Aims: Protein tyrosine phosphatases (PTPs) play an important role in regulating a wide range of cellular processes. Understanding the role of PTPs within these processes has been hampered by a lack of potent and selective PTP inhibitors. Generating potent and selective probes for PTPs remains a significant challenge because of the highly conserved and positively charged PTP active site that also harbors a redox-sensitive Cys residue. Results: We describe a facile method that uses an appropriate hydroxyindole carboxylic acid to anchor the inhibitor to the PTP active site and relies on the secondary binding elements introduced through an amide-focused library to enhance binding affinity for the target PTP and to impart selectivity against off-target phosphatases. Here, we disclose a novel series of hydroxyindole carboxylic acid-based inhibitors for receptor-type tyrosine protein phosphatase beta (RPTPβ), a potential target that is implicated in blood vessel development. The representative RPTPβ inhibitor 8b-1 (L87B44) has an IC50 of 0.38 μM and at least 14-fold selectivity for RPTPβ over a large panel of PTPs. Moreover, 8b-1 also exhibits excellent cellular activity and augments growth factor signaling in HEK293, MDA-MB-468, and human umbilical vein endothelial cells. Innovation: The bicyclic salicylic acid pharmacophore-based focused library approach may provide a potential solution to overcome the bioavailability issue that has plagued the PTP drug discovery field for many years. Conclusion: A novel method is described for the development of bioavailable PTP inhibitors that utilizes bicyclic salicylic acid to anchor the inhibitors to the active site and peripheral site interactions to enhance binding affinity and selectivity. Antioxid. Redox Signal. 20, 2130–2140.
Introduction
Protein tyrosine phosphorylation is a major post-translational modification by which many cellular pathways are regulated. In living cells, the proper level of protein tyrosine phosphorylation is fine-tuned and dynamically controlled by the balanced activity of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs), which are crucial for cell growth, differentiation, metabolism, motility, and death (16, 29). Understandably, perturbation of this balance leads to many human diseases, including diabetes, cancer, and immune dysfunctions (16, 17, 27, 39), implicating both enzyme classes as highly attractive targets for the development of novel therapeutics (8, 27). Indeed, nearly two dozen of small-molecule drugs targeting PTKs have been approved for the clinic over the past decade, and many more are undergoing clinical trials (7). However, the therapeutic potential of modulating the activity of PTPs is underexplored despite the fact that several PTPs have been identified as highly promising targets (17, 39). This is rather surprising, as both PTKs and PTPs are equally important for the regulation of cellular functions.
Although the protein tyrosine phosphatases (PTPs) have been garnering attention as potential therapeutic targets, they remain largely an untapped resource. In fact, PTPbased drug discovery programs have historically been shrouded with difficulty in inhibitor selectivity and bioavilability. This study describes a chemistry platform based on bicyclic salicylic acid pharmacophores that are sufficiently polar to bind the PTP active site, yet remain capable of crossing cell membranes, offering PTP inhibitors with both high selectivity and cellular efficacy. Potent and specific PTP inhibitors could serve as valuable tools to illuminate the druggability of PTPs and may constitute valuable therapeutics for many human diseases.
Among the reasons that PTP-based drug discovery still remains an untapped resource is that the PTP active site presents a number of challenges to developing small-molecule inhibitors. First, the PTPs feature an invariant active site cysteine for nucleophilic catalysis (40). Due to the highly positively charged environment in the PTP active site, the side chain of this Cys residue has an extremely low pKa (∼5) (41) in comparison to that of a typical thiol group in proteins (∼8.5). Thus, at physiological conditions, the catalytic Cys exists as a thiolate anion, which not only enhances its nucleophilicity but also renders it susceptible to oxidation. The sensitivity of the active site Cys to oxidation makes the PTPs prime targets of stimulus-induced production of reactive oxygen species (ROS) (2, 9, 20, 24), which inactivate the PTPs, thereby potentiating tyrosine phosphorylation-mediated signaling (14, 19, 25, 33). Thus, the oxidative inactivation of the PTPs by ROS offers a dynamic mechanism of PTP regulation. Unfortunately, the reactivity of the active site Cys also causes serious problems in high throughput screening campaigns that are aimed at the discovery of PTP inhibitory agents, as highly reactive oxidizing and alkylating agents often surface as hits (4, 30), which are undesirable as therapeutics due to poor safety and selectivity profiles. Furthermore, because of the positively charged PTP active site evolved to complement the negatively charged pTyr substrate, the brute-force screening of large compound collections usually leads to the identification of negatively charged molecules that suffer from poor bioavailability. Finally, the PTP active site is structurally very well conserved, so it is not trivial to obtain compounds that can inhibit single PTPs with good selectivity.
One way of circumventing the inhibitor specificity issue is through focused library approaches that link a nonhydrolyzable pTyr mimetic to appropriately functionalized moieties to engage both the PTP active site and its nearby peripheral binding pockets which are less conserved (28, 40). To improve the pharmacological properties of PTP inhibitory agents, we discovered that bicyclic hydroxybenzofuran and hydroxyindole carboxylic acid scaffolds can serve as effective nonhydrolyzable pTyr mimics and are sufficiently polar to bind the PTP active site, yet remain capable of crossing cell membranes (35, 38, 42). We demonstrated that hydroxybenzofuran or indole-based carboxylic acids bind the PTP active site in an analogous fashion as pTyr, but additional diversity elements attached to the benzofuran/indole core interact with unique secondary pockets in the vicinity of the active site and, therefore, render the inhibitors PTP isozyme selective. Click chemistry was initially utilized to tether an alkyne-containing hydroxybenzofuran/indole carboxylic acid scaffold with a large number of azide-containing amines and hydrazines in order to target adjacent secondary binding pockets in the vicinity of the PTP active site. This led to the identification of several PTP inhibitors (35, 38, 42). However, despite the highly efficacious cellular activity, the potency and selectivity displayed by these inhibitors are relatively modest, and, therefore, are not adequate for chemical biological investigation and therapeutic development. The reasons that the Click chemistry approach did not produce better results may be as follows: (i) the hydroxybenzofuran/indole carboxylic acid cores were not optimized for the intended target PTPs; (ii) the triazole linker resulting from the Click reaction was not most favorable for ligand interactions with the PTPs; and (iii) the presence of cuprous ion used in the Click reaction may compromise on the quality of the identified hits.
To avoid the problems associated with the Click chemistry approach and to develop more potent and selective PTP inhibitors, we decide to first identify the most efficient hydroxyindole carboxylic acid core for the target PTP, which will then be combined with a large and diverse number of carboxylic acids through the well-established amide chemistry in order to target peripheral pockets in the vicinity of the active site. We describe the application of this strategy to the development of potent and selective inhibitors of receptor-type tyrosine protein phosphatase beta (RPTPβ) with efficacious cellular activity.
RPTPβ, encoded by the PTPRB gene in humans, is expressed primarily in endothelial cells that form the protective lining of blood vessels. RPTPβ is composed of an extracellular domain with multiple fibronectin type III repeats, a single transmembrane segment, and one cytoplasmic catalytic domain (18). Mice expressing a truncated form of VE-PTP (mouse homolog of human RPTPβ) and VE-PTP null mice undergo vasculogenesis but die embryonically as a result of defects in angiogenesis, indicating an important role for VE-PTP in blood vessel development (3, 10). RPTPβ can negatively regulate the activation of Tie-2, an endothelial cell-specific receptor PTK, whose activation has been implicated in the development of collateral blood vessels that restore blood flow to ischemic tissue, vascular vessel maintenance, and integrity (6, 11, 12, 32). Antibodies against the extracellular domain of RPTPβ mimic the effects of VE-PTP gene deletion, triggering the activation of Tie-2 and leading to enhanced endothelial cell proliferation and enlargement of vascular structures through activation of Erk1/2 (31). A nonspecific phosphatase inhibitor bis(maltolato)-oxovanadium (IV) that inhibits RPTPβ augments collateral blood flow in a rat model of vascular insufficiency (5). All together, these data suggest that RPTPβ plays an important role in blood vessel growth and maintenance, and RPTPβ may be a potential target for tumor growth, occlusive cardiovascular disease, vascular leaking syndrome, and other vascular-related diseases. Understandably, there is intense interest in targeting RPTPβ for therapeutic development. However, although several RPTPβ inhibitors have been reported (1, 15, 21, 26), none exhibit activity inside the cell.
Results
We previously identified a hydroxyindole carboxylic acid as a pTyr mimic for PTPs. Through Click chemistry, a peripheral binding element was attached to the hydroxyindole carboxylic acid core, yielding an SHP2 inhibitor (II-B08) with highly efficacious activity in a number of cell lines as well as in a mouse model bearing KIT/D814V-induced mast cell leukemia (22, 38). Given the favorable pharmacological properties of hydroxyindole carboxylic acid-based PTP inhibitors, we decided to take a focused library approach to transform the hydroxyindole carboxylic acid scaffold into a highly potent and selective RPTPβ inhibitor with excellent cellular activity.
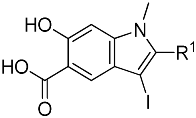
To identify a suitable hydroxyindole carboxylic acid core for RPTPβ, we introduced a diverse series of substituents to the 2-position of hydroxyindole carboxylic acid. Specifically, compounds 1a-l were designed, synthesized (37) and their inhibitory activity toward RPTPβ was evaluated at pH 7 and 25°C using p-nitrophenyl phosphate (pNPP) (Table 1). Compound 1a bearing a biphenyl group surfaced as the most potent hydroxyindole carboxylic acid core (IC50=3.6±0.8 μM) for RPTPβ. Unfortunately, compound 1a, bearing a biphenyl ring with no functional group on it, was not easily amendable for further chemical modification at the 2-position. However, compound 1f (IC50=25±5 μM), endowed with a free amino group, can be easily modified. As a result, we chose compound 1f as a starting core for further optimization.
IC50 Values (μM) of 1a-l for RPTPβ
Compd
R1
IC50 (μM)
1a
(1,1′-biphenyl)-4-yl
3.6±0.8
1b
Cyclohexyl
9.6±0.6
1c
4-CF3OPh
16±2
1d
4-CF3Ph
24±1
1e
Ph
24±2
1f
3-NH2Ph
25±5
1g
3-CF3Ph
27±2
1h
4-FPh
40±4
1i
3-FPh
56±5
1j
2-CF3Ph
70±10
1k
3-OHPh
90±20
1l
2-FPh
113±4
The IC50 values for RPTPβ were evaluated at pH 7.0 and 25°C using pNPP as a substrate.
RPTPβ, receptor-type tyrosine protein phosphatase beta; pNPP, p-nitrophenyl phosphate.
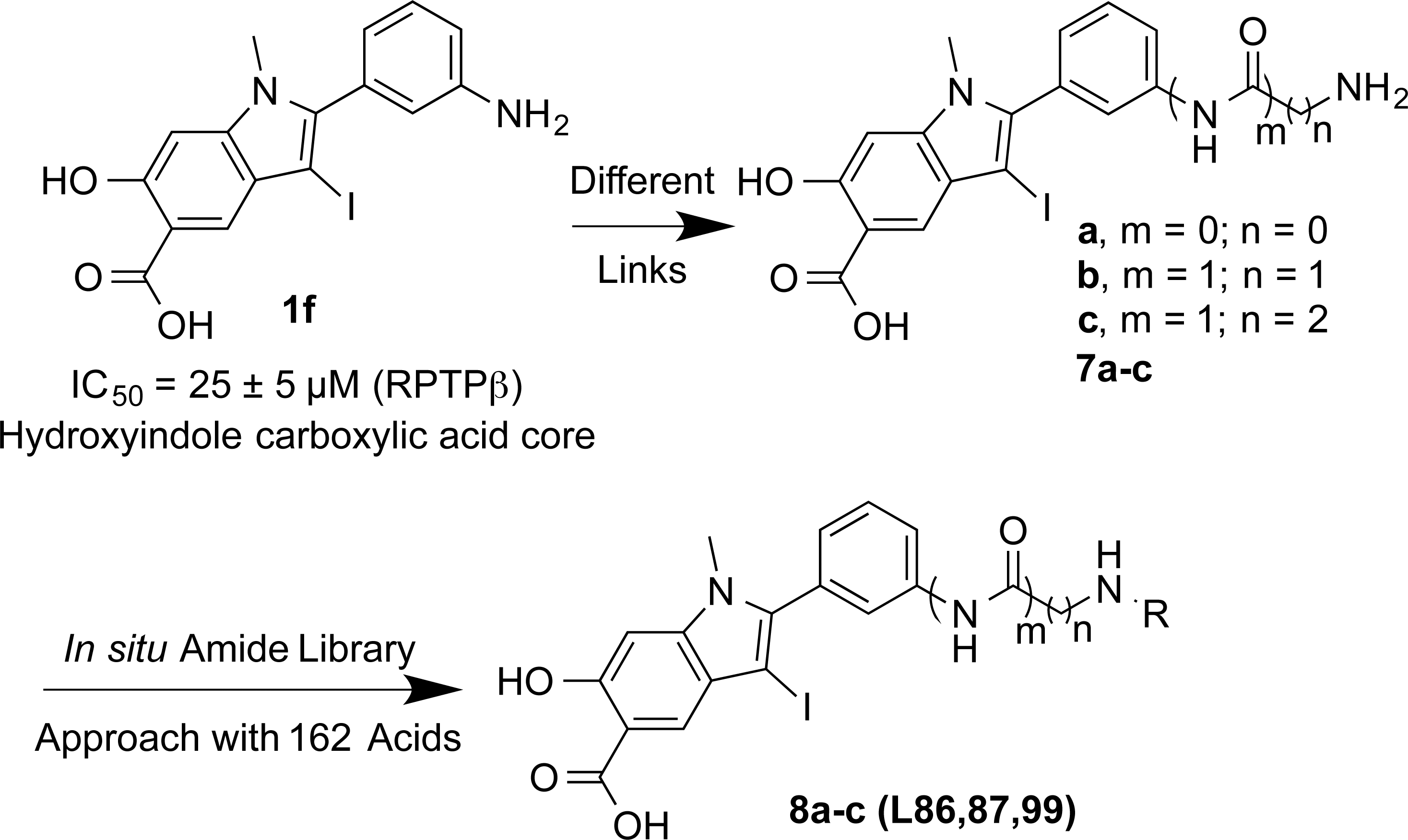
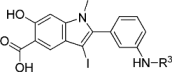
To increase the potency and selectivity of compound 1f for RPTPβ, we sought to introduce molecular diversity to the phenyl ring at the 2-position of the hydroxyindole carboxylic acid core in order to engage peripheral binding pockets adjacent to the active site (Fig. 1). As discussed earlier, the rationale originates from our earlier observations that diversity elements appended to the β-position of the core enhance both inhibitor potency and selectivity (35, 38, 42), likely through added interactions with secondary binding pockets adjacent to the PTP active site. However, the enhancement in binding affinity through this approach has been rather modest, likely due to the rigidity of the triazole linker formed as a result of the Click reaction. Indeed, no significant contact was observed between the triazole linker and the PTPs (35, 38). Here, we decided to employ amide chemistry, because amide bond formation is one of the most efficient and reliable methods for library constructions, and it enables the use of the most common and commercially available amines and carboxylic acids as reactants. In addition, amide chemistry can be carried out in solution in the absence of deleterious reagents, thus enabling direct screening and identification of hits from the library. Finally, appropriately structured amide linker may impart flexibility that is necessary for optimal interactions with the enzyme.
Strategy and design of hydroxyindole carboxylic acid-based RPTPβ inhibitor. RPTPβ, receptor-type tyrosine protein phosphatase beta.
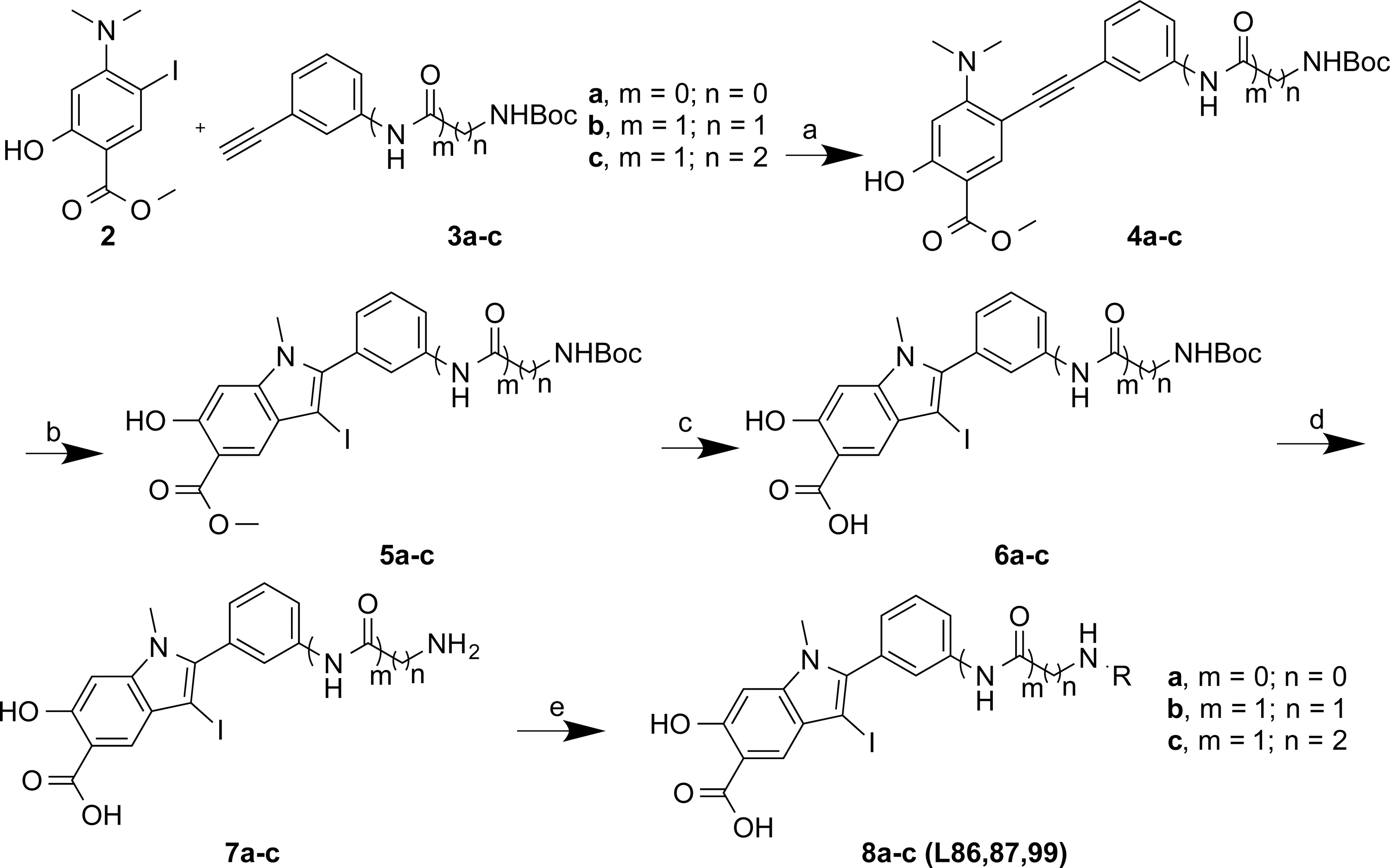
As shown in Figure 2, compound 2 methyl 4-(dimethylamino)-2-hydroxy-5-iodobenzoate (36, 37) was coupled with the corresponding alkynes 3a-c by Sonogashira coupling to afford compound 4a-c in high yield. Electrophilic cyclizations of 4a-c by I2 provided iodides 5a-c in 85%–95% yield. After hydrolysis of 5a-c in 5% LiOH for 2 days, 6a-c was obtained. 6a-c was treated with 10% of trifluoroacetic acid (TFA) in dichloromethane (DCM) for 1 h to afford 7a-c by high performance liquid chromatography (HPLC) in 35%–40% yield with more than 95% purity. Compounds 7a-c each contain an amine linker with a different length, attached to the meta-position of the phenyl group at the 2-position of the indole ring. A structurally diverse and commercially available set of 162 carboxylic acids were tethered to 7a-c to generate three focused libraries aimed at capturing additional interactions with adjacent pockets surrounding the active site. The amide libraries 8a-c were assembled in 96-well plates in the presence of HOBT, HBTU, and DIPEA in DMF. The reactions were randomly monitored by liquid chromatography–mass spectrometry (LC-MS). The products were determined to be at least 60%–80% yield and were used directly for screening without further purification.
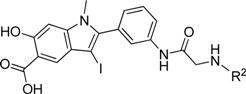
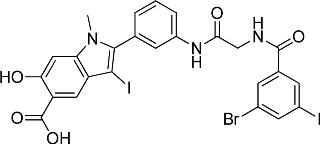
The ability of the library compounds to inhibit the RPTPβ-catalyzed hydrolysis of pNPP was assessed in situ at 10 μM. The linker size appears to be of significant importance because of the hits identified from the screen; the majority (24) were from library 8b, only five were from 8a, and none were from 8c. These initial hits were resynthesized, purified and their IC50 values for RPTPβ were determined (Tables 2 and 3). The most potent compound 8b-1 (L87B44), bearing a 3-bromo-5-iodo substituted phenyl, has an IC50 value of 0.38±0.02 μM for RPTPβ. Moreover, compound 8b-1 is 11 times more potent than the other di-halogen substituted phenyl molecules (e.g., 8b-7, 8b-13, and 8b-21). 3-Bromo or iodo substituted compounds are thrice more potent than 4-bromo or iodo substituted analogues. For example, we could compare 8b-15 (IC50=7.0 μM) versus8b-23 (IC50=32.0 μM) and 8b-17 (IC50=8.0 μM) versus8b-22 (IC50=28.0 μM). The IC50 values of the hits from library 8a range from 3.0 to 5.4 μM. Interestingly, compounds 8b-1 and 8a-4 have the same 3-bromo-5-iodobenzamido scaffold but different linkers; however, compound 8b-1 is 12 times more potent than 8a-4 (IC50=0.38 μMvs. 4.6 μM). We can draw the conclusion that both a proper linker and a scaffold can significantly influence the binding affinity of RPTPβ inhibitors.
IC50 Values (μM) of Hits Identified from Library 8b (L87) for RPTPβ
ID
R2
IC50 (μM)
ID
R2
IC50 (μM)
7b (Core 87)
H
94
8b-1 (L87B44)
0.38±0.02
8b-13 (L87B42)
6.1±0.8
8b-2 (L87A87)
1.99±0.06
8b-14 (L87A60)
6.4±1.7
8b-3 (L87A72)
2.2±0.2
8b-15 (L87A51)
7.0±0.8
8b-4 (L87A61)
2.7±0.1
8b-16 (L87A43)
7.8±0.8
8b-5 (L87B34)
3.4±0.3
8b-17 (L87A12)
8.0±1.0
8b-6 (L87A57)
3.4±0.4
8b-18 (L87A52)
8.0±1.0
8b-7 (L87B39)
4.3±0.2
8b-19 (L87B02)
8.2±1.5
8b-8 (L87A59)
4.9±0.1
8b-20 (L87A88)
9.6±1.6
8b-9 (L87A53)
5.1±0.2
8b-21 (L87A54)
14.5±7.0
8b-10 (L87A27)
5.2±0.5
8b-22 (L87B09)
28.0±4
8b-11 (L87A79)
5.3±0.6
8b-23 (L87B05)
32.0±4
8b-12 (L87B16)
5.6±0.4
8b-24 (L87A66)
40±20
The IC50 values for RPTPβ were evaluated at pH 7.0 and 25°C using pNPP as a substrate.
IC50 Values (μM) of Hits from Library 8a (L86) for RPTPβ
ID
R3
IC50 (μM)
7a (Core 86)
H
25.9±5.8
8a-1 (L86A45)
3.0±0.1
8a-2 (L86B34)
3.1±0.2
8a-3 (L86A53)
4.2±0.3
8a-4 (L86B44)
4.6±0.6
8a-5 (L86A37)
5.4±0.3
The IC50 values for RPTPβ were evaluated at pH 7.0 and 25°C using pNPP as a substrate.
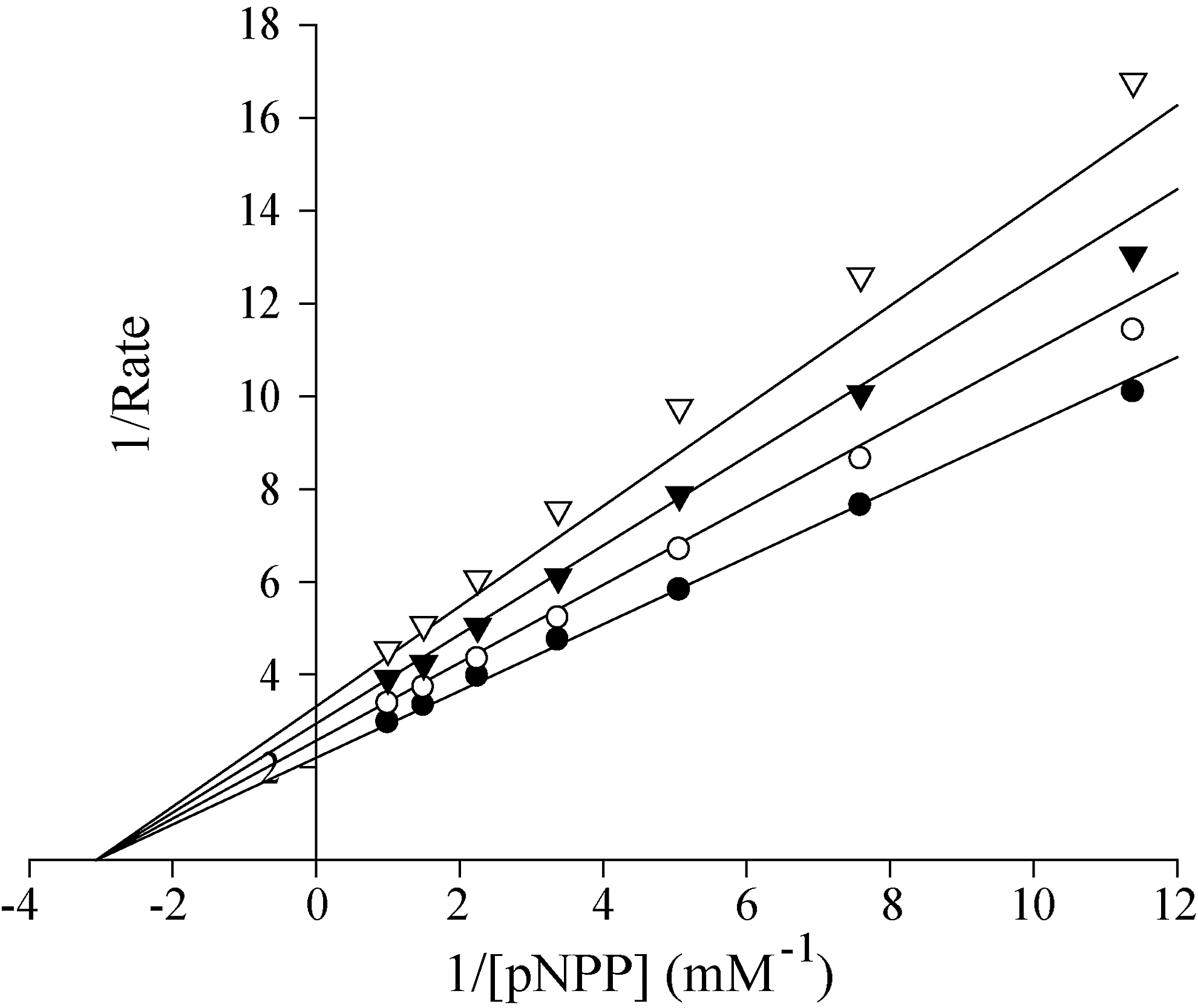
Further kinetic analysis indicated that 8b-1 is a reversible and noncompetitive inhibitor for RPTPβ with a Ki of 0.48±0.08 μM (Fig. 3). Selectivity profiling showed that compound 8b-1 displays more than 14-fold selectivity for RPTPβ over 18 other PTPs, including receptor-like PTPs, CD45, PTPμ, LAR PTPɛ, DEP1, PTPσ, and PTPγ, cytosolic PTPs, PTP1B, FAP-1, SHP2, PTP-PEST, and PTP-MEG2, the Yersinia PTP YopH, the CTD phosphatase SSu72, and the dual specificity phosphatase VHR, MKP5, VHZ, and Laforin (Table 4). Together, the results show that 8b-1 is among the most potent and specific of the RPTPβ inhibitors reported to date.
Lineweaver–Burk plot for Compound 8b-1 (L87B44) mediated RPTPβ inhibition. Compound 8b-1 concentrations were 0 (•), 0.1 μM (O), 0.2 μM (▼), and 0.3 μM (▿), respectively.
Selectivity of 8b-1 (L87B44) Against a Panel of PTPs
PTP
IC50 (μM)
RPTPβ
0.38±0.02
FAP-1
5.2±0.3
SHP2
5.6±0.5
VHR
5.8±0.4
PTP1B
5.5±1.6
PTP-PEST
7.4±1.0
YopH
8.5±0.8
CD45
8.6±0.8
PTP-MEG2
8.2±1.7
MKP5
>10
SSu72
9.7±1.2
VHZ
10±2
PTPμ
15±2
Laforin
16±3
LAR
>25
PTPɛ
>25
DEP1
23±4
PTPσ
32±2
PTPγ
60±20
The IC50 values for the PTPs were evaluated at pH 7.0 and 25°C using pNPP as a substrate. The kcat (s−1) and Km (mM) values for the PTPs at pH 7 and 25°C with pNPP as a substrate are RPTPβ (55.3 s−1 and 0.18 mM), FAP-1 (5.5 s−1 and 0.56 mM), SHP2 (6.7 s−1 and 3.6 mM), VHR (1.8 s−1 and 5.8 mM), PTP1B (19.9 s−1 and 1.6 mM), PTP-PEST (0.32 s−1 and 3.2 mM), YopH (24.6 s−1 and 2.1 mM), CD45 (5.6 s−1 and 4.9 mM), PTP-MEG2 (5.5 s−1 and 6.2 mM), MKP5 (1.4 s−1 and 12.7 mM), SSu72 (0.42 s−1 and 10.9 mM), VHZ (0.10 s−1 and 15.4 mM), PTPμ (0.61 s−1 and 2.8 mM), Laforin (0.36 s−1 and 3.1 mM), LAR (0.32 s−1 and 2.3 mM), PTPɛ (5.3 s−1 and 5.8 mM), DEP1 (331 s−1 and 2.2 mM), PTPσ (0.31 s−1 and 2.3 mM), and PTPγ (16.8 s−1 and 5.3 mM).
PTP, protein tyrosine phosphatase.
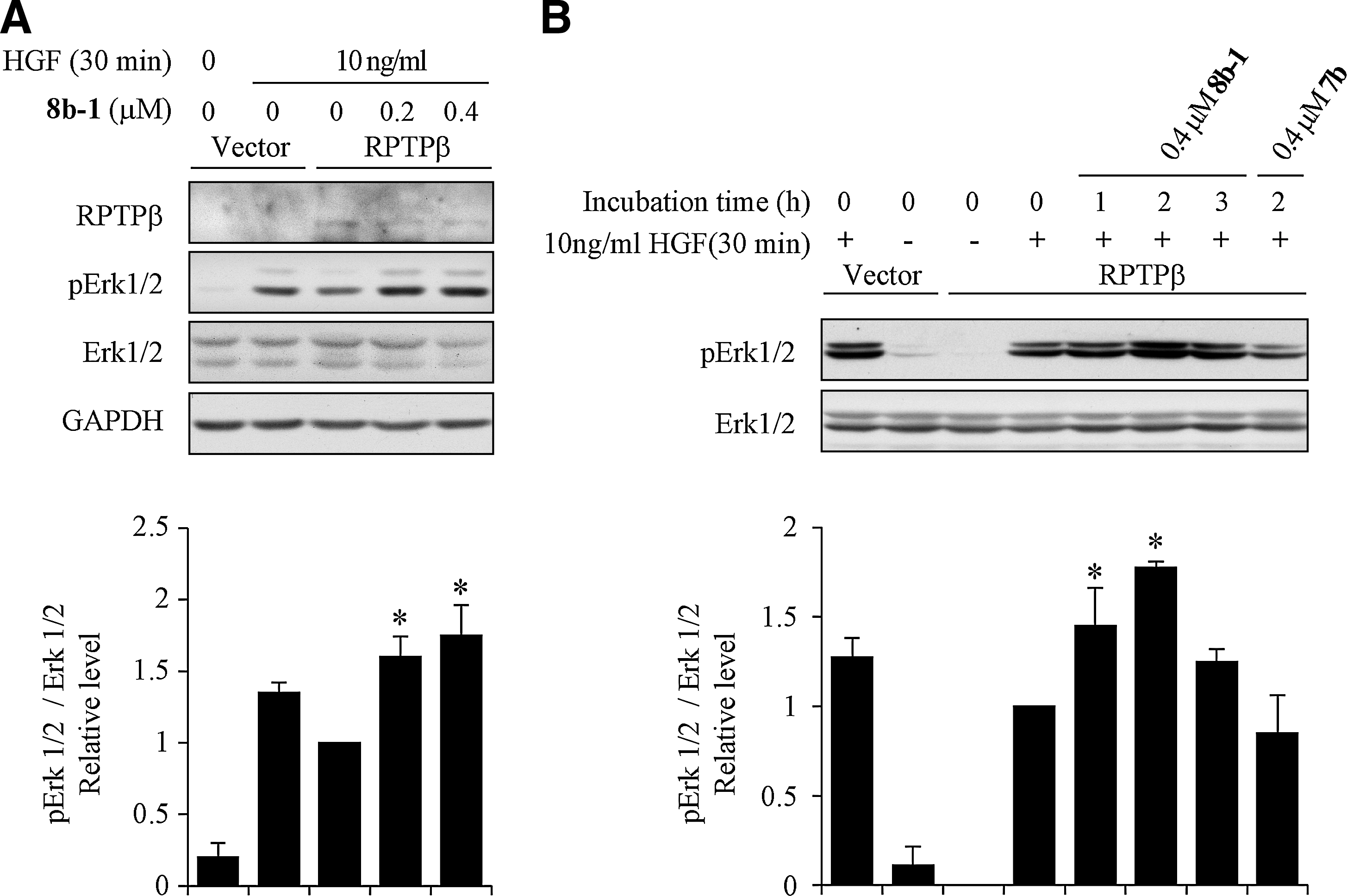
Given the excellent potency and selectivity of 8b-1 toward RPTPβ, we proceeded to evaluate its cellular efficacy. It has been reported that RPTPβ can suppress Met functions by inhibiting the Met-induced Erk1/2 phosphorylation in HEK293 cells (34). To assess the ability of compound 8b-1 to block the activity of RPTPβ inside the cell, we examined the effect of compound 8b-1 treatment on phospho-Erk1/2 signaling in HEK293 cells. Briefly, RPTPβ or vector-expressing HEK293 cells were starved overnight and treated with compound 8b-1 for 2 h, followed by stimulation with 10 ng/ml hepatocyte growth factor (HGF) for 30 min, or left unstimulated. Cell lysates were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and the levels of phospho-Erk1/2 were examined. As shown in Figure 4A, overexpression of RPTPβ reduces the phospho-Erk1/2 level, which is consistent with the reported finding that RPTPβ reduced HGF-induced Erk1/2 activation. As expected, treatment with RPTPβ inhibitor 8b-1 led to a concentration-dependent enhancement in Erk1/2 phosphorylation level (1.6±0.1 and 1.8±0.2-fold increase, respectively, at 0.2 and 0.4 μM compound 8b-1 concentrations). We also determined the optimal time required for the compound to take effect. RPTPβ-expressing cells were treated with 0.4 μM8b-1 for various times, and then stimulated with 10 ng/ml HGF for 30 min. As shown in Figure 4B, the treatment of cells with 8b-1 for 1 h leads to 1.5±0.2-fold increase in Erk1/2 phosphorylation. The enhancement of Erk1/2 phosphorylation peaks (1.8±0.1-fold) after treatment for 2 h, while prolonged treatment (3 h) only results in a modest increase in Erk1/2 activation (1.3±0.1-fold). Finally, to ensure that the cellular activity displayed by 8b-1 is not due to nonspecific effects, a structurally related but inactive compound 7b (Table 2, Core 87, IC50 of 94 μM against RPTPβ) was also evaluated. As expected, compound 7b showed no measurable effect on Erk1/2 activation (Fig. 4B, Lane 8) when it was incubated with RPTPβ-expressing cells for 2 h.
The cellular activity of compound 8b-1 (L87B44) on HGF-induced Erk1/2 activation in HEK293 cells. (A) Concentration-dependent enhancement of HGF-induced Erk1/2 activation by Compound 8b-1. (B) Time course of compound 8b-1 on HGF-mediated Erk1/2 activation. Bar graph represents the fold change of relative pErk1/2/Erk1/2 level. The results are shown as mean±SD (n=3). * Represents the significant difference of p<0.05 compared with the control that was stimulated with HGF without compound treatment. HGF, hepatocyte growth factor.
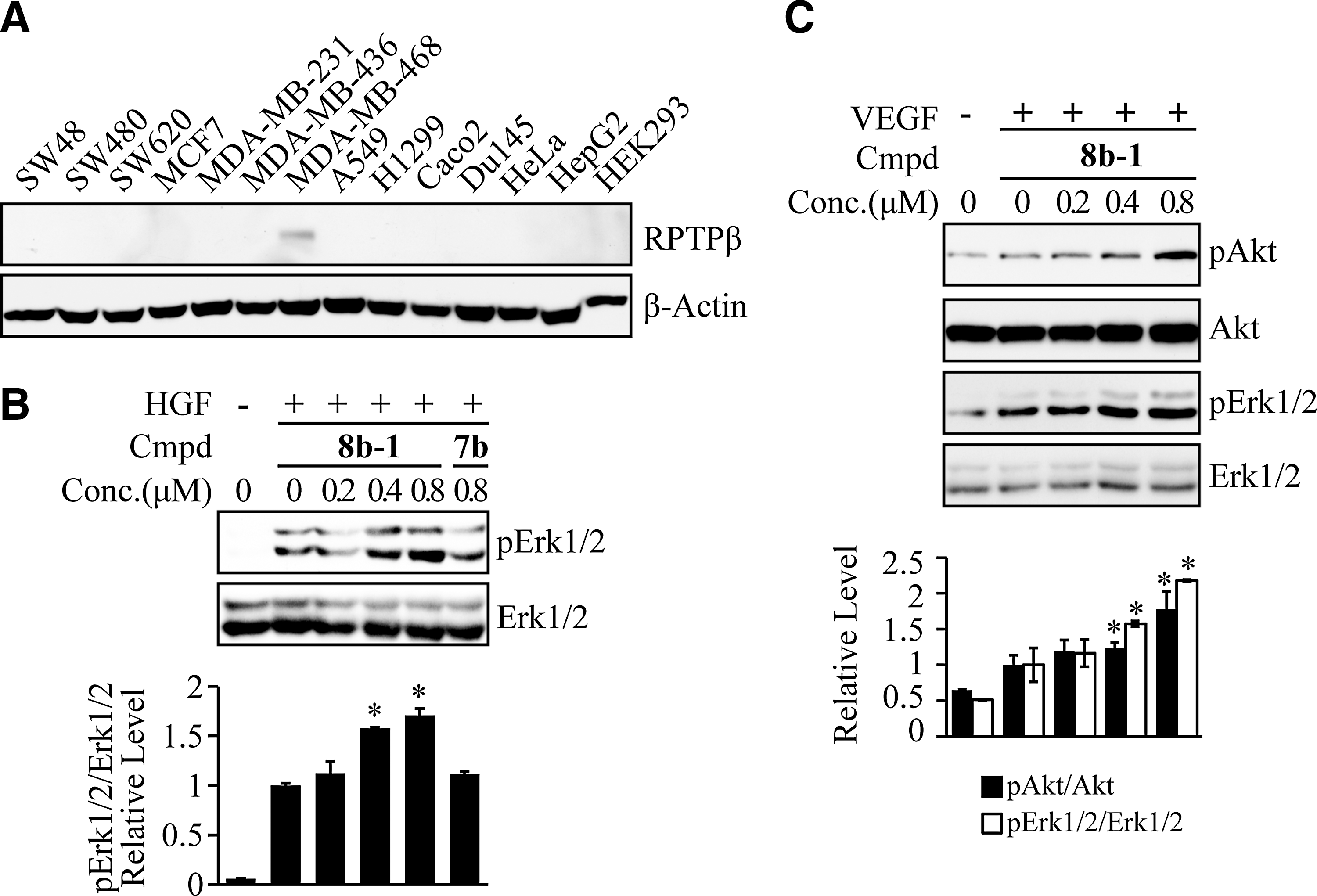
To determine whether 8b-1 could inhibit endogenous RPTPβ activity, we surveyed a number of tumor cell lines for RPTPβ expression. We found that RPTPβ is endogenously expressed in human metastatic breast adenocarcinoma MDA-MB-468 cells (Fig. 5A). To examine the effect of 8b-1 on Erk1/2 activation, MDA-MB-468 cells were serum starved overnight and treated with compound 8b-1 or a negative control 7b for 2 h, followed by stimulation with 10 ng/ml HGF for 30 min. Cell lysates were collected and resolved by SDS-PAGE, Phospho-Erk1/2 and total Erk1/2 level were determined by western blot. As shown in Figure 5B, treatment with RPTPβ inhibitor 8b-1 increased Erk1/2 activation in a dose-dependent manner (1.57±0.01 and 1.71±0.07-fold increase at 0.4 and 0.8 μM inhibitor 8b-1 concentration), while 0.8 μM7b had no effect on Erk1/2 activation, again suggesting that 8b-1 specifically inhibits RPTPβ activity and enhances HGF-induced Erk1/2 activation.
The cellular activity of compound 8b-1 (L87B44) on cells expressing endogenous PTPβ. (A) Endogenous PTPβ expression pattern in cancer cell lines. (B) Concentration-dependent enhancement of HGF-induced Erk1/2 activation by Compound 8b-1 in MDA-MB-468 cells. Bar graph represents the mean fold change of relative pErk1/2/Erk level. (C) Concentration-dependent enhancement of VEGF-induced Akt and Erk1/2 activation by Compound 8b-1 in HUVECs. Bar graph represents the fold change of relative pAkt/Akt and pErk1/2/Erk1/2 level. The results are shown as mean±SD (n=3). *Represents the significant difference of p<0.05 compared with the control that was stimulated with growth factor without compound treatment. PTP, protein tyrosine phosphatase; HUVEC, human umbilical vein endothelial cell; VEGF, vascular endothelial growth factor.
To further evaluate the effect of 8b-1 on RPTPβ-mediated cellular events in a more physiological setting, we note that RPTPβ expression is restricted to endothelial cells (3). In addition, RPTPβ has been shown to play a role in vascular endothelial growth factor (VEGF)/VEGFR signaling (13), and RPTPβ phosphatase activity is required for the de-activation of VEGFR-induced downstream Erk1/2 and Akt signaling pathways (23). To investigate whether RPTPβ inhibitor 8b-1 could enhance VEGFR signaling, we measured the VEGF-induced Erk1/2 and Akt activation in the presence of 8b-1 in human umbilical vein endothelial cells (HUVECs). HUVECs were serum starved overnight and treated with compound 8b-1 for 2 h, followed by stimulation with 20 ng/ml VEGF for 30 min. Cell lysates were collected and resolved by SDS-PAGE; phospho-Erk1/2 and phospho-Akt levels and total Erk1/2 and Akt levels were determined by western blot. As shown in Figure 5C, treatment with RPTPβ inhibitor 8b-1 at 0.4 μM leads to a 1.23±0.08-fold increase in Akt activation and a 1.57±0.04-fold increase in Erk1/2 activation, and this fold increase enlarged to 1.78±0.25-fold for Akt activation and 2.18±0.01-fold for Erk1/2 activation when 8b-1 concentration was increased to 0.8 μM, suggesting that 8b-1 significantly extended VEGFR activation in a dose-dependent manner. Taken together, the data show that 8b-1 is highly efficacious in cell-based assays, specifically inhibits RPTPβ activity, and increases HGF/Met signaling in both HEK293 and MDA-MB-468 cells and VEGF/VEGFR signaling in HUVECs.
Discussion
Protein tyrosine phosphorylation is an essential post-translational modification that modulates the function of proteins involved in many important signaling pathways. PTPs work in concert with PTKs to control the tyrosine phosphorylation status of target proteins. Not surprisingly, the activity of the PTPs is tightly regulated as a part of the cellular mechanisms for controlling the intensity and duration of responses to various stimuli. One important mechanism for the regulation of PTPs involves covalent modification of the PTP active site Cys thiolate group by endogenously produced ROS. Thus, the oxidative inactivation of the PTPs by ROS offers a dynamic mechanism of PTP regulation that can directly impact the level of tyrosine phosphorylation inside the cell. Thus, it is of great interest to understand the roles that PTPs play in redox signaling. Small-molecule inhibitors can be used as powerful research tools to interrogate the function of the PTPs in redox biology. Unfortunately, there is a general lack of potent, selective, and cell-permeable inhibitors for the PTPs. This study describes a novel approach toward the development of high-affinity and bioavailable PTP inhibitors. As a proof of concept, we report the most potent and selective inhibitor for RPTPβ, which has been implicated as a potential target for tumor growth and vascular-related diseases. Importantly, the RPTPβ inhibitor 8b-1 possesses highly efficacious cellular activity and inhibits its target in intact cells with similar potencies as those toward the isolated enzyme, whereas previous PTP inhibitors experience 100–10,000-fold loss of potency from biochemical to cellular assays. This inhibitor not only provides a potential lead for tumor and vascular-related diseases but also serves as a useful probe for redox signaling that is regulated by RPTPβ.
In conclusion, we report a method that uses an appropriate hydroxyindole carboxylic acid to anchor the inhibitor to the PTP active site and relies on the secondary binding elements introduced through an amide-focused library approach to enhance binding affinity for the target PTP and to impart selectivity against off-target phosphatases. Using the method, we disclose a novel series of hydroxyindole carboxylic acid-based inhibitors for RPTPβ, which has been implicated in blood vessel development. The representative RPTPβ inhibitor 8b-1 has an IC50 of 0.38 μM and at least 14-fold selectivity for RPTPβ over a large panel of PTPs. Moreover, compound 8b-1 also exhibits excellent cellular activity and can increase HGF-induced Erk1/2 phosphorylation in both HEK293 and MDA-MB-468 cells. Importantly, 8b-1 can also inhibit RPTPβ activity and augment VEGF/VEGFR signaling in HUVECs. Thus, compound 8b-1 serves as a promising candidate for the development of novel agents for tumor growth, occlusive cardiovascular disease, vascular leaking syndrome, and other vascular-related diseases. The results give further support that the bicyclic salicylic acid pharmacophore chemistry platform may provide a potential solution to overcome the inhibitor specificity and bioavailability issues that have plagued the PTP drug discovery field for many years. Given the ease and versatility of the amide chemistry-based library approach, we expect that the strategy can be used to identify cell-permeable, potent, and selective inhibitors for other members of the PTP superfamily.
Experimental Section
Materials and general procedures
pNPP was purchased from Fluke Co. For organic synthesis, reagents were used as purchased (Aldrich, Acros, Alfa Aesar, TCI), except where noted. 1H and 13C NMR spectra were obtained on Brucker 500 spectrometers with TMS or residual solvent as a standard. All column chromatography was performed using Dynamic Adsorbents 230–400 mesh silica gel (SiO2) with the indicated solvent system unless otherwise noted. TLC analysis was performed using 254 nm glass-backed plates and visualized using UV light (254 nm); low-resolution mass spectra and purity data were obtained using an Agilent Technologies 6130 Quadrupole LC/MS. High-resolution mass spectrum data were collected on Agilent 6520 Accurate-Mass Q-TOF LC/MS. HPLC purification was carried out on a Waters Delta 600 that was equipped with a Sunfire Prep C18 OBD column (30×150 mm, 5 μm) with methanol-water (both containing 0.1% TFA) as a mobile phase. The preparative HPLC gradient started at 50% methanol in water and ended at 100% methanol after 40 min with 0.1% of TFA. The purity of all final tested compounds was established to be >95% by Agilent Technologies 6130 Quadrupole LC/MS (UV, λ=254 nm). To analyze compound purity, the analytical HPLC gradient started at 0% methanol in water and ended at 100% methanol after 8 min with 0.1% of TFA. Details on compound synthesis and characterization are described in Supplementary Data (Supplementary Data are available online at www.liebertpub.com/ars).
Protein expression and purification
Briefly, RPTPβ was used to transform into Escherichia coli BL21/DE3 and grown in LB medium containing 50 μg/ml kanamycin at 37°C to an OD600 of 0.4. After the addition of IPTG to a final concentration of 20 μM, the culture was incubated at 20°C with shaking for an additional 16 h. The cells were harvested by centrifugation at 5000 rpm for 5 min at 4°C. The bacterial cell pellets were resuspended in 20 mM Tris, pH 7.9, 500 mM NaCl, and 5 mM imidazole, and were lysed by passage through a French press cell at 1200 psi twice. Cellular debris was removed by centrifugation at 16,000 rpm for 30 min at 4°C. The protein was purified from the supernatant using standard procedures of Ni-nitrilotriacetic acid-agarose (Qiagen) affinity purification. The protein eluted from Ni-NTA column was concentrated with an Amicon Ultra centrifugal filter device (Millipore), and the buffer was changed to 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT. Protein concentration was determined using the Bradford dye binding assay (Bio-Rad) diluted according to the manufacturer's recommendations with bovine serum albumin as a standard. The purified RPTPβ were made to 30% glycerol and stored at −20°C.
Inhibition study
The inhibition assays were performed at 25°C in 50 mM 3,3-dimethylglutarate buffer, pH 7.0, containing 1 mM EDTA with an ionic strength of 0.15 M adjusted by NaCl. The salicylic acid-based library was screened in a 96-well format at a 10 μM compound concentration. The reaction was started by the addition of 50 μl of the enzyme (20 nM) to 150 μl of reaction mixture containing the final Km value of pNPP and various concentrations of the inhibitor in a 96-well plate. The reaction was quenched after 2 min by the addition of 50 μl of 5 N NaOH, and then, this reaction mixture was detected for absorbance at 405 nm by a Spectra MAX340 microplate spectrophotometer (Molecular Devices). IC50 values were calculated by fitting the absorbance at 405 nm versus inhibitor concentration to the following equation:\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document} \begin{align*}A_1 / A_0 = { \rm IC}_{50} / ( { \rm IC}_{50} + [ { \rm I} ] ) ,\end{align*} \end{document}
where AI is the absorbance at 405 nm of the sample in the presence of inhibitor; A0 is the absorbance at 405 nm in the absence of inhibitor; and [I] is the concentration of the inhibitor.
The inhibition constants (Ki) for the inhibitor for RPTPβ were determined at pH 7.0 and 25°C. The mode of inhibition and Ki value were determined in the following manner. At various fixed concentrations of the inhibitor, the initial rate at a series of pNPP concentrations was measured by following the production of p-nitrophenol as describe earlier, ranging from 0.4- to 5-fold of the apparent Km values. The data were fitted to appropriate equations using SigmaPlot-Enzyme Kinetics to obtain the inhibition constant and to assess the mode of inhibition.
For selectivity studies, the PTPs, including the catalytic domains of FAP-1, PTP1B, SHP2, VHR, PTP-PEST, PTP-MEG2, YopH, CD45, VHZ, SSu72, LAR, PTPɛ, Laforin, DEP1, PTPμ, PTPσ, PTPγ, and MKP5, were expressed and purified from E. coli. The inhibition assay for these PTPs was performed under the same conditions (pH 7.0 and 25°C) as RPTPβ, except using a different pNPP concentration corresponding to the Km of the PTP studied.
Cell culture and immunoblot analysis
HEK293 cells were cultured at 37°C and 5% CO2 in Dulbecco's-modified Eagle's medium (DMEM) (Corning) that was supplemented with 10% fetal bovine serum (FBS) (HyClone). pShuttle mammalian expression vector subcloned with full-length human RPTPβ cDNA (a kind gift from Dr. Yiru Xu in the University of Michigan) or the empty vector was transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen). For biochemical studies, 24 h post-transfection, cells were serum starved overnight followed by treatment with vehicle or compound 8b-1 for an indicated time, and then left either stimulated or unstimulated with 10 ng/ml HGF (Sigma) for 30 min. MDA-MB-468 cells were grown in DMEM that was supplemented with 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin to 80% confluence, starved with serum-free medium overnight followed by treatment with vehicle, compound 8b-1 or 7b for 2 h, and then left either stimulated or unstimulated with 10 ng/ml HGF (Sigma) for 30 min. HUVECs were maintained in EBM-2 medium (Lonza) that was supplemented with endothelial growth medium 2 (EGM-2) kit (FCS, hydrocortisone, hFGF-B, VEGF, R3-IGF-1, ascorbic acid, hEGF, GA-1000, and heparin), according to the manufacturer's instructions, then serum starved overnight followed by treatment with vehicle or compound 8b-1 for 2 h, and left either stimulated or unstimulated with 20 ng/ml VEGF for 30 min. Cells were lysed in lysis buffer [1.0% Nonidet P-40, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 2 mM NaVO3, and 10 mM NaF] plus a protease inhibitor mixture (Roche) and clarified in a microcentrifuge. Lysates were electrophoresed on a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with antiphospho-Erk1/2 (Cell Signaling), anti-Erk1/2 (Cell Signaling), antiphospho-Akt (Cell signaling), anti-Akt (Cell signaling), anti-GAPDH (Santa Cruz), and anti-PTPβ (Santa Cruz) followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The blots were developed by the enhanced chemiluminescence technique using the SuperSignal West Pico Chemiluminescent substrate (Pierce). Data shown are a representation of multiple repeat experiments.
Footnotes
Acknowledgment
This work was supported in part by the National Institutes of Health Grant RO1CA152194 to Z.-Y.Z.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
1.
AmarasingheKKD, EvidokimovAG, XuK, ClarkCM, MaierMB, SrivastavaA, ColsonAO, GerweGS, StakeGE, HowardBW, PokrossME, GrayaJL, and PetersKG. Design and synthesis of potent, non-peptidic inhibitors of HPTPβ. Bioorg Med Chem Lett, 16: 4252–4256, 2006.
2.
BarrettWC, DeGnoreJP, KengYF, ZhangZY, YimMB, and ChockPB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem, 274: 34543–34546, 1999.
3.
BaumerS, KellerL, HoltmannA, FunkeR, AugustB, GampA, WolburgH, Wolburg-BuchholzK, DeutschU, and VestweberD. Vascular endothelial cell–specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood, 107: 4754–4762, 2006.
4.
BovaMP, MattsonMN, VasileS, TamD, HolsingerL, BremerM, HuiT, McMahonG, RiceA, and FukutoJM. The oxidative mechanism of action of ortho-quinone inhibitors of protein-tyrosine phosphatase alpha is mediated by hydrogen peroxide. Arch Biochem Biophys, 429: 30–41, 2004.
5.
CarrAN, DavisMG, Eby-WilkensE, HowardBW, TowneBA, DufresneTE, and PetersKG. Tyrosine phosphatase inhibition augments collateral blood flow in a rat model of peripheral vascular disease. Am J Physiol Heart Circ Physiol, 287: 268–276, 2004.
6.
ChaeJK, KimI, LimST, ChungMJ, KimWH, KimHG, KoJK, and KohGY. Coadministration of angiopoietin-1 and vascular endothelial growth factor enhances collateral vascularization. Arterioscler Thromb Vasc Biol, 20: 2573–2578, 2000.
7.
CohenP and AlessiDR. Kinase drug discovery—what's next in the field?. ACS Chem Biol, 8: 96–104, 2013.
8.
De MunterS, KohnM, and BollenM. Challenges and opportunities in the development of protein phosphatase-directed therapeutics. ACS Chem Biol, 8: 36–45, 2013.
9.
DenuJM and TannerKG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide, evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry, 37: 5633–5642, 1998.
10.
DominguezMG, HughesVC, PanL, SimmonsM, DalyC, AndersonK, Noguera-TroiseI, MurphyAJ, ValenzuelaDM, DavisS, ThurstonG, YancopoulosGD, and GaleNW. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci USA, 104: 3243–32482007.
11.
FachingerG, DeutschU, and RisauW. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the Angiopoietin receptor Tie-2. Oncogene, 18: 5948–5953, 1999.
12.
GaleNW and YancopoulosGD. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev, 13: 1055–1066, 1999.
13.
HayashiM, MajumdarA, LiX, AdlerJ, SunZ, VertuaniS, HellbergC, MellbergS, KochS, DimbergA, KohGY, DejanaE, BeltingHG, AffolterM, ThurstonG, HolmgrenL, VestweberD, and Claesson-WelshL. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun, 4: 1672, 2013.
14.
HenebergP, DráberováL, BambouskováM, PompachP, and DráberP. Down-regulation of protein-tyrosine phosphatases activates an immune receptor in the absence of its translocation into lipid rafts. J Biol Chem, 285: 12787–12802, 2010.
15.
HuangP, RamphalJ, WeiJ, LiangC, JallalB, McMahonG, and TangC. Structure-based design and discovery of novel inhibitors of protein tyrosine phosphatases. Bioorg Med Chem, 11: 1835–1849, 2003.
16.
HunterT. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol, 21: 140–146, 2009.
17.
JulienSG, DubéN, HardyS, and TremblayML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer, 11: 35–49, 2011.
18.
KaplanR, MorseB, HuebnerK, CroceC, HowkR, RaveraM, RiccaG, JayeM, and SchlessingerJ. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc Natl Acad Sci USA, 87: 7000–7004, 1990.
19.
KarischR, FernandezM, TaylorP, VirtanenC, St-GermainJR, JinLL, HarrisIS, MoriJ, MakTW, SenisYA, ÖstmanA, MoranMF, and NeelBG. Global proteomic assessment of the classical protein-tyrosine phosphatome and “Redoxome”. Cell, 146: 826–840, 2011.
20.
LeeSR, KwonKS, KimSR, and RheeSG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem, 273: 15366–15372, 1998.
21.
LundIK, AndersenHS, IversenLF, OlsenOH, MøllerKB, PedersenAK, GeY, HolsworthDD, NewmanMJ, AxeFU, and MøllerNPH. Structure-based design of selective and potent inhibitors of protein-tyrosine phosphatase β. J Biol Chem, 279: 24226–24235, 2004.
22.
MaliRS, MaP, ZengLF, MartinH, RamdasB, HeY, SimsE, GoshJ, NabingerS, LiS, MunugalavadlaV, CraigAW, BuntingKD, FengGS, ChanRJ, ZhangZY, and KapurR. Role of SHP2 phosphatase in KIT-induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood, 120: 2669–2678, 2012.
23.
MellbergS, DimbergA, BahramF, HayashiM, RennelE, AmeurA, WestholmJO, LarssonE, LindahlP, CrossMJ, and Claesson-WelshL. Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J, 23: 1490–1502, 2009.
24.
MengFG and ZhangZY. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim Biophys Acta, 1834: 464–469, 2013.
25.
MengTC, FukadaT, and TonksNK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell, 9: 387–399, 2002.
26.
Noren-MullerA, Reis-CorreaI, PrinzH, RosenbaumC, SaxenaK, SchwalbeHJ, VestweberD, CagnaG, SchunkS, SchwarzO, SchieweH, and WaldmannH. Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. Proc Natl Acad Sci USA, 103, 10606–10611, 2006.
27.
ÖstmanA, HellbergC, and BöhmerFD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer, 6: 307–320, 2006.
28.
PuiusYA, ZhaoY, SullivanM, LawrenceDS, AlmoSC, and ZhangZY. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proc Natl Acad Sci USA, 94: 13420–13425, 1997.
29.
TonksNK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol, 7: 833–846, 2006.
30.
WangQ, DubéD, FriesenRW, LeRicheTG, BatemanKP, TrimbleL, SangharaJ, PollexR, RamachandranC, GresserMJ, and HuangZ. Catalytic inactivation of protein tyrosine phosphatase CD45 and protein tyrosine phosphatase 1B by polyaromatic quinones. Biochemistry, 43: 4294–4303, 2004.
31.
WinderlichM, KellerL, CagnaG, BroermannA, KamenyevaO, KieferF, DeutschU, NottebaumAF, and VestweberD. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol, 185: 657–671, 2009.
32.
WrightMB, SeifertRA, and Bowen-PopeDF. Protein-tyrosine phosphatases in the vessel wall differential expression after acute arterial injury. Arterioscler Thromb Vasc Biol, 20: 1189–1198, 2000.
33.
XuD, RoviraII, and FinkelT. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell, 2: 251–252, 2002.
34.
XuY, XiaW, BakerD, ZhouJ, ChaHC, VoorheesJJ, and FisherGJ. Receptor-type protein tyrosine phosphatase β (RPTP-β) directly dephosphorylates and regulates hepatocyte growth factor receptor (HGFR/Met) function. J Biol Chem, 286: 15980–15988, 2011.
35.
YuX, SunJP, HeY, GuoXL, LiuS, ZhouB, HudmonA, and ZhangZY. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci USA, 104: 19767–19772, 2007.
36.
YueD and LarockRC. Synthesis of 3-iodoindoles by electrophilic cyclization of N,N-dialkyl-2-(1-alkynyl)anilines. Org Lett, 6: 1037–1040, 2004.
37.
ZengLF, XuJ, HeY, HeR, WuL, GunawanAM, and ZhangZY. A facile hydroxyindole carboxylic acid-based focused library approach for potent and selective inhibitors of mycobacterium protein tyrosine phosphatase B. ChemMedChem, 8: 904–908, 2013.
38.
ZhangX, HeY, LiuS, YuZ, JiangZX, YangZ, DongY, NabingerSC, WuL, GunawanAM, WangL, ChanRJ, and ZhangZY. Salicylic acid-based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2). J Med Chem, 53: 2482–2493, 2010.
39.
ZhangZY. Protein tyrosine phosphatases: prospects for therapeutics. Curr Opin Chem Biol, 5: 416–423, 2001.
40.
ZhangZY. Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol, 42: 209–234, 2002.
41.
ZhangZY and DixonJE. Active site labeling of the Yersinia protein tyrosine phosphatase: The determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry, 32: 9340–9345, 1993.
42.
ZhouB, HeY, ZhangX, XuJ, LuoY, WangY, FranzblauSG, YangZ, ChanRJ, LiuY, ZhengJ, and ZhangZY. Targeting mycobacterium protein tyrosine phosphatase B for anti-tuberculosis agents. Proc Natl Acad Sci USA, 107: 4573–4578, 2010.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.