
Abstract
Introduction
T
NADPH oxidase (Nox) and Dual oxidase (Duox) enzymes are an evolutionarily conserved family that has diversified to seven members in mammals (Nox1–5 and Duox1–2) (15, 80, 147). NOX enzymes typically catalyze the reduction of molecular oxygen (O2) to superoxide (O2 •−), the primary product of the enzymatic reaction in most cases (14, 89). Depending on the microenvironment or cellular compartment in which it is produced, spontaneous or superoxide dismutase (SOD)-catalyzed reduction of O2 •− to hydrogen peroxide (H2O2) may occur in association with the generation of other reactive oxygen species (ROS). ROS function as signaling molecules and regulators of cell function when they are generated in a compartmentalized and regulated manner (159). Here, we examine roles of these ROS-generating enzymes in normal cellular physiology of the lung and in the pathogenesis of selected lung diseases.
Biochemistry and Structure of Nox Enzymes
The Nox enzymes are encoded by seven genes in humans and six in mice (which lacks Nox5) (29, 89, 146). Nox1, Nox3, and Nox4 encode proteins that are similar in size and domain structure to Nox2. They consist of a C-terminal flavoprotein domain containing an NADPH-binding region and a flavin adenine dinucleotide binding region; the N-terminal hydrophobic domain consists of six transmembrane α helices that contain two heme-binding sites (90). Nox5 includes two major forms; the structure of the short form (Nox5-S) is similar to Nox1, Nox3, and Nox4, while the long form consists of the same domains along with an N-terminal extension containing a calcium-binding domain. Duox1 and Duox2 build on the Nox5 structure with an N-terminal extension consisting of a peroxidase homology region. Nox5, Duox1, and Duox2 are activated by calcium as predicted by their calcium-binding domains.
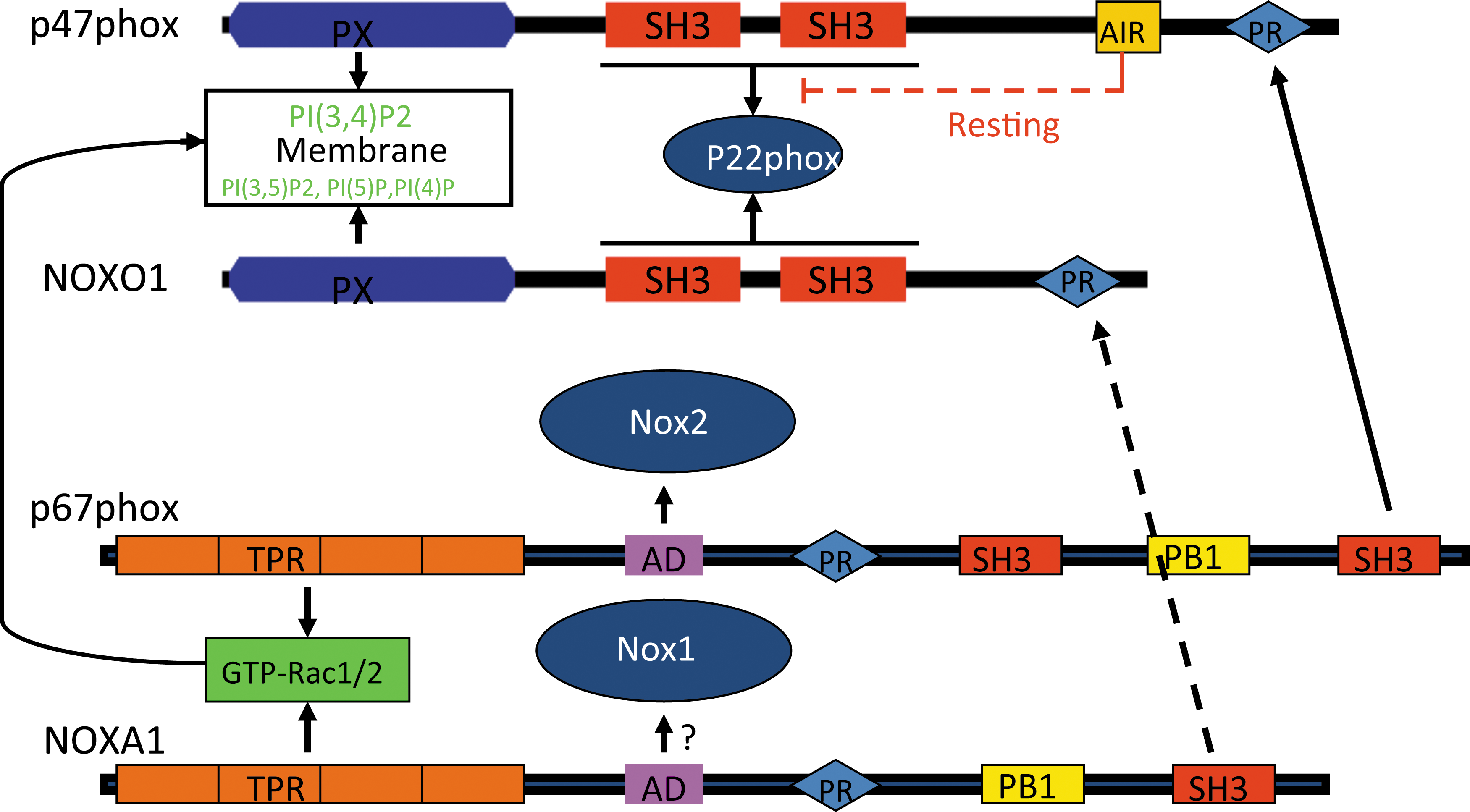
The activation of Nox2 by regulatory subunits has been extensively studied and has been reviewed elsewhere (14, 89). Nox1 is the first identified homolog of Nox2 (146). Nox1-dependent ROS generation can be reconstituted in cells by co-transfection with the regulatory subunits NoxO1 and NoxA1 (12, 30, 31, 152) (Fig. 1). Nox3 is primarily expressed in the kidney and inner ear (13, 29), although it may be induced in the lung (186). Similar to Nox1 and Nox2, it is also associated with p22 phox in biological membranes and is regulated by regulatory subunits. However, Nox3 activation reveals more flexibility. For example, NoxO1 alone is sufficient to activate Nox3; p67 phox further potentiates the effect of NoxO1 on Nox3 activation (32). The combination of NoxA1 and p47 phox may also mediate Nox3 activation, suggesting variable mechanisms for activation of Nox3. Nox4 is more ubiquitously expressed. Similar to Nox1, 2, and 3, it functionally associates with p22 phox and is generally considered a constitutively activated Nox enzyme. Interestingly, Nox4 is unique among the Nox1–5 isoforms in generating H2O2, not O2 •− (106, 136, 170 –172), although mechanisms for this and its biological significance remain to be elucidated.
Anatomic and Cellular Localization of Nox Enzymes
The lung is a complex organ and comprises more than forty cell types, including immune cells (18). It extends from the proximal conducting airways involving the trachea, bronchi, and bronchioles to the distal gas-exchanging alveolo-capillary units. Nox/Duox enzymes have been localized to various anatomic structures of the lungs and to specific cell types (Fig. 2).
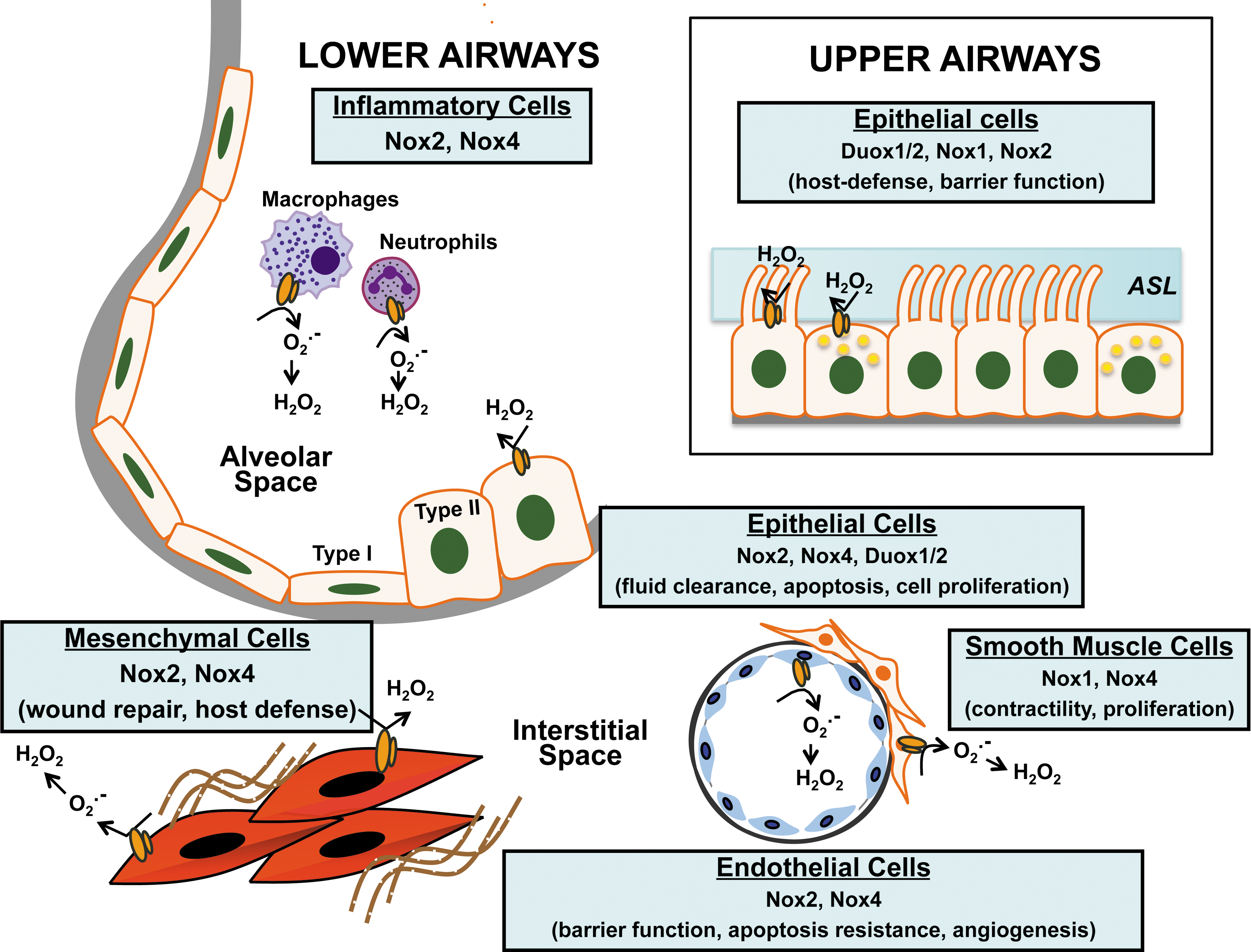
Nox expression and function in the upper respiratory tract
The combined action of resident immune cells and the secretory products of epithelial cells maintain the sterility of the airways. Nox/Duox enzymes participate in innate immunity and host defense of the lung (89). Duox1/2 localize to the surface of tracheal epithelial cells where they produce H2O2 and, in the presence of lumenal lactoperoxidase, generate antimicrobial hypothiocyanite (OSCN−) (128). A deficiency of this antimicrobial system has been implicated as an important host defense mechanism against Staphylococcus aureus and Pseudomonas aeruginosa infections in cystic fibrosis (115). This Duox-dependent antimicrobial system is induced by virulence factors such as flagellin (for Duox2), and the anti-inflammatory cytokines, interferon γ (INF-γ) (for Duox2), interleukin (IL)-4, and IL-13 (for Duox1) (68). In addition to microbicidal products such as OSCN−, the airway surface liquid (ASL) covering the upper airways contains complex polysaccharide mucins that trap pathogens and particulates from entering the lower airway. Secretion of Mucin-5 Subtype AC (MUC5AC) by human bronchial epithelial cells has been shown to be dependent on Duox1 activation by neutrophil elastase via PKCdelta/PKC (137); this supports Duox1 as a therapeutic target in chronic inflammatory airway diseases that are characterized by mucus hypersecretion. The ASL anti-microbial property depends partly on the maintenance of its pH. Secretion of H+ by tracheal epithelial cells has been shown to be mediated by an extracellular H2O2-dependent mechanism and is correlated with the expression of Duox1/2, p22 phox , p40 phox , p47 phox , and p67 phox (135). Viral infection of the airways triggers secretion of anti-inflammatory cytokines by immune cells and epithelial cells. Silencing of Nox2 expression/activity in human bronchial epithelial cells by small interfering RNA (siRNA) or the scavenging of ROS with antioxidants blocks the up-regulation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and cytokine production in response to the respiratory syncytial virus and the sendai-virus (51). In contrast, Nox1-derived ROS has been reported to contribute to barrier dysfunction and transmigration of rhinoviruses by disruption of the zona occludins of the apical tight junctions (33).
Nox expression and function in the lower airways
Two morphologically distinct cell populations constitute the alveolar epithelium: type I (which covers 95% of the epithelial surface) and type II cells. Efficient alveolar gas exchange is dependent on alveolar fluid clearance via the action of epithelial sodium channel (ENaC) that is expressed on the surface of type I and type II cells (46). Nox2/Rac1-dependent production of ROS has been shown to control the activity of ENaC in mouse and rat epithelial cells; lack (Nox2-deficient mice) or decreased Nox2 activity (pharmacologic blockade of Nox2 with Rac-1 inhibitor NSC23766) as well as ROS scavenging with TEMPO resulted in alveolar fluid retention in an in-vivo model of lipopolysaccharide-induced lung injury (63). Up-regulation of Nox2 and Rac-1 in the mouse alveolar epithelium after chronic exposure to alcohol has been linked to ENaC hyperactivity, suggesting a potential mechanism underlying the increased incidence of acute respiratory distress syndrome (ARDS) observed in the alcoholic population (47). In addition, Nox2 has been implicated in cell cycle control of alveolar epithelial cells; PPAR-γ activation through Nox2-derived ROS promotes cell-cycle progression from G0/G1 into S and G2/M phases (162). Nox4 expression is induced in alveolar type II cells in response to lung injury, and Nox4-derived ROS have been shown to induce apoptosis of alveolar epithelial cells and promote lung fibrosis (24). Duox1/2 are expressed by type II epithelial cells and Duox-generated H2O2 has been suggested to control acid release during lung development in mice (53, 54).
Nox expression by lung fibroblasts
The generation of ROS by NAD(P)H-like enzymes in fibroblasts was described well before the cloning and identification of the Nox gene family (108, 157, 158). The primary Nox isoform expressed in fibroblasts is Nox4, although p67 phox and p47 phox are also co-expressed in the absence of Nox2 (43). Nox4-derived ROS appears to mediate signaling events in cell that regulate IL-8 secretion (43), fibroblast migration (5), and myofibroblast differentiation (5, 37, 71). Deficiency or silencing of Nox4 protects against the development of experimental lung fibrosis in mice (24, 71), and this enzyme is also highly expressed in the lungs of patients with idiopathic pulmonary fibrosis (5, 71).
Nox expression in the pulmonary endothelium
The pulmonary endothelium serves as a tightly controlled barrier to prevent plasma exudation into the interstitium and alveolar space. The primary Nox isoforms expressed by vascular endothelial cells are Nox2 and Nox4. In P. aeruginosa lung infection, Nox4 and Nox2 play distinct roles in regulating lung inflammation, apoptosis, and permeability; Nox2 was critical in regulating inflammation, while Nox4 mediated apoptosis of endothelial cells and vascular permeability (58). Vascular cell adhesion molecule-1 signals activation of Nox2, which then mediates ROS-dependent activation of PKCα, protein tyrosine phosphatase 1B (PTP1B), and extracellular-signal-regulated kinases 1/2 (ERK1/2) and initiation of leucocyte migration (1, 34). Nox4-dependent ROS regulates endothelial cell motility and angiogenesis; RNAi-mediated silencing of Nox4 or pretreatment with N-acetylcysteine attenuates hyperoxia-induced endothelial cell migration and capillary tube formation (124). Hyperoxia has been shown to induce Nox4 expression via nuclear factor (erythroid-derived 2)-like 2 (Nrf2) binding to antioxidant response elements on the Nox4 promoter (125). Hyperoxia has also been shown to stimulate phosphorylation of myosin light chain (MLC) and to recruit phosphorylated and nonphosphorylated cortactin, MLC, Src, and p47 phox to caveolin-enriched microdomains (CEMs), which are essential for Nox activation (165). This process involves c-Abl-mediated dynamin 2 phosphorylation that is required for the recruitment of p47 phox to CEMs (139). Nox2-mediated ROS in pulmonary artery endothelial cells has been implicated in the induction of autophagy, which contributes to impaired angiogenesis in persistent pulmonary hypertension in fetal lambs (154).
Nox expression by pulmonary smooth muscle cells
Smooth muscles cells (SMCs) are found in the medial layer of the vasculature, where their primary function is to control pulmonary perfusion. SMCs are also found underlying the tracheal and bronchial epithelium, where their contraction is stimulated in response to inflammation. Several reports point to a key role of Nox4 in the contractility and proliferation of airway/vascular SMCs induced by pro-fibrotic cytokines or hypoxia (145, 148). Hypoxia induces Nox4 expression in SMCs via a hypoxia-inducible transcription factor HIF-1α (44, 112). Hypoxia-induced mitochondrial ROS production has been shown to activate protein kinase C-ξ (PKCξ) and Nox, providing a positive feedback mechanism to further increase intracellular ROS, calcium-induced calcium-release, and SMC contraction (129, 175). The pro-fibrotic cytokine, transforming growth factor-β1 (TGF-β1), induces the expression of Nox4 in human pulmonary artery SMCs via an Smad2/3-dependent pathway, which mediates ROS-dependent ERK1/2 phosphorylation and cellular proliferation (145).
Nox expression by immune cells
The prototypical Nox isoform, Nox2, has been well characterized in phagocytic cells as a critical component of the innate immune response [reviewed in Nauseef (117)]. However, in addition to its conserved role in combating invading pathogens, Nox2 appears to mediate additional (paradoxical) roles in suppressing inflammation (62, 181, 188). Nox1 and Nox4 are also expressed in monocyte/macrophage populations (164, 182). Metabolic stress (low-density lipoprotein and high D-glucose) induces Nox4 expression, which mediates monocyte chemotaxis in response to monocyte chemoattractant protein (MCP)-1, thereby contributing to vascular injury (93, 164). Further studies are required to determine the function of nonclassical Nox isoforms in immune cells.
In summary, Nox isoforms are expressed in the different compartments of the human lung, where they regulate several critical functions. The production of H2O2 by Duox/Nox enzymes by epithelial cells of the upper and lower airways as well as by immune cells is of primary importance in the lung host defense system. Indeed, H2O2 serves as a substrate to produce OSCN−, regulates chemotaxis and the production of other host defense molecules. Nox-generated H2O2 also contributes to airway epithelial cells proliferation and differentiation by activating signaling cascades that modulate gene transcription. Transcription factors such as NF-kB, p53, and activator protein 1 (AP-1), which are redox sensitive, provide the link between oxidative stress and gene expression. Finally, there is an emerging role of Nox enzymes in lung remodeling (ECM production, angiogenesis) by controlling mechanisms such as cell differentiation, motility, and apoptosis.
Subcellular Compartmentalization and Function of Nox Isoforms
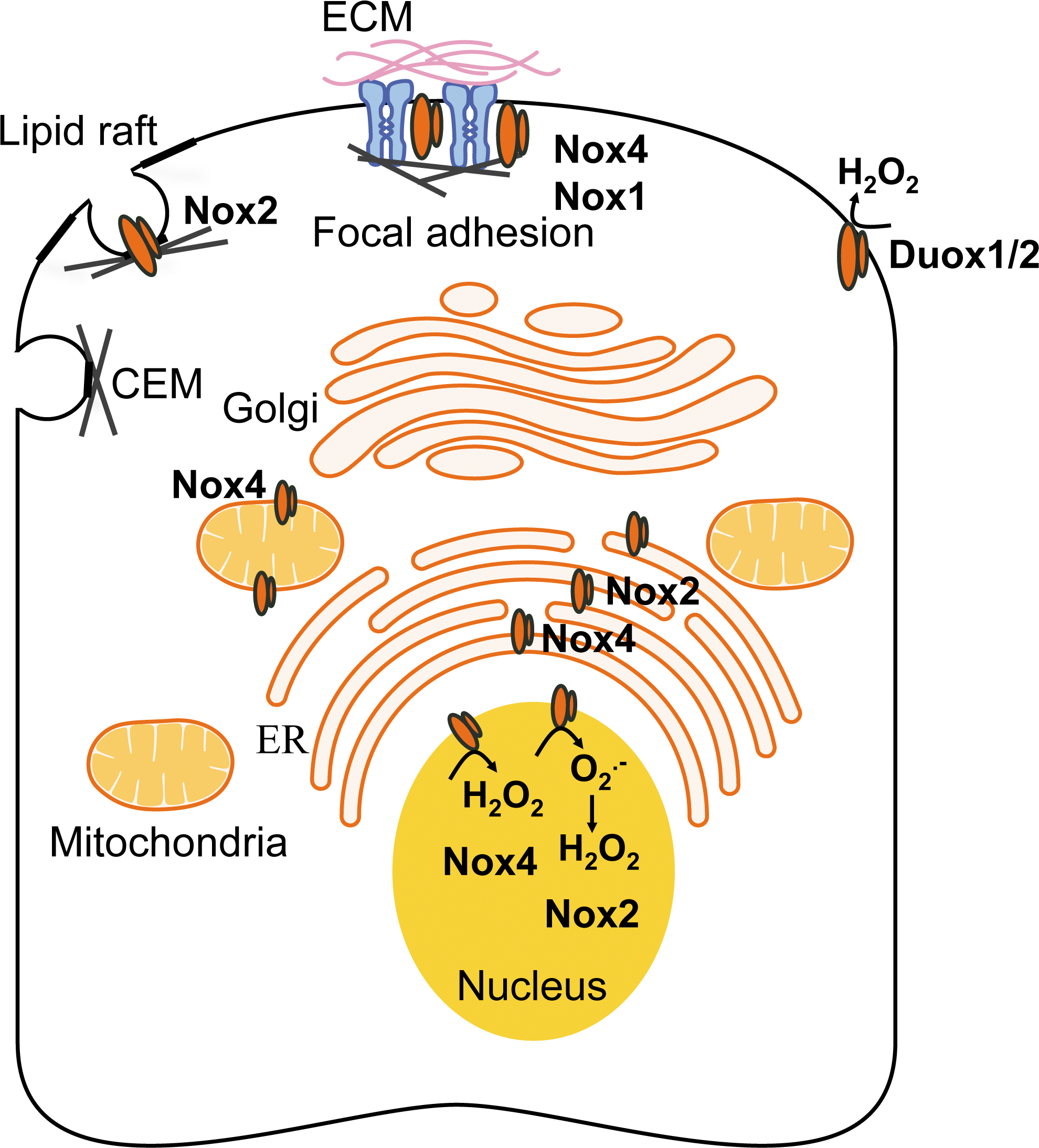
The signaling functions of Nox enzymes are likely to be controlled by their subcellular localization, which dictates signaling specificity, reactivity, and half lives of ROS (Fig. 3). The study of Nox subcellular localization remains challenging due to the lack of isoform-specific inhibitors. Chimera studies in which the N-terminus or the cytoplasmic tail of different Nox isoforms is interchanged indicate that the Nox amino-terminal tail likely determines Nox subcellular localization (73, 172).
Nox/Duox at the plasma membrane
Nox1, Nox2, and Nox4 have been localized to the plasma membrane. Their localization often associates with specific signaling domains, such as lipid rafts, caveolae, or focal adhesions, where they facilitate signaling of cellular proliferation, differentiation, and migration (166). For instance, lipid raft-generated ROS stimulate proliferation and migration of endothelial cells by activating vascular endothelial growth factor receptor-2 (VEGFR2) and downstream p38 mitogen-activated protein kinases (MAPK) activation (122). Nox2-generated ROS in caveolae regulates blood–brain barrier function through the modulation of occludin expression via an ERK1/2-dependent signaling cascade (123). Nox4 has been co-localized with vinculin at the site of focal adhesions in vascular SMCs; in association with polymerase delta-interacting protein 2 (Poldip2), Nox4 participates in stress fiber formation (102). Nox1-dependent ROS have been reported to regulate stress fiber assembly at the site of focal adhesions in Ras-transformed-Swiss3T3 fibroblasts (138).
The Duox isoform was first identified at the apical membrane of thyrocytes, where they generate H2O2, the substrate for follicular thyroperoxidase that catalyzes oxidization of iodide and formation of thyroxine (65, 114). Duox2 is mainly expressed by thyrocytes, while the apical surface of the airway epithelial cells is the primary expression site of Duox1 (54, 56). Production of Duox-generated H2O2 is necessary to confer anti-microbial properties to the ASL (135). Functional Duox1/2 also localize at the leading edge of migrating lung cancer or epithelial cells, suggesting a key role of Duox-generated H2O2 in metastasis and wound repair (101, 177).
Nox in the endoplasmic reticulum
The first evidence of an endoplasmic reticulum (ER) localization of Nox to the ER came from studies using overexpression of tagged or chimeric Nox in HEK293 cells that are designed to study Nox-antibodies specificity or domains within the Nox sequence which are responsible for their localization (183). A signaling function for ER Nox-dependent ROS production has been reported in the acute myeloid leukemia (AML) cell line, MV4–11, on stimulation of the tyrosine-like kinase receptor, Fms-like tyrosine kinase 3 (FLT3) (180). Mutations of the FLT3 are associated with the development of AML. Inhibition of the FLT3 receptor or knocking down the expression of p22 phox abolishes ER-generated H2O2, while preserving mitochondrial-ROS; ER-associated H2O2 production has been suggested to promote the synthesis of the proto-oncogene Pim-1 via signal transducer and activator of transcription 5 (STAT5) signaling, thus promoting the oncogenic process (180). In macrophages, the antimicrobial function of Nox2-depend ROS is executed, in part, by the translocation of Nox2 and p22 phox from intracellular compartments (ER, endosomes) to the plasma membrane after stimulation with INF-γ (27).
Several lines of evidence link ER stress-induced apoptosis to increased cytosolic production of ROS, although further studies are warranted to determine the precise source(s) of ROS (36, 96). Another proposed function of Nox-dependent ROS in the ER relates to the control of protein trafficking. Through inactivation of PTP1B, Nox4-dependent ROS negatively regulates the trafficking of the endothelial growth factor (EGF) receptor to the plasma membrane, thus terminating EGF signaling in endothelial cells (28). Cytosolic ROS also contributes to regulation of muscle contraction in skeletal muscle cells. Nox2, Nox4, and p22 phox have been reported to co-localize with transverse tubules and the sarcolemma in murine skeletal muscle cells; muscle contraction induces the recruitment of the activator and regulator subunits p67 and p40 phox and activates ROS-production (131).
Nox in the nucleus
Nox2 and Nox4 have been localized to the nucleus/peri-nuclear region of certain cell types. Nuclear Nox2-dependent ROS has been implicated in apoptosis of endothelial cell death that is triggered by exposure to homocysteine (140). In this study, 3D-digital imaging showed the accumulation of Nox2 and p47 phox to the nucleus of human umbilical vein endothelial cells on homocysteine in association with the formation of nitrotyrosine, suggesting O2 •− production, and induced cleaved-caspase-3 activity (140). In contrast, another study indicated Nox4, but not Nox2, in the nucleus of human pulmonary artery endothelial cells in which it mediated hyperoxia-induced cell migration and capillary tube formation (124). In murine embryo fibroblasts (NIH3T3 cells), nuclear Nox4 mediates TGF-β1-induced plasminogen activator inhibitor-1 (PAI-1) gene expression, at least in part through oxidative modification and inhibition of MAPK phosphatase-1 (MKP-1), a nuclear phosphatase (99).
Nox in the mitochondria
So far, Nox4 is the only member of the Nox gene family to have been reported functionally expressed in the mitochondria. Mitochondrial Nox4-produced ROS have been implicated in the etiology of several pathologies through the modulation of senescence, apoptosis, and oncogenicity (17, 64). A 73-amino-acid long domain within Nox4 amino-terminal tail, identified by sequence analysis using the Mitoprot program combined with mutagenesis (64), targets Nox4 to the mitochondria. One of the first evidence for the functional expression of mitochondrial Nox4 came from a study performed in a rat model of diabetes (17). Using subcellular fractionation assays combined with microscopy and measurement of mitochondrial activity, this study showed that Nox4 up-regulation in the cortex of diabetic rat kidney was linked to glucose-induced mitochondrial ROS (17). More recently, angiotensin II has been shown to increase mitochondrial Nox4-dependent ROS (O2 •− and H2O2) via mitochondrial membrane depolarization in a model of kidney tubular cells; this increase in mitochondrial ROS activated the intrinsic pathway of apoptosis with release of cytochrome c and apoptosis-inducible factor (82).
Mitochondrial Nox4 has been reported as a major source of O2 •− in the failing heart (3, 86); these studies employed cardiac-specific Nox4 conditional knockout mice (86) or cardiac Nox4 over-expressing mice (2). Up-regulation of Nox4-dependent mitochondrial O2 •− mediates cardiac myocyte apoptosis, fibrosis, and heart failure after pressure overload (86). Breast cancer has also been linked to oxidative stress; although Nox1, Nox4, and Nox5 are expressed in breast tissue, mitochondrial Nox4-dependent H2O2 production appears critical in the tumorigenic process (64). It remains to be determined whether Nox4 and potentially other Nox isoforms are localized to the mitochondria of specific lung cells; their functional roles in such cells will also need to be elucidated.
In summary, Nox/Duox isoforms are known to be localized to the plasma membrane in specialized structures such as lipid rafts, caveolae, or focal adhesions. The cell surface localization of Nox enzymes, while essential in host defense and phagocytosis, is critical for their emerging roles in mediating intracellular signaling, endocytosis, cellular adhesion, and migration. Nox enzymes are also found in biological membranes of intracellular organelles such as the ER, nucleus, and mitochondria. Although much work needs to be done on the functional roles of Nox enzyme in specific organelles, there is a growing recognition of their participation in the mediating stress responses in each of these subcellular compartments.
Nox Enzymes in Lung Health
The evolutionary conservation and diversification of Nox enzymes suggest essential and adaptive roles of Nox-generated ROS in human physiology. Physiological roles of ROS include signal transduction (8, 146), angiogenesis (7), and innate immunity (61). Nox2 generates high levels of ROS in neutrophils as a central mechanism of host defense against microbial infection, including infection of airways and lung, and has been reviewed elsewhere (49, 84, 169). A functional role of Nox2 role in innate immunity is well exemplified by its loss-of-function mutations in chronic granulomatous disease (CGD). CGD is caused by an inherited mutation of Nox2 or its subunits. Patients with CGD suffer recurrent infections of multiple organs, and pulmonary infection is the leading cause of death (9, 143).
Other Nox/Duox isoforms have been proposed to function in host defense and innate immunity. Studies in gastric mucosal cells support a role for Nox1 in antimicrobial host defense (81, 155), although a similar role for NOX1 in the lung is yet to be demonstrated. Duox1 and Duox2 in upper airway epithelium generate H2O2 to mediate lactoperoxidase-catalyzed generation of microbicidal oxidants (61, 115, 179). The expression of Duox1 and Duox2 in salivary, tracheal, and bronchial epithelium supports a broad role of these isoforms in host defense functions at these epithelial surfaces (56, 61, 115, 128, 135).
Nonphagocytic Nox isoforms generate ROS at lower levels and in a regulated manner, participate in signaling of immune responses and other cellular functions (89). Nox enzymes may be induced by a variety of bacteria and viruses and participate in host inflammatory responses. For example, airway instillation of P. aeruginosa significantly increases the expression of Nox2 and Nox4 in lung microvascular endothelial cells, where they mediate distinct signaling functions (58). Nonimmune mediated functions of Nox-derived ROS include, but are not limited to cell growth, proliferation, angiogenesis, apoptosis, and autophagy (22, 48, 52, 89, 133).
Nox Enzymes in Lung Diseases
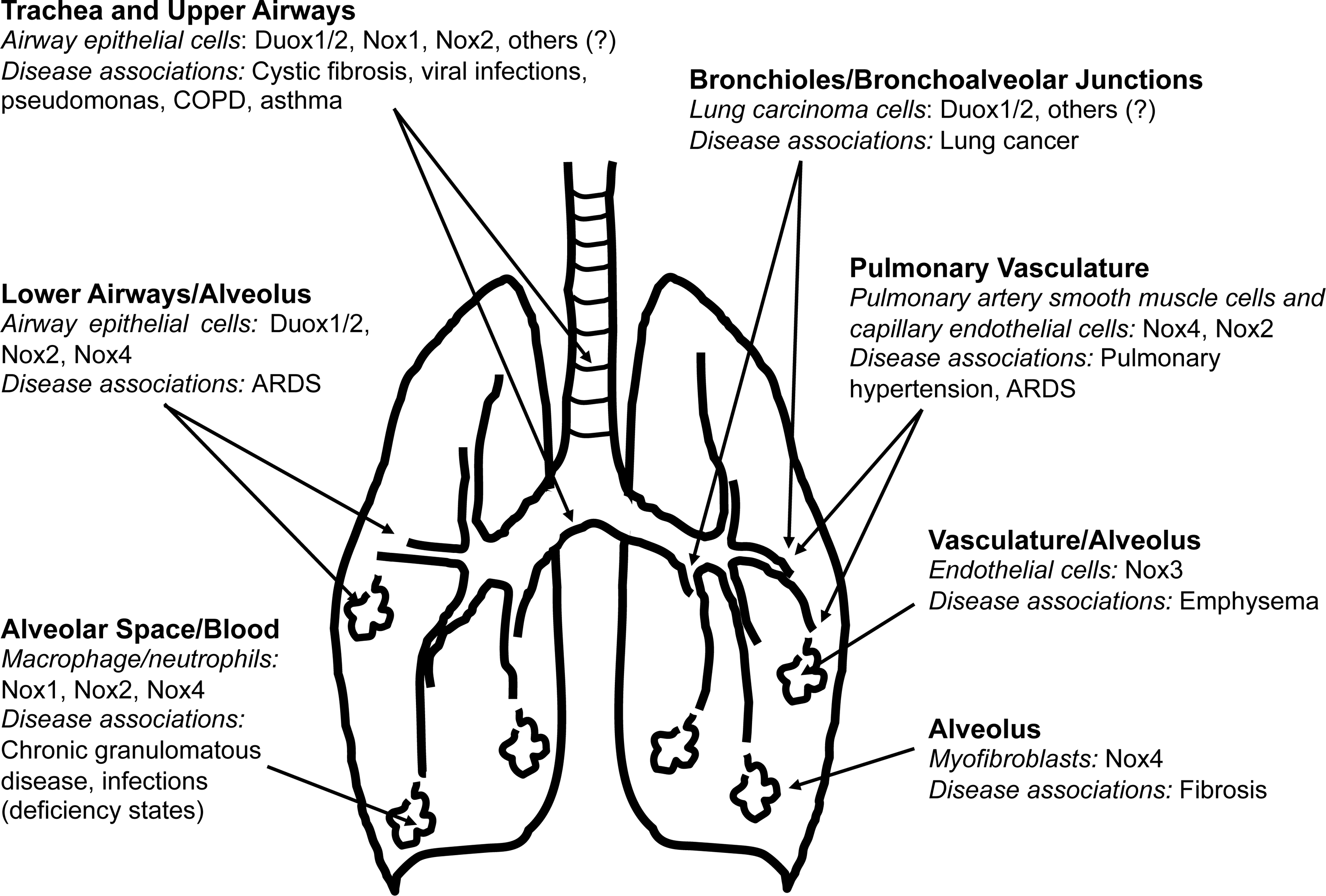
Nox/Duox enzymes contribute to a variety of lung diseases, either due to a loss of function or due to heightened expression/activity of specific isoforms (Fig. 4). In this section, we consider a select group of lung diseases in which Nox enzymes have been implicated in pathogenesis.
Pulmonary infections and inflammatory diseases
A central theme with many of the Nox family genes is their participation in various aspects of host defense against pathogens. Indeed, Nox2 is well established as an archaic antimicrobial defense mechanism that is conserved across multiple species (45, 117). In addition to the predicable susceptibility to infections with loss of function of Nox2, there is also evidence for a homeostatic role of Nox2 in controlling inflammation.
CGD, characterized by susceptibility to recurrent pyogenic infections, is the prototypical example of a human disease that is associated with inherited loss-of-function of genes encoding components of the Nox2 enzymatic complex. Initially characterized as a fatal granulomatous disease of childhood, the clinical course of CGD is marked by recurrent, suppurative infections (21). CGD can be associated with a defect in any of the subunits of the multicomponent Nox2 enzyme (45, 95, 118, 130, 153). Emerging data suggest a shift in the most common infecting organisms from staphylococci and enteric bacteria to other pathogens, including Aspergillus pneumonia and Burkholderia cepacia (78).
An unexpected role for Nox2 in suppressing neutrophilic inflammation has also been suggested (35). Similar findings of a putative “anti-inflammatory” role for p47 phox /Nox2 are reported in mice challenged with intra-peritoneal live Escherichia coli to induce sepsis (60), and in murine models of emphysema (181), pneumococcal pneumonia (105), influenza pneumonia (141), and disseminated Cryptococcus neoformans infection (142). A deficiency in neutrophil cytosolic factor-1, required for activation of Nox2, protects from virus-induced acute lung injury (ALI) (76). These studies support a homeostatic role Nox2 (and potentially other p47 phox - and/or p22 phox - requiring Nox enzymes) in modulating the host inflammatory responses.
Asthma
Systemic and airway-associated oxidative stress is increased in asthmatic patients compared with healthy individuals; this has been correlated to the degree of airflow obstruction and airway hyperresponsiveness (AHR) (41, 121, 148). Serum levels of damaged lipid and carbonylated proteins are increased in asthmatic children compared with controls (121). Airway SMCs of asthmatics demonstrate an increase in stress-induced DNA damage and ROS production; compounds such as apocynin diphenylene iodonium (DPI) that are known to inhibit Nox activity, albeit not specific, decreased agonist-induced airway smooth muscle contraction, which underlines AHR (148). Apocynin has also been shown to inhibit the production of anti-inflammatory cytokines (tumor necrosis factor α [TNF-α], IL-1β, and IL-6) by the airway mucosa, as well as the migration of macrophages and eosinophils (83). The effect of apocynin on cytokine synthesis is likely due to the regulation of gene expression by oxidative stress-responsive sensitive transcription factors (159).
The activity of Nox2 and Nox4 isoforms are primarily implicated in the increased oxidative stress that is associated with asthmatic airways, both in humans and in experimental murine models. Studies using Nox2 knockout mice sensitized with ovalbumin suggest a role of Nox2 in the cross-talk between T-lymphocytes and macrophages to limit the inflammatory response and to restrain acute allergic reactions (11). Accordingly, Nox2 deficiency resulted in enhanced recruitment of inflammatory cells to the airways and cytokine production, which worsens the asthmatic phenotype compared with wild-type (WT) mice (10). Alternatively, Nox2 has also been suggested to promote asthmatic airway inflammation, primarily by enhancing recruitment of eosinophils (1, 34). While Nox2 modulates inflammatory responses in asthma, Nox4-generated ROS have been suggested to underlie AHR by mediating airway smooth muscle hypercontractility. Indeed, Nox4 expression is enhanced in primary airway smooth muscle derived from asthmatic patients, and the silencing of its expression abrogates agonist-induced contraction of airway smooth muscle (148). Therefore, targeting of Nox2 and Nox4 may offer therapeutic benefits in specific phenotypes of asthma.
Acute lung injury
ALI and the ARDS represent clinical syndromes of varying severity and diverse causes that present with a set of defined clinical-physiologic-radiologic criteria (55). The common pathophysiologic feature involves disruption of the alveolo-capillary membrane, resulting in diffuse bilateral infiltrates on chest radiographs and severe arterial hypoxemia (176). The generation of ROS by enzymatic and nonenzymatic mechanisms, including activation of NOX enzymes, may contribute to the pathobiology of ALI/ARDS (26).
Nox1 is an important contributor to ROS production, epithelial cell death, and disruption of the alveolo-capillary barrier during hyperoxia; this oxidative stress-induced ALI involves activation of JNK and ERK pathways (25). Vascular endothelial cells function is also critical in maintaining the alveolo-capillary barrier function. Both Nox2 and Nox4 are expressed in pulmonary vascular ECs and contribute to hyperoxia-induced ROS generation (124). Hyperoxia induces pulmonary edema and neutrophil influx into the alveolar space of WT mice, effects that are attenuated in Nox2-deficient mice. The observed protection is incomplete, suggesting the potential involvement of other Nox isoforms, including Nox4, in alveolo-capillary barrier dysfunction (124). Interestingly, baseline levels of Nox4 mRNA expression are increased in Nox2-deficient mice compared with WT mice, suggesting the existence of a compensatory mechanism for ROS production. Nox2 promotes NF-κB-dependent acute inflammatory responses, neutrophil influx, and tissue injury specifically in the lungs, but not other organs, in response to systemic TNF-α administration (185). In response to acid aspiration, however, Nox2 appears to reduce neutrophil accumulation, while Nrf2 decreases ALI without affecting neutrophil influx (39); these observations suggest distinct functions of Nox2 and Nrf2 in modulating inflammation and injury in the lung.
Pulmonary arterial hypertension
Chronic hypoxia is the most common risk factor for pulmonary arterial hypertension (PAH), characterized by vascular remodeling and enhanced vasoreactivity. Specific Nox isoforms, in particular Nox2 and Nox4, have been implicated in hypoxia-induced pulmonary hypertension (50, 57, 98, 112). Nox2 has been implicated in hypoxia-induced endothelial dysfunction (57). Hypoxia-induced PAH in mice has been linked to increased Nox4 expression in pulmonary artery (SMCs) (112), suggesting a key role for Nox4 in vascular remodeling that is associated with hypoxia-induced PAH. TGF-β-induced Nox4 expression and ROS production have been shown to mediate proliferation of human pulmonary artery SMCs (77, 145). Targeting Nox1/4 with a pharmacological inhibitor, GKT137831, attenuates hypoxia-induced pulmonary SMC proliferation, vascular remodeling, and the development of PAH (66).
Nox-derived ROS may directly cause extracellular Ca2+ influx by inhibiting voltage-dependent K+ (KV) channels and the opening of store-operated Ca2+ channels, as well as intracellular Ca2+ release by activating ryanodine receptors, leading to an increase in intracellular Ca2+ concentration and associated SMC contraction (175). The Nox inhibitor and ROS scavenger, apocynin, as well as Nox4 siRNA reverses the hypoxia-induced decrease in Kv current density, whereas the protein levels of the channels remain unaffected by Nox4 silencing; this effect appears to be related to, at least in part, direct effects of Nox4-derived ROS in cysteine oxidation of the Kv1.5 channel (111). Finally, in addition to the intima and media, remodeling of the vascular adventitia may contribute to hypoxia-induced PAH. For example, hypoxia has also been shown to up-regulate Nox4 expression in pulmonary artery adventitial fibroblasts in association with increased ROS levels and increased cellular proliferation, effects that are abrogated by siRNA silencing of Nox4 (97).
Emphysema
Emphysema is the most common cause of chronic obstructive pulmonary disease (COPD), and it is primarily related to cigarette smoking. Human subjects with COPD and cigarette smokers exhibit differential Duox1 and Duox2 depending on smoking status and type of lung epithelium sampled; for example, airway epithelium of current smokers express decreased Duox1 and increased Duox2 compared with never smokers, whereas former smokers with COPD demonstrate reduced levels of both Duox isoforms (116, 127). In contrast, alveolar epithelial Duox1 and Duox2 were expressed at low levels and were unchanged regardless of smoking or COPD status (116). The precise role of Duox enzymes in COPD pathogenesis requires further studies.
The role of Nox2 in emphysema has been studied in knockout mouse models. Mice deficient in p47 phox or Nox2 (gp91 phox−/−) exhibit increased cigarette smoke (CS)-induced lung inflammation and emphysema despite decreased ROS production; this was associated with increased production of pro-inflammatory cytokines/chemokines via a toll-like receptor 4 (TLR4)-NFκB pathway, suggesting that Nox2 may mediate anti-inflammatory functions by restraining TLR4 activation (181). It is interesting to note that aging gp91 phox−/− mice (>6 months) exhibit spontaneous emphysema (79); in this study, basal levels of oxidative stress markers were not altered in p47 phox or Nox2-deficient mice, indicating stress-mediated responses. Interestingly, CS-exposed p47 phox or Nox2-deficient mice, despite a compensatory increase in Nox4 expression, demonstrate reduced CS-induced release of ROS, lipid peroxidation, and DNA damage. In contrast, a pro-inflammatory role for p47 phox -containing Nox enzyme(s) was suggested as p47 phox null mice develop less inflammation with lower levels of IL-6, keratinocyte-derived chemokine (KC/CXCL1) and monocyte chemoattractant protein-1 (MCP1/CCL2) in lung lavage specimens after CS exposure in comparison to wild-type mice (87). Gene profiling studies in lung tissues from CS-exposed mice recently revealed up-regulation of NoxO1, which primarily regulates Nox1 activation, indicating that specific roles of other Nox isoforms in emphysema remain to be determined (109).
An unexpected role for Nox3 and TLR4 regulation in emphysema has been elucidated (186). Mice deficient in TLR4 develop age-dependent emphysema, a phenotype that is ameliorated with chemical Nox inhibitors or Nox3 siRNA, suggesting a role for Nox3-generated ROS in emphysema (186). Lung endothelial cells from TLR4-deficient mice were identified as the primary source of increased ROS production, which potentiates matrix degrading enzymatic activity (186). It is possible that Nox3, due to its potential pro-oxidant effects in the lung, requires tight suppression (by TLR4) under homeostatic conditions; however, pathologic states of TLR4 deficiency may allow for unrestrained Nox3 activity and pro-oxidant effects that contribute to emphysema. Indeed, aging and CS exposure are associated with depressed TLR4 function in human subjects (103, 167), supporting the theory of a disrupted TLR-Nox3 axis in human emphysema.
Pulmonary fibrosis
Pulmonary fibrosis is a chronic “scarring” disease of the lung that may result from known causes (e.g., environmental exposures, drugs, and connective tissue diseases) or unknown etiology (i.e., idiopathic). In almost all cases, the fibrotic reponse is characterized by the accumulation of activated myofibroblasts and the deposition of extracellular matrix (ECM) (161). Myofibroblast differentiation is mediated by soluble factors, primarily TGF-β1 (42, 160), and by ECM factors, primarily tissue stiffness (75, 187). In addition to its multiple fibrogenic actions, myofibroblasts generate ROS in response to TGF-β1 (37, 158, 173). Although the cellular localization/compartmentalization of Nox4 has not been clarified in myofibroblasts, a unique feature of NOX4 activity is its capacity for constitutive generation of extracellular H2O2 (106, 136, 170 –172). Extracellular generation of H2O2 by lung myofibroblasts may mediate additional fibrogenic effects by inducing apoptosis of adjacent lung epithelial cells (173), or by inducing matrix cross-linking reactions (91), potentially contributing to tissue stiffness.
A pro-fibrogenic role for Nox4 in a number of organs systems has been proposed, including kidney fibrosis (16, 120, 151, 178), vascular remodeling/fibrosis associated with chronic hypertension (4), cardiac fibrosis (74, 86, 144, 174), pancreatic fibrosis (107), liver cirrhosis (6, 40, 132), and lung fibrosis (24, 71). Interestingly, recent studies also suggest a protective effect of Nox4, primarily in the kidney (119) and cardiovascular system (23, 134, 184).
In addition to Nox4, other Nox isoforms have been implicated in lung fibrosis. A p47 phox -requiring Nox isoform is required for the development of fibrosis in a murine lung injury model; this bleomycin injury model is inflammation dependent and the observed protection in p47 phox−/− mice may be related to modulation of neutrophilic inflammation and/or matrix metalloproteinase-9 activity in the bronchoaveolar lavage of these deficient animals (104). Studies of the gelatinase activities of lung fibroblasts from p47 phox−/− mice will provide further insights into the extent by which p47 phox -dependent ROS function in fibrotic lung remodeling. Nox1 has not been implicated in lung fibrosis, although there is evidence for a role of this isoform in liver fibrosis (38). Importantly, a pharmacologic inhibitor against Nox1/4 appears to be effective as an anti-fibrotic agent in preclinical models (59, 88).
Lung cancer
Nox enzymes may participate in several of the key events in the multi-step development of human cancers (67, 69). Studies more than two decades ago indicated the generation of ROS by an Nox-like flavoenzyme in several different cancer cells, although the identity of the enzymatic source(s) was not known at that time (150). Since then, specific Nox isoforms have been identified in a variety of human malignancies, including colon (92, 126, 149), gastric (163), pancreatic (94, 113, 168), and prostate (20) cancers. The tumorigenic potential of Nox1 was demonstrated by showing that Nox1-transfected cells produce phenotypically aggressive tumors in athymic mice (8, 146). Nox1 silencing in K-Ras transformed cells abrogates anchorage-independent cell growth and capacity for tumor formation in vivo (110). In fact, Nox1 was originally referred to as the “mitogenic oxidase” (8, 146); however, its effects on specific cell types are likely contextual and include other cellular functions. A number of reports support a role for Nox1 in angiogenesis (7). Vascular endothelial growth factor (VEGF) functions as a key mediator of neovascularization within tumors. Ras-induced VEGF transcription is mediated by an NOX1/Ras/ERK-MAPK pathway (85).
Resistance to apoptosis is another hallmark of cancer cells (67). Nox4 promotes apoptosis resistance in pancreatic cancer cells (168). Nox5 has been reported to mediate cell proliferation and resistance to apoptosis in prostate cancer cells (20). A potential mechanism by which the Nox isoforms promote apoptosis resistance may involve ROS-mediated inactivation of PTPs (94); alternatively or in parallel, activation of the phosphoinositide-3-phosphate (PI3K)/AKT and apoptosis signal-regulating kinase 1 pathway may contribute to Nox4-induced pro-survival signaling (113). Specific roles for Nox4 in lung cancer are yet to be determined. However, Duox1 and Duox2 and their maturation factors are reported to be down-regulated by promoter methylation in primary lung carcinomas (100). Restoration of functional Duox1 reverses the phenotype of the lung cancer cells lines, supporting epigenetic mechanisms involving Duox1 in lung carcinogenesis (100). Further studies are required to clearly define the precise roles of Nox enzymes in various aspects of cancer development and progression.
Conclusion
Nox family enzymes serve many homeostatic and host defense functions in the lung. Our current understanding of the physiological roles of Nox/Duox enzymes in the lung is still in its very early stages. A clearer understanding of these roles and their aberrant function in disease pathogenesis will expedite the eventual development and testing of Nox inhibitors in specific lung diseases (70, 156). All of the Nox enzymes are likely to be contextual in their actions and, thus, defining their “disease context” is critical in designing informative preclinical and successful clinical studies. Animal models of most chronic lung diseases have significant limitations, and this highlights the importance of validating the expression and localization of Nox isoforms in lung cells/tissues derived from patients with specific lung disorders. The utility of Nox enzymes as biomarkers in disease expression and progression deserves further studies.
Footnotes
Acknowledgments
This work is supported in part by Veterans Administration Health System grant, 1IK2BX001477 (to L.H.); American Heart Association grant, 12GRNT12040409 (to G.C.); National Institutes of Health grants, K08 HL094666 (to T.R.L.); R01 HL086836 and R21 AI101642 (to G.C.); and R01 HL067967, R01 HL094230, and P50 HL107181 (to V.J.T.).