, and integrin αIIbβ3 activation, in response to convulxin. Given that collagen can cause problems for flow cytometry by crosslinking with itself and aggregating platelets (21), we used convulxin, a GPVI activator (26). Convulxin-induced surface expression of P-selectin from α granules was greater in platelets from GPx1−/−Cat−/− mice than WT mice (Fig. 5A). Integrin αIIbβ3 activation by convulxin was also greater in the GPx1−/−Cat−/− platelets than the WT platelets (Fig. 5B). These results suggest that GPVI-induced H2O2 production promotes degranulation and inside–out activation of integrin αIIbβ3 by the increased cytosolic calcium resulting from PLCγ2 activation following oxidative inactivation of SHP-2.
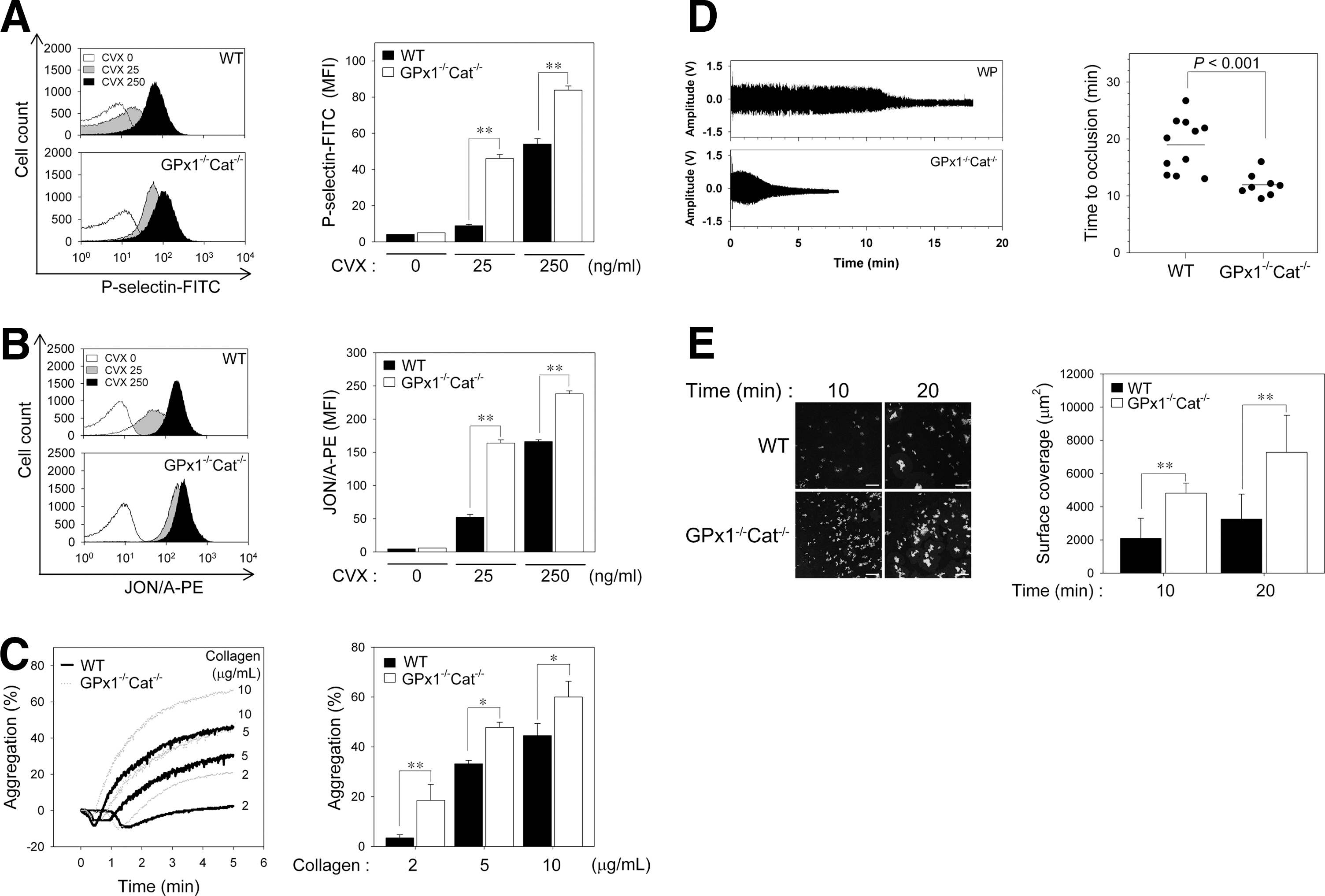
To assess the functional consequences of GPx1/catalase double deficiency, we measured aggregation in response to graded concentrations of collagen (Fig. 5C). The concentrations of collagen that elicited low, intermediate, and high levels of responsiveness from the WT platelets elicited a significantly greater response from the GPx1−/−Cat−/− platelets, indicating that H2O2 functions as a stimulatory molecule for collagen-induced platelet aggregation through oxidative inactivation of SHP-2.
GPx1/catalase double deficiency accelerates thrombotic response in injured carotid arteries
To investigate the consequences of GPx1/catalase double deficiency on arterial thrombus formation, we induced carotid artery thrombi with FeCl3, and the time to arterial occlusion was monitored. Carotid occlusion in the WT mice occurred at a mean of 19.4 min, whereas in GPx1−/−Cat−/− mice, the mean was shortened to 11.9 min (p<0.001, Fig. 5D). To exclude the effects of other components in hemostasis/thrombosis, we analyzed thrombus formation using the ex vivo flow chamber assay (Fig. 5E). Compared to WT platelets, we found a marked increase in GPx1−/−/Cat−/− platelets adhesion to a collagen-coated surface under shear-flow conditions. These results further suggest that H2O2 promotes platelet activation through SHP-2 oxidation-mediated upregulation of tyrosine phosphorylation-based signal transduction, which accelerates arterial thrombus formation.
Discussion
Tyrosine phosphorylation is required for appropriate cell signaling and activation, and this phosphorylation is coordinated by the balanced and specific action of protein–tyrosine kinases and PTPs. Receptor-mediated ROS production has emerged as an important regulator of phosphorylation-dependent signal transduction (8, 50). Collagen-induced platelet activation is specifically caused by the binding of collagen to the GPVI receptor (43), signaling through which requires tyrosine phosphorylation of various signaling molecules (43, 56). Although the tyrosine phosphorylation response to a variety of physiological stimuli can be enhanced by ROS-dependent oxidation and PTP inactivation (8, 40, 50, 51), specific PTP regulation by ROS in collagen-stimulated platelets has not been understood. This study demonstrated that the PTP SHP-2 is transiently oxidized in platelets by ROS produced by collagen stimulation. Furthermore, we identified Syk, Vav1, and Btk as putative substrates of SHP-2 in collagen-stimulated platelets. The data suggest that oxidative inactivation of SHP-2 promotes collagen-induced phosphorylation of Syk, Vav1, Btk, and PLCγ2 in the LAT signaling complex and that these changes enhance granule secretion, integrin αIIbβ3 activation, platelet aggregation, and thrombosis.
SHP-1 and SHP-2 are structurally homologous PTPs possessing tandem SH2 domains. Similar to previous studies reporting that the ROS generation induced by activating platelet-derived growth factor or the T-cell receptor leads to SHP-2 but not SHP-1 oxidation (32, 38), our results also demonstrated that SHP-2 is selectively sensitive to ROS-mediated oxidation of the catalytic cysteine compared to SHP-1 in platelets. Although the intrinsic oxidation susceptibility of SHP-1 and SHP-2 has not yet been established, higher oxidation susceptibility for SHP-2 compared to SHP-1 in human T-cell blasts and NIH3T3 mouse fibroblasts treated with different H2O2 concentrations has recently been demonstrated (29, 32). Structural analyses of both PTPs have revealed that in the basal state the N-terminal SH2 domain folds into the catalytic domain and simultaneously hinders access to the substrates (5). Regarding SH2 domain-mediated regulation, the SH2 domain-mediated occlusion of the active site might obstruct the access for the ROS and thereby decrease oxidation susceptibility and play a significant protective role against oxidation in SHP-1's SH2 domain (57).
The current data suggest that after GPVI stimulation, SHP-2 associates with multiple proteins in the LAT signaling complex in which the oxidized SHP-2 is localized. The association of oxidized SHP-2 with proteins may be caused by prolonged binding to phosphorylated substrates (17, 32, 38). In platelets pretreated with an antioxidant, anti-SHP-2 immunoprecipitates showed decreased LAT association. Similarly, SHP-2 immunoprecipitation also revealed a limited association of SHP-2 with tyrosine-phosphorylated proteins primarily involved in a LAT signaling complex. Notably, our study suggests that Syk, Vav1, and Btk are SHP-2 substrates in collagen-stimulated platelets. However, we still cannot completely exclude the possibility that their tyrosine phosphorylations can be indirectly regulated by SHP-2 and that other PTPs are also involved. Accumulating data show that several PTPs, including LMW-PTP, SHIP-1, and PTEN, play a negative role in platelet activation (10, 37, 58) by dephosphorylation of multiple substrates. Although PTP-1B has been reported as a positive regulator of platelet signaling (2, 31), the possibility that PTP-1B oxidation promotes platelet activation by upregulating tyrosine phosphorylation-based signal transduction can be proposed.
Collagen-induced GPVI stimulation results in platelet aggregation, which is associated with subsequent ROS production by Nox. Previous studies with Nox inhibitors have indicated that Nox is responsible for the ROS production in GPVI-stimulated platelets (4, 7).
Because many mammalian cell types produce H2O2 for intracellular signaling in response to stimulation through various cell surface receptors (12, 50), in platelets after GPVI stimulation, the electrons generated by Nox, cyclooxygenase, or the mitochondrial electron transport chain appear to mediate the reduction of molecular oxygen to a superoxide anion (O2 •−), which then undergoes spontaneous or enzyme-catalyzed dismutation to H2O2 (12, 50). H2O2 is a nonpolar nonradical molecule and can diffuse freely across membranes. This characteristic allows H2O2 to function as an efficient signaling molecule and thereby an intracellular mechanism for eliminating H2O2, which is an important factor in redox-dependent cell signaling (12, 50, 53). In mammalian cells, GPx and catalase are the major enzymes responsible for eliminating H2O2. Collagen-induced platelet aggregation has been shown to be closely associated with H2O2 production, and catalase has been demonstrated to inhibit H2O2 formation and platelet aggregation (13, 49). Deficiency of H2O2 eliminating systems would be expected to promote H2O2 accumulation by reducing the cellular capacity to remove the H2O2 generated in GPVI-stimulated platelets. In support of this notion, we found that after collagen stimulation, platelets isolated from the GPx1−/−Cat−/− mice had more ROS compared to WT mice. Although we did not directly detect the level of intracellular H2O2 in the GPx1−/−Cat−/− platelets, the higher level of intracellular ROS seems to be mainly due to more amount of H2O2. In addition, subsequent SHP-2 oxidation in the GPx1−/−Cat−/− platelets was greater than the WT platelets, implying the important functions of intracellular H2O2 in platelet activation via redox regulation of SHP-2. On the other hand, it can be possible that superoxide is more efficient ROS than H2O2 for inactivating SHP-2 as in the case of PTP1B described by Barrett et al. (6). Further analysis of the GPx1−/−Cat−/− platelets demonstrated enhanced tyrosine phosphorylation of Syk, LAT, Vav1, Btk, and PLCγ2 compared to the WT platelets, strongly suggesting that augmented intracellular H2O2 may be critical for modulating tyrosine phosphorylation-induced platelet activation.
Our data suggest that H2O2 modulates platelet α-granule release in activated platelets. Increased intracellular H2O2 resulting from the inhibition of GPx1 and catalase also promoted P-selectin surface expression in GPVI-stimulated platelets. Similarly, the GPx mimetic ebselen or the general antioxidant NAC inhibited P-selectin upregulation in convulxin-stimulated platelets (4). Taken together, the data support a model in which platelet-derived P-selectin surface expression is redox regulated in response to GPVI stimulation. Our present study demonstrates that depleting GPx1/catalase augments the inside–out activation of integrin αIIbβ3 in response to GPVI stimulation, which is consistent with a previous study that showed integrin αIIbβ3 activation is accompanied by a concomitant increase in ROS production in convulxin-stimulated platelets (4, 7). Thus, the data suggest that intracellular H2O2 regulates integrin αIIbβ3 activation in GPVI-stimulated platelets. It is unclear whether the molecular mechanism of α-granule secretion and integrin activation is directly involved in SHP-2 oxidation following ROS production; however, our results strongly suggest the involvement of intracellular ROS in regulating P-selectin and integrin. Given that intracellular calcium contributes to the reorganization of the actin cytoskeleton, which is necessary for degranulation and integrin αIIbβ3 activation (43, 56), the mechanism via SHP-2 oxidation is partly explained by the increased intracellular calcium resulting from PLCγ2 activation following oxidative inactivation of SHP-2.
A study illustrating the link between antioxidant proteins and platelet aggregation reported that the platelet GPx and catalase activities in patients with coronary heart disease were significantly lower than those in healthy subjects, suggesting that GPx and catalase in platelets may affect thrombotic processes in patients (9). Our loss-of-function studies also revealed that GPx1 and catalase are physiological regulators of endogenous H2O2-mediated signaling in platelets. Furthermore, GPx1/catalase double deficiency accelerated the thrombotic response in injured carotid arteries, indicating that H2O2 promotes platelet activation through a redox-dependent pathway and that GPx1 and catalase play a preventive role in platelet-mediated arterial thrombosis.
In conclusion, the present study has identified SHP-2 as a biological target of collagen-induced ROS in platelets. Furthermore, the loss-of-function study with GPx1−/−Cat−/−platelets suggests that the redox regulation of SHP-2 modulates collagen-induced aggregation and oxidant-stimulated arterial thrombosis; these results imply that increased ROS levels during inflammation or vessel damage may promote platelet aggregation and vascular thrombosis. Further studies on the signaling pathways regulated by ROS and antioxidant systems will help to develop a strategy for preventing platelet hyperactivation under pathological oxidative conditions.
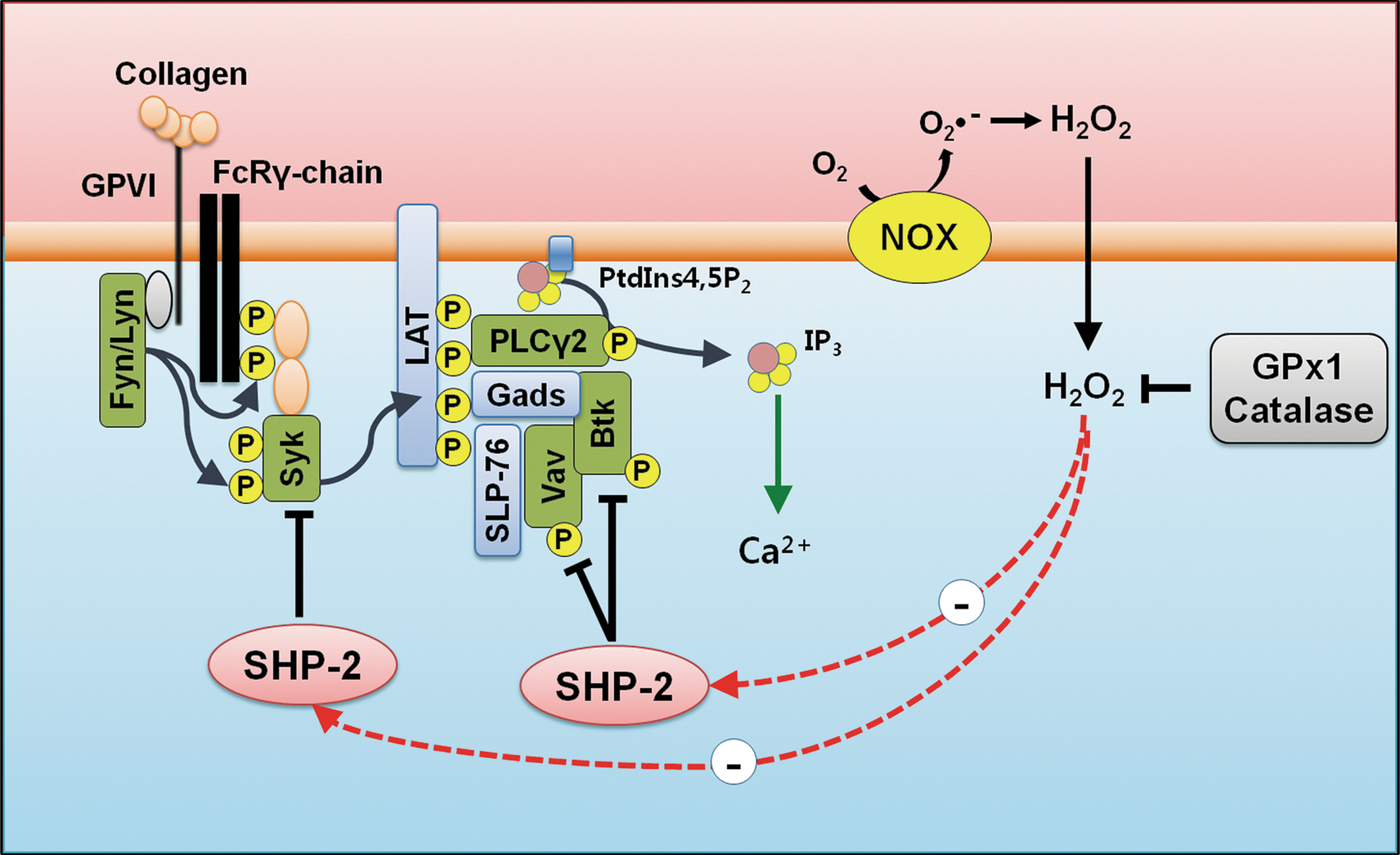
In summary, SHP-2 is oxidized in platelets by ROS produced upon collagen stimulation. The oxidative inactivation of SHP-2 leads to the enhanced tyrosine phosphorylation of Syk, Vav1, and Btk in the LAT signaling complex, which promotes the tyrosine phosphorylation-mediated activation of PLCγ2, which subsequently leads to increase in cytosolic calcium levels, platelet aggregation, and thrombosis. Antioxidants, GPx1 and catalase, decrease cellular H2O2 levels and SHP-2 oxidation in response to collagen. Finally, it leads to attenuation in platelet aggregation and thrombosis through decrease of PLCγ2 activity and intracellular calcium levels (Fig. 6).
Materials and Methods
Antibodies and reagents
CM-H2DCFDA and Fluo-3AM were from Molecular Probes. Monoclonal antibodies to SHP-2 and anti-CD62P-FITC were from BD-Biosciences. JON/A-PE was from Emfret Analytics. Polyclonal antibodies to GPx1 and catalase were from LabFrontier. Polyclonal antibodies to SHP-2, Btk, phospho-Vav (Tyr174), Syk, and monoclonal antibodies to α-tubulin were from Santa Cruz Biotechnology. Monoclonal anti-oxPTP antibody specific to the conserved active site of classical PTPs oxidized to the sulfonic acid (VHCSO3HSAG) (48) was from R&D Systems. Polyclonal antibody to phospho-Btk (Tyr551) was from Biosource. Polyclonal antibody to phospho-Syk (Tyr525/526) was from Cell Signaling Technology. Monoclonal antibodies to phosphotyrosine antibody (4G10) and Vav1, and polyclonal antibodies to ADAP, LAT, and Gads were from Upstate Biotechnology, Inc. Polyclonal antibodies to PLCγ2, phospho-PLCγ2 (Tyr753), and phospho-PLCγ2 (Tyr759) were kindly gifted by Dr. S.G Rhee (Ewha Womans University). Horseradish peroxidase (HRP)-conjugated streptavidin was from Pierce. Convulxin was from Alexis Biochemicals.
Experimental animals
C57BL/6 WT, glutathione peroxidase 1-deficient (GPx1−/−), catalase-deficient (Cat−/−), and GPx1/catalase double-deficient (GPx1−/−Cat−/−, Fig. 4A) mice on the C57BL/6 background (22, 23, 27) were housed under specific pathogen-free conditions at the Ewha Womans University. The animal handling and experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee. The animals were used from 6 to 8 weeks of age.
Human platelet preparation
Human blood was obtained from healthy drug-free volunteers and anticoagulated with acid/citrate/dextrose (22.0 g sodium citrate, 24.5 g dextrose, 7.3 g citric acid per 1 L). Platelet-rich plasma, which was obtained by centrifugation for 15 min at 150g, was further centrifuged for 10 min at 300g to concentrate the platelets. The platelet pellet was then suspended in a washing buffer containing Tyrode's buffer (10 mM HEPES [pH 7.4], 129 mM NaCl, 0.8 mM KH2PO4, 8.9 mM NaHCO3, 2.8 mM KCl, 0.8 mM MgCl2, and 5.6 mM glucose), 2 mM EDTA, 10% acid citrate dextrose solution, and 1 μM PGE1 and was centrifuged again. The platelets were finally resuspended at a concentration of 3×108 platelets/ml in Tyrode's buffer. Purity of the isolated human platelets was assessed by flow cytometry as shown in Supplementary Figure S8A. All of the studies using human platelets were approved by the Institute Faculty Ethics Committee at the Ewha Womans University.
Mouse platelet preparation
Mouse blood was collected from the abdominal aorta using a syringe containing 1 volume of acid/citrate/dextrose for 10 volume of blood under isofluran anesthesia. The blood was diluted with an equal volume of washing buffer. Platelet-rich plasma, which was obtained by centrifugation for 15 min at 50g, was further centrifuged for 10 min at 300g to concentrate the platelets. The platelet pellet was then suspended in washing buffer and spun once more. Platelets were finally resuspended at a concentration of 3×108 platelets/ml in Tyrode's buffer. Purity of the isolated mouse platelets was assessed by flow cytometry as shown in Supplementary Figure S8B.
Aggregation study
Washed platelets in Tyrode's buffer containing 0.35% bovine serum albumin were preincubated with 1 mM CaCl2 for 2 min before adding collagen, thrombin (Chrono-Log), U46619 (Calbiochem), or ADP (Sigma-Aldrich). Platelet aggregation was measured in a siliconized glass cuvette under continuous stirring at 1000 rpm at 37°C using a four-channel aggregometer (Chrono-Log).
Analysis of intracellular ROS and cytosolic calcium
Washed platelets suspended in phosphate-buffered saline (PBS) were incubated with 5 μM CM-H2DCFDA or 1 μM Fluo-3 AM for 15 min at 37°C in the dark. Then, the excess dye was removed, and the platelets were resuspended in Tyrode's buffer containing 1 mM CaCl2. After the dye-loaded platelets were stimulated with collagen, thrombin, U46619, or ADP under continuous stirring at 1000 rpm at 37°C, the intracellular ROS level at 495 nm excitation and 525 nm emission and the intracellular calcium level at 488 nm excitation and 525 nm emission were measured using a spectrofluorophotometer (Shimadzu).
Immunoblotting and immunoprecipitation
After stimulation, the platelets were lysed in cell extraction buffer (20 mM HEPES [pH 7.0], 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, 2 mM EGTA, 20 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 mM AEBSF). The cell debris was removed by centrifugation at 12,500g for 10 min. For the western blot and immunoprecipitation analyses, equal amounts of cell lysates were subjected to analysis using specific antibodies, as indicated.
Detection of oxidized SHP-2 with anti-oxPTP antibodies
As previously described (29, 48, 57), platelets were lysed using an oxygen-free buffer (50 mM HEPES [pH 6.5], 1% NP-40, 150 mM NaCl, 10% glycerol, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 mM AEBSF) with 100 mM iodoacetamide in an anoxic chamber (<1% oxygen; Thermo Forma). Cell debris was removed by centrifugation at 12,500g for 10 min. SHP-2 was immunoprecipitated with antibody specific for SHP-2 and protein G-Sepharose 4B (Amersham) at 4°C. The beads were washed three times in lysis buffer to remove excess iodoacetamide. Reversibly oxidized SHP-2 was reduced with 10 mM dithiothreitol for 30 min and then further washed three times in 20 mM HEPES (pH 7.4) at room temperature. To oxidize SHP-2 to sulfonic acid (-SO3H), the reduced SHP-2 was finally incubated with 100 μM pervanadate for 1 h at 4°C. Hyperoxidized SHP-2 (SHP-2-SO3H) was then detected by immunoblotting with anti-oxPTP antibody (R&D Systems).
Biotinylation to detect oxidized thiols in SHP-2
As previously described (32), platelets were lysed using the oxygen-free buffer in an anoxic chamber (<1% oxygen). Cell debris was removed by centrifugation. The lysates were incubated with 1 mM PEO-iodoacetyl-biotin (Pierce) for 3 h under continuous shaking at 800 rpm at room temperature in the dark. SHP-2 was immunoprecipitated with specific antibody and protein G-Sepharose 4B for 2 h at 4°C. Biotin incorporation into the immunoprecipitated proteins was detected by immunoblot analysis with (HRP)-conjugated streptavidin (Pierce).
Flow cytometry
After being stimulated with convulxin (25 ng/ml), the platelets were incubated with FITC-conjugated anti-P-selectin (CD62P-FITC, 0.5 mg/ml) or PE-conjugated anti-active-integrin αIIbβ3 (JON/A-PE, 0.5 mg/ml) for 5 min in the dark. The reaction was stopped by adding ice-cold PBS. A FACSCalibur flow cytometer (Becton Dickinson) was used for all of the analyses with a minimum of 5×104 cells per sample for each measurement. The surface expression of P-selectin and active-integrin αIIbβ3 on the platelets was measured at 530 (FL1) and 585 nm (FL2), respectively. The relative change in fluorescence was analyzed using WinMDI software.
Assessment of arterial thrombosis after ferric chloride exposure
Thrombosis was induced in mice using a previously described carotid artery injury model (15). After an intraperitoneal injection containing 1.0 ml/kg Zoletil (Virbac Animal Health Co.) and 0.7 ml/kg Rompun (Bayer Korea Co.) for anesthesia, the left common carotid artery was exposed. Vascular injury was induced by applying a filter paper (1×1 mm) that had been saturated with 20% FeCl3 proximal to the carotid artery. The blood volume changes in the carotid artery downstream of the injury site were measured by the photoplethysmography method using a minimized OxiPulse probe (Hurev, Inc.) in transmission mode (1). USB 6009 (National Instrument, Inc.) and LabVIEW 7.1 (National Instrument, Inc.) were used to acquire and analyze the changes in photoplethysmography waveforms. The amplitudes representing the differences between the peaks and valleys of each waveform were averaged to obtain the mean blood volume changes in the carotid artery. The time to thrombotic occlusion was defined as the time required for >90% loss of the initial blood volume.
Ex vivo flow chamber assay
Washed platelet (1×108/ml) was incubated with 1 μM of the fluorescent dye DiOC6 (Sigma-Aldrich) for 10 min at 37°C as described (42). Collagen-coated coverslip (Neuvitro) was mounted on custom-made flow chamber (Chamlide CF, from LCI Korea). The fluorescently labeled platelet was then perfused over a matrix of collagen at 150 s−1 using a syringe pump (Harvard Apparatus, Inc.) (11). Nonadherent platelets in the chamber were washed with PBS. Adherent platelets were fixed with cold 4% paraformaldehyde for 15 min and then washed with PBS. Thrombus formation was visualized with a 40×long working distance objective for confocal microscopy (Nikon A1R). Flow chamber surface coverage by the thrombi was calculated using ImageJ software.
Statistical analysis
For the statistical analysis, all of the experiments were repeated at least three times, and the data were analyzed using Student's t-test.
Footnotes
Acknowledgments
We thank Dr. H.A. Woo for catalase knockout mice. This study was supported by Bio R&D Program (M10642040002-07N4204-00210 to T.-S.C.) and Basic Science Research Program (2007-0053827 to T.-S.C.) from the National Research Foundation (NRF) grant funded by the Korea government (Ministry of Education, Science and Technology [MEST]), the Brain Korea 21 Scholars Program (to J.Y.J., J.H.M., Y.H.C., J.Y.B., S.B.W., and S.J.P.) and the Brain Korea 21 Plus Program (to J.Y.J. and S.B.W.).
Author Disclosure Statement
The authors declare no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.