
Abstract
Introduction
C

Thus, cells respond to a broad spectrum of physical stresses, analogous to their response to chemical agents. Among the physical forces that play a role in biology are gravity, adhesion, pressure, turgor, and shear stress. Of particular importance in mammalian systems are the mechanical (hemodynamic) forces associated with blood flow, that is, fluid shear stress and distending pressure. Shear stress acts at the blood–endothelial cell (EC) interface and plays a major role in physiological and pathophysiological vascular responses. The “sensing” of the shear force by ECs produces a signal in which redox-sensitive processes play a major role; this signal acts on cellular targets that modify cell structure, metabolism, and gene expression (42, 91). Although the components of this signaling pathway are now reasonably well understood, the mechanism for coupling between the sensor of shear stress and transduction of the stimulus to signaling systems has not yet been established.
Apart from the ECs, vascular tissue consists of smooth muscle cells (SMCs), fibroblasts, and various other cell types. Unlike ECs that are in contact with the blood, other cells are not normally exposed directly to shear stress except possibly through interstitial fluid flow. However, shear stress may be indirectly conveyed to SMCs and fibroblasts through signals arising from endothelium (endothelial–smooth muscle communication), possibly through the release of mediators, such as nitric oxide (NO) or transmission via functional complexes (148).
This presentation will focus on sensing and response to shear stress in the vascular endothelium. We will provide an overview of the endothelial mechanotransduction machinery, including the initiation, transduction, transmission, and reception of the shear signal with emphasis on the role of reactive oxygen species (ROS).
Shear Stress in the Vasculature
The endothelium by virtue of its location serves as a dynamic interface; elements on the endothelium act as mechanotransducers of altered shear associated with fluid flow. Shear stress in the vasculature is the force that liquid flow exerts per unit area of the vessel wall and can be expressed in units of dyn/cm2. Depending on the pattern of flow, shear stress may be classified as: (a) steady or laminar, (b) pulsatile, (c) oscillatory, or (d) turbulent. In the case of laminar flow, where the orderly motion of the fluid is parallel to the vessel wall, the shear stress is directly proportional to the blood flow (more specifically, the velocity of blood flow). Thus in regions of laminar flow, a change in fluid flow rate corresponds to a change in shear stress. However, under conditions of disturbed flow, the relationship between flow and shear stress is complex and shear (especially in areas of narrowing or partial obstruction) is not proportional to bulk flow rate. Although blood flow in smaller vessels is typically laminar, in larger vessels, it can be a combination of laminar and nonlaminar depending on possible obstructions to the vessel lumen and the phase of the cardiac cycle. Shear patterns and magnitude are particularly relevant as these give rise to specific signaling cascades with different outcomes; laminar flow helps maintain vascular homeostasis while turbulent or oscillatory flow may be either the result of or associated with the pathogenesis of vascular diseases.
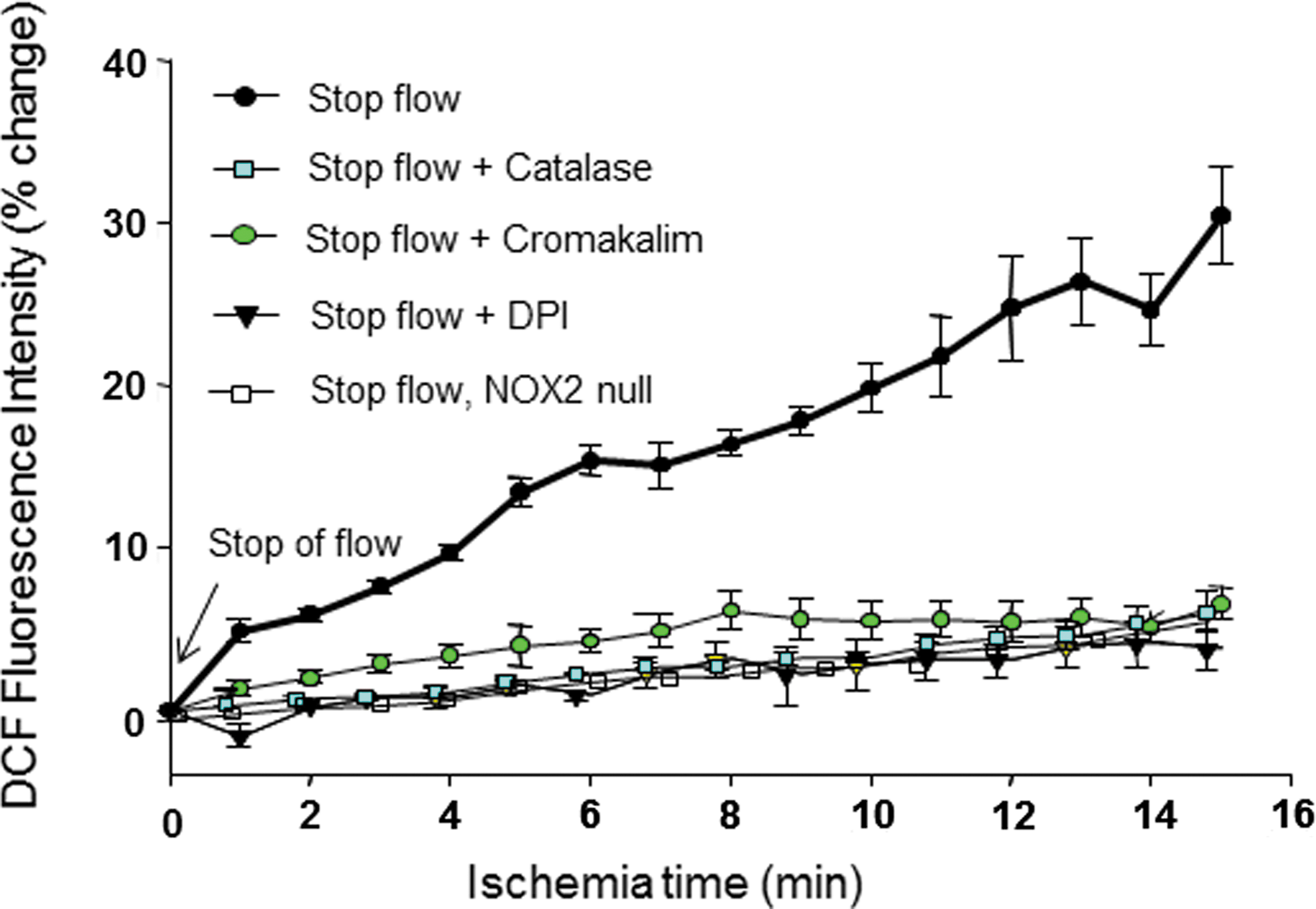
The magnitude of shear experienced by ECs varies depending on the vascular bed. In the systemic vasculature, shear stress ranges from 5 to 25 dyn/cm2 and in normal conditions is either laminar or pulsatile (144). An increase in shear stress occurs with increased blood flow such as transiently with exercise or chronically with diseases associated with increased cardiac output. Vascular narrowing in the face of unchanged bulk flow results in increased blood flow velocity and increased shear, although commonly the flow rate in obstructed vessels is decreased, thereby restoring normal shear. In stenotic arteries, the flow pattern may become turbulent or oscillatory and shear values increase to 30–40 dyn/cm2 or higher. Traditionally, the flow rate resulting in a change from laminar to turbulent flow has been estimated by calculation of the Reynolds number (the ratio of inertial to viscous forces). Shear stress in the pulmonary arteries is similar to that in systemic vessels, although it is probably lower in the pulmonary capillaries where the rate of blood flow is relatively slow. Shear stress is decreased chronically in conditions of compromised cardiac output, such as heart failure or primary pulmonary hypertension (132), and can abruptly decrease to essentially zero with cardiac arrest, vascular obstruction, or during surgeries, such as organ transplant (76, 124). In the rat lung, a decrease in shear stress of ∼90% was required to activate cell signaling (4).
Models for Study of Shear-Dependent Mechanotransduction
Onset of flow and flow adaptation
Mechanosignaling has been extensively studied using various “onset of flow” models (42, 46, 132). Most of the studies have been carried out with cells in culture that are grown under zero shear stress conditions (i.e., in static cell culture) and then subjected to flow. These studies have used special flow chambers, either a parallel plate or a cone and plate chamber, where cells are subjected to controlled shear stresses in flow channels or circuits, which can be monitored for endothelial responses (35, 119, 140), including gene expression and protein production (37, 110, 120, 135). Alternatively, isolated intact vessels are cannulated and connected to a flow system. These in vitro/in situ models have been used to study the effect of the onset or incremental change in shear stress (97, 109, 113, 152).
The sequence of events that characterizes the response of cultured cells to the onset of laminar flow can be classified as immediate, short term, and long term (adaptive). The process of full adaptation of cells to shear requires ∼24–48 h; the cells then reach a new equilibrium with their environment. An essentially immediate response (within seconds) to the onset of flow with cells in culture is ion channel (specifically K+ channel) activation that results in membrane hyperpolarization, generation of ROS, calcium influx, possibly through newly opened “store-operated” calcium channels (88, 107), activation of kinases, and release of NO (83, 143). Short-term changes that occur within 1–2 h include the activation of transcription factors (NFκB and AP-1) and matrix proteins, such as focal adhesion kinases. The more sustained (long-term) responses to an increase in laminar shear stress include a modest increase in expression of cell adhesion molecules (intercellular adhesion molecule [ICAM] and vascular endothelial cell adhesion molecule [VCAM]) and decreased expression of endothelin-1 (11, 43, 45, 145). In the quiescent state, the cytoskeleton provides mechanical support and maintains cell shape and topology of the endothelial monolayer. The cell–cell junction proteins and cortical cytoskeleton are linked by actin filaments to the adhesion matrices on the basal side of the cell. Prolonged exposure to shear stress causes cells to reorganize their actin cytoskeleton and focal adhesions and subsequently align with their long axes in the direction of flow (46, 57, 78, 136). Presumably, this flow-adapted state is the physiological condition of ECs that are normally exposed continuously to blood flow in vitro.
The presence of promoter sequences termed shear stress response elements (SSREs) has been proposed to mediate the responsiveness of EC genes to shear stress. Although a single SSRE (the sequence GAGACC) was reported to encode shear stress responsiveness in endothelium (121, 131), promoter regions of genes may encode for multiple SSREs (100, 127). On the other hand, more recent studies have shown that transcriptional changes seen with flow also may be independent of SSREs (136).
Although much of the blood flow in vessels is laminar, flow around branch points and in regions distal to stenosis may become random and unpredictable. This is disturbed flow and could either be turbulent (irregular flow forming vortices and eddies) or oscillatory (to and fro movement of blood in response to an oscillating pressure gradient). Disturbed flow is studied using in vitro parallel plate chambers that have uniquely shaped flow channels (with cuts on the sides of the channel) or an obstacle in the flow path or backward shaped step to replicate aortic artery bifurcation geometries and flow profiles (102, 104, 128, 133, 138). Oscillatory or turbulent shear upregulates endothelin-1 (ET-1) expression and results in a significant increase in secretion of proinflammatory cytokines. ECs exposed to disturbed flow when subjected to transcription profiling analyses show increased expression of proinflammatory molecules, such as the transcription factor NFκB and cell adhesion molecules, ICAM-1, VCAM-1, monocyte chemoattractant protein (MCP), and E-selectin (33, 145). These changes with disturbed flow appear to drive an atherogenic phenotype (56), as supported by the observation that atherosclerotic plaques are predominately located at branch points of the vascular tree, which experience disturbed flow (21, 42a). However, whether the correlation of atherogenesis with disturbed flow represents a response to cell signaling or some other pathophysiological mechanism has not been resolved as interpretation of these studies has been hampered by the nonphysiological conditions of using nonflow adapted cells as the “control” state.
Studies with the onset of flow have been interpreted to reflect the response to an increase in blood flow: laminar, oscillatory, or disturbed. However, one of the limitations of these studies is the implicit assumption that cells cultured under static conditions will respond to increased flow similar to a flow-adapted cell. This assumption is unlikely since there is considerable change in gene expression, of cells in vitro during the process of flow adaptation. Furthermore, the pattern of gene expression reflects the type of flow used for flow adaptation (52, 95, 117). Under physiological conditions, ECs are expected to be flow adapted. Thus, additional studies are necessary where cells are flow adapted (similar to physiological conditions) and then subjected to increased shear stress.
Stop of flow: ischemia
Our research group has studied cell models where mechanotransduction is initiated by an abrupt decrease in shear stress. These latter studies have utilized flow adapted cells as the control state and can be considered representative of in vivo conditions that occur with various obstructive pathologies. Loss of shear stress occurs in vivo with cardiac arrest or vascular obstruction of blood flow. Use of the intact lung provides a methodological advantage over other organs for “stop of flow” studies since oxygenation of cells can be maintained through lung ventilation, thereby avoiding the complicating effects of tissue hypoxia (3, 4, 20, 25, 27, 28a, 29, 48, 148, 149). The tissue pO2 values remained constant (pO2 ∼140 mmHg), and there was no decline in ATP levels with lung ischemia. Similarly, oxygen delivery was maintained during the study of cells in vitro by using an artificial capillary chamber with side ports that allow the delivery of oxygenated medium to cells when flow is stopped (24, 29, 148, 149). ECs when grown in culture under continuous laminar flow for 24–48 h became adapted to flow as described above, and upon cessation of flow show mechanosignaling responses similar to ECs in intact lungs (53, 148, 149). This response is described below.
Incremental increase or decrease in flow
Studies on the effect of fluctuating flow, in the form of relatively slow and steady changes, abnormal regimens such as impulse and reverse impulse flow, or step and ramp flow, show differential endothelial responses. Fluctuating flow conditions can occur in vivo during exercise where shear stress increases incrementally in inactive limbs during acute exercise or during phases of the cardiac cycle. In vitro models have been used to investigate “step up” flow (instantaneous shear stress increase from 0 to 16 dyn/cm2 followed by steady shear for a sustained period); “ramp” flow (shear stress increased from 0 to 16 dyn/cm2 during 10 min and then sustained for a certain period); “impulse” flow (a 3-s pulse of 16 dyn/cm2); and “reverse impulse” flow (a step increase to 16 dyn/cm2, sustained for 3 s, followed by a ramped decrease to 0 dyn/cm2 during 10 min). Overall such systems demonstrate that it is the change in shear stress rather than steady levels of shear stress that is the stimulus for shear-sensitive signaling. The threshold for change with increased shear has not been adequately evaluated. However, incremental decrease of shear with isolated lungs showed a threshold of ∼90% reduction in shear stress to activate endothelial production of ROS. Thus, signaling occurs predominantly related to a relatively large change in shear stress with essentially no signaling associated with the steady state.
The Role of ROS in Mechanotransduction
Generation of ROS with altered shear stress
ECs in pulmonary vessels and aortic endothelium in situ and in flow chambers in vitro where oxygenation of cells is maintained at normal levels responded to stop of flow by generating ROS (1, 3, 18, 25, 48, 99, 129, 149, 161). ROS generation is essentially immediate and continuous over an observation period over several minutes. The onset of laminar as well as oscillatory and turbulent shear stress also elicit endothelial ROS production (61, 66, 67); these results were obtained with cells cultured under static conditions and therefore were not flow adapted. It is indeed interesting that stop and increase of flow elicit a similar response and emphasize that ECs are responding to a change from the “steady state” rather than the absolute value of shear stress.
The generation of ROS in these models was detected with the use of fluorescent probes. These include the chlorinated fluorescein derivative H2DCF, which is susceptible to photo enhancement of the fluorescence signal and the fluoro compound H2DFF that exhibits improved photostability and cell retention (106). However, neither of these compounds shows ROS specificity. The fluorescein derivatives are used as the diacetate to ensure membrane permeability. Dihydroethidium (DHE) with the detection of the product 2-hydroxyethidium has been used as an O2 •−-specific probe. Amplex red has been used to detect the appearance of H2O2 in the vessel lumen as, unlike the other fluorophores, it is membrane impermeable. The important caveats for the use of these probes to reflect ROS production must be considered in the interpretation of results (106).
Sources of ROS
The NADPH oxidase (NOX) enzymes are a major source of regulated ROS production in endothelium. The NOX family comprises seven homologs, NOX1–5 and DUOX1 and 2. NOX1, 2, 4, and 5 are present cells of the vasculature; NOX2 and 4 and possibly NOX1 are in expressed in endothelium. ROS production in ECs with abrupt cessation of shear is abolished by the deletion of NOX2 (106, 161). These results indicate that NOX2 is primarily responsible for the immediate burst of ROS generation associated with altered shear stress. NOX2 produces O2 •− that rapidly dismutes, either spontaneously or catalyzed by superoxide dismutase, to H2O2. Thus, H2O2 is the primary agent for signal transmission in the ROS-mediated signaling cascade. NOX2 is present in the EC membrane where it functions to transfer an electron from cytoplasmic NADPH to extracellular O2. Thus, O2 •− (and its dismutation product, H2O2) are generated outside the cell. H2O2 is freely diffusible and able to reenter the cell where it can perform its physiological role. NOX4 is constitutively active and its role in ROS generation is dependent on the expression level of the protein. An increase of NOX4 expression with a change in shear stress requires induction of the message and protein synthesis, a relatively slow event. It is possible that the response to altered shear over a long-term result in increased ROS production by NOX4 or by NOX1. Sorescu et al. showed that exposure of aortic endothelium to oscillatory shear for 24 h resulted in a downstream effect of ROS (monocyte adhesion) that was abolished by the depletion of NOX1 (129a). The role of NOX proteins in mechanotransduction is discussed further in the reviews by Brandes et al. and Ward et al. (16, 147) in this Forum.
Apart from NOX proteins, mitochondria represent another source of ROS production with altered shear. These organelles release ROS during cyclic strain in umbilical vein endothelium (6). However, the conclusions from these studies were based on the use of inhibitors (such as diphenyleneiodonium, ebselen, and diethyldithiocarbamate) that are not specific for mitochondrial ROS as such, so that a role for mitochondria in the initial response to shear is tenuous. This conclusion is consistent with the absence of ROS production in endothelium from NOX2 null mice (105, 106, 161).
However, mitochondrion can serve as a secondary source of ROS. One possible mechanism for the activation of mitochondrial ROS production is a response to extracellular O2 •− generated by NOX2, a response that is dependent on the cell membrane chloride channel-3 (60).
Using ROS generation as a starting point, investigations have evaluated the upstream events that sense flow and the signaling pathways that lead to NOX2 activation as well as the downstream pathways that represent the physiological response to ROS-initiated signaling.
Flow Sensing
Candidate flow sensors
Various cellular microdomains and membrane proteins on the EC surface have been proposed to sense mechanical stimuli, that is, to act as mechanosensors. However, no universal sensor has been identified definitively; indeed, multimeric complexes of several entities are now proposed to act in concert to sense and transduce mechanical stimuli. In this section, several of these putative mechanosensitive complexes are discussed.
Tensegrity
The cell as a tensegrity structure. This theory proposes that a cell with its membrane and cytosolic components acts as an integrated structure to sense physical forces through tensegrity (tensional integrity). The tensegrity theory posits that mechanical stresses are conveyed through load-bearing cytoskeletal elements into the cytoplasm and nucleus (68, 146). Consistent with this theory, studies have shown that the cellular response to mechanical stress applied to the cell surface depends on molecular connectivity of the cell membrane to actin, microfilaments, microtubules, and intermediate filaments (69). The actin filaments associate with myosin filaments and generate mechanical tension that is distributed to all elements of the cell, as well as to the external extracellular matrix, probably via integrin contact points on the cell.
Cytoskeleton
This is a modification of the tensegrity theory. The cellular cytoskeleton along with other structural components can “sense” deformation at the cell surface and transmit it as a cytoskeletal tension via focal adhesion sites, integrins, cellular junctions, and extracellular matrix (43). The cytoskeleton is composed of three major types of protein filaments: microtubules, microfilaments, and intermediate filaments. This scaffold can transmit tensional forces that are generated to other parts of the cell or to the extracellular matrix and other cells (43, 45).
Integrins
Integrins are transmembrane receptors that link cytoskeletal proteins with components of the EC matrix. Focal adhesions contain multiple actin-associated proteins, such as talin, vinculin, paxillin, and zylin (54, 55). Mechanical forces such as shear may be transferred by the cytoskeleton to integrins that distribute the force by rearranging its interlinked actin microfilaments, microtubules, and intermediate filaments. This reorganization occurs via the activation of focal adhesion kinase and c-Src kinases. One of the major flow-sensitive integrins in vascular ECs may be αVβ3; indeed, pretreatment with anti-αVβ3 antibody decreased shear-induced signaling (73).
Platelet endothelial cell adhesion molecule-1
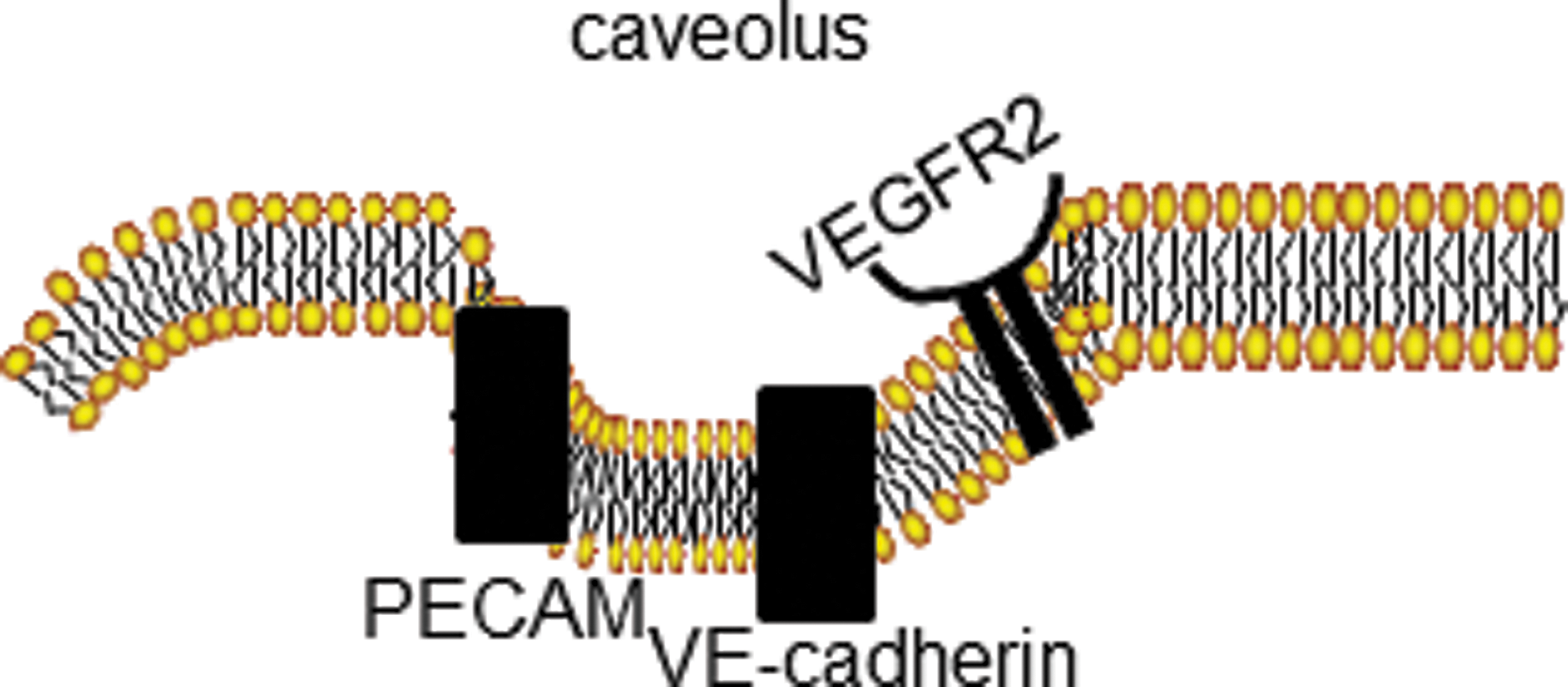
Platelet endothelial cell adhesion molecule (PECAM), an adhesion molecule expressed on the surface of ECs, platelets, and leukocytes, is found in cell–cell junctions and is reportedly bound to several other structural moieties, namely integrin αvβ3 and vascular endothelial growth factor receptor 2 (VEGFR2) (137). PECAM, along with the tyrosine-specific phosphotransferase Fyn, has been reported to form a mechanosensory complex (34, 108). A multimeric complex comprising PECAM-1, VEGFR2, and vascular endothelial (VE)-cadherin was sufficient to confer responsiveness to shear stress in cells (137). Studies on a hind limb ischemia model showed impaired collateral formation and reduced remodeling in PECAM null compared to wild-type mice (30). PECAM null mice showed an impaired response to reduction of shear stress in an in vivo model of reduced shear achieved by partial carotid artery ligation, (31). Isolated lungs from PECAM null mice showed a marked reduction in ROS production with stop of flow compared to wild type (105, 106). These observations provide strong evidence for an important role of PECAM in the response of ECs to altered shear stress.
Caveolae
Caveolae, lipid-rich invaginations on the EC surface, are reported to sense or transduce hemodynamic changes into biochemical signals that regulate vascular function; indeed, exposure to shear stress over a long duration altered the expression of caveolin-1, the major coat protein of caveolae, and the distribution and density of caveolae that were associated with enhanced sensing of hemodynamic forces by pulmonary endothelium (122, 123). Studies with caveolin null mice indicated a role for this protein in vasodilation and remodeling of the carotid artery with blood flow (159). Our studies on stop of flow show markedly decreased mechanosignaling in ECs and intact lungs depleted of caveolae by molecular engineering (98, 148). It is not clear how caveolae act as mechanosensors, but it may be functionally linked to other mechanosensing molecules, such as β1 integrins (118, 123), or may provide a scaffold for flow sensitive or signaling elements. We have shown recently that PECAM-1 is colocalized with caveolae as part of the mechanosensing machinery that generates superoxide with loss of shear (106).
The mechanosome
The mechanosome can be defined as a network of mechanosensors and transducers that transmit a physical force into a biochemical and transcriptional activity that alters cellular structure and function. This term has been applied previously in studies of bone (13, 14, 111). We posit that the mechanosome on the EC membrane consists of caveolae, PECAM, VEGFR, VE-cadherin and possibly other elements (Fig. 2).
Transducers of the Response to Mechanical Stress
Modulation of ion channel activity: an early event in mechanotransduction
Although ion channels have been proposed in the past as a primary sensor of altered shear, it is more likely that alterations of ion channel activity represent an early response in the signaling cascade, at least in the case of mechanosensing by the endothelium (3, 9, 29, 63, 107). Indeed, the earliest detectable endothelial responses upon the onset of flow are the opening or activation of an inwardly rectified K+ channel (KIR) (63, 72, 107). Flow simultaneously activates an outwardly rectifying Cl− channel that antagonizes the K+channel-mediated hyperpolarization (9, 88, 116). The molecular identity of either of these channels has not been determined. A shear-sensitive inward rectifying current has been observed when the KIR2.1 channel was expressed in Xenopus oocytes (63), but its significance is not clear. Ion channels appear to respond differently to different types of flow. For instance, an oscillatory shear stress (1 Hz) of±10 dyn/cm2 activates both hyperpolarizing and depolarizing currents, but this effect was not observed at a higher oscillation frequency (5 Hz) (10).
In pulmonary endothelium, loss of flow causes the closure of an inwardly rectifying K+ channel resulting in depolarization of the EC membrane as assessed by membrane potential sensitive dyes and the patch clamp technique. The pulmonary EC membrane channel responsible for EC depolarization with ischemia is a KATP channel consisting of a KIR6.2 channel pore and SUR regulatory subunit. Whether this is the same channel associated with the response to increase shear stress has not been studied. Cromakalin, a KATP channel agonist, prevented membrane depolarization with ischemia (5, 94, 134, 161), whereas glybenclamide, a KATP channel antagonist, resulted in depolarization during continuous flow (29, 134). Isolated perfused lungs and ECs from mice with “knock-out” of KIR6.2 showed almost no change in cell membrane potential with stopped flow (76, 98). Based on these results, we have proposed that a KATP channel of lung endothelium is responsible for maintaining membrane potential with normal shear and is inactivated by loss of shear leading to EC membrane depolarization as an important component of the cell signaling cascade (29). This channel on pulmonary microvascular ECs is induced during flow adaptation; cells exposed to flow for 24–48 h showed increased KATP channel expression (mRNA and protein) and activity (inwardly rectified K+ current) compared to statically cultured cells (24). This increased expression was inhibited by pretreatment with cycloheximide indicating that shear stress results in increased KATP channel synthesis (24). The KIR6.2 gene promoter region contains a putative “shear stress” response element (GAGACC) (24), which could account for its activation by flow, although this has not yet been tested experimentally. Induction of this channel by shear suggests a flow responsive element in the EC that is upstream and independent of the channel.
Ion channels also have been reported to respond to other mechanical stimuli. Transient receptor potential (TRP) channels, the potassium TREK channels (TREK-1 or K2P channel), and the amiloride-sensitive ENaC/ASIC channels (12, 49, 64) are activated upon membrane stretch. TREK-1 channels when expressed in either Xenopus oocytes or COS-7 cells showed robust mechanosensitive K+ currents (77, 154); TRP channels (TRPC channels, TRPC1 and 6) when expressed in CHO cells are also reported to be mechanosensitive (93, 130). Other TRP channels such as TRPA1 (38, 39, 80) and transient receptor potential V4 (TRPV4) are implicated in mechanosensation (74). The stretch-activated MscCa has been widely reported to be mechanosensitive (36, 59, 112). Stretch in the heart (114) affect the ryanodine receptor calcium channels causing a rapid burst of calcium (Ca2+ sparks).
ECs sensing flow can communicate with the smooth muscle layer immediately below it and may result in the activation of the large conductance Ca2+-activated K+ (BKCa) channel in vascular smooth muscle cells (75). Myoendothelial communication occurs through gap junctions and myoendothelial electrical coupling facilitates the spread of hyperpolarization. Smooth muscle hyperpolarization drives opening of store-operated Ca2+ channels or TRPV4 channels; the resultant entry and elevation of intracellular Ca2+ causes endothelial NOS (eNOS) activation, NO release, and vasodilatation (8).
It is not clear how ion channels are inactivated/activated in response to mechanical force? There are currently several postulates: the first is that there is a direct physical interaction between force on the cell surface and the ion channel, that is, the channel is either pushed open or closed by the onset or removal of a stimulus, such as blood flow (10). In this scenario, ion channels could be considered a “sensor” of altered shear stress. However, loss of the ion channel response to shear with knock-out of caveolin-1 or PECAM-1 indicates that ion channels are not the primary sensor of altered flow in endothelium (98). Furthermore, induction of KATP channels with increased shear argues for a different primary sensor (24). A second postulate is that mechanical forces change the tension on the cytoskeleton, thus activating or inactivating ion channels that are tethered to the cell membrane (81). A third postulate is that forces acting on cells change cell membrane fluidity (as for example, via cholesterol sequestration); such alterations have been reported to change channel function and activity (47). Finally, the response of ion channels may be secondary to the activation of cellular proteins (e.g., kinases), which then modulate ion channel activity. At this time, the mechanism for the modulation of ion channel activity with altered shear is not clear, and although they are an early part of the signaling cascade, they do not appear to be primarily responsible for sensing alterations of shear.
Kinases and phosphatases
Kinases, phosphatases, and other signaling molecules provide a link between the sensing of shear stress and the transduction of this information to a physiological response. Candidate-transducing molecules include mitogen-activating protein kinases (MAPKs) (ERK, p38MAPK, JNK) (82, 84, 101), tyrosine kinases (22, 151), NOXs (79, 114), transcription factors (NFκB, AP-1) (17, 62, 139), growth factors (PDGF) (7, 115), chemoattractants (MCP-1), cytokines (162), NO synthase (NOS), ET-1, and ROS as described above (103, 125). The shear stimulus (onset, cessation), shear profile (laminar, oscillatory), and shear magnitude may determine which of these transducers is expressed and participates in signaling.
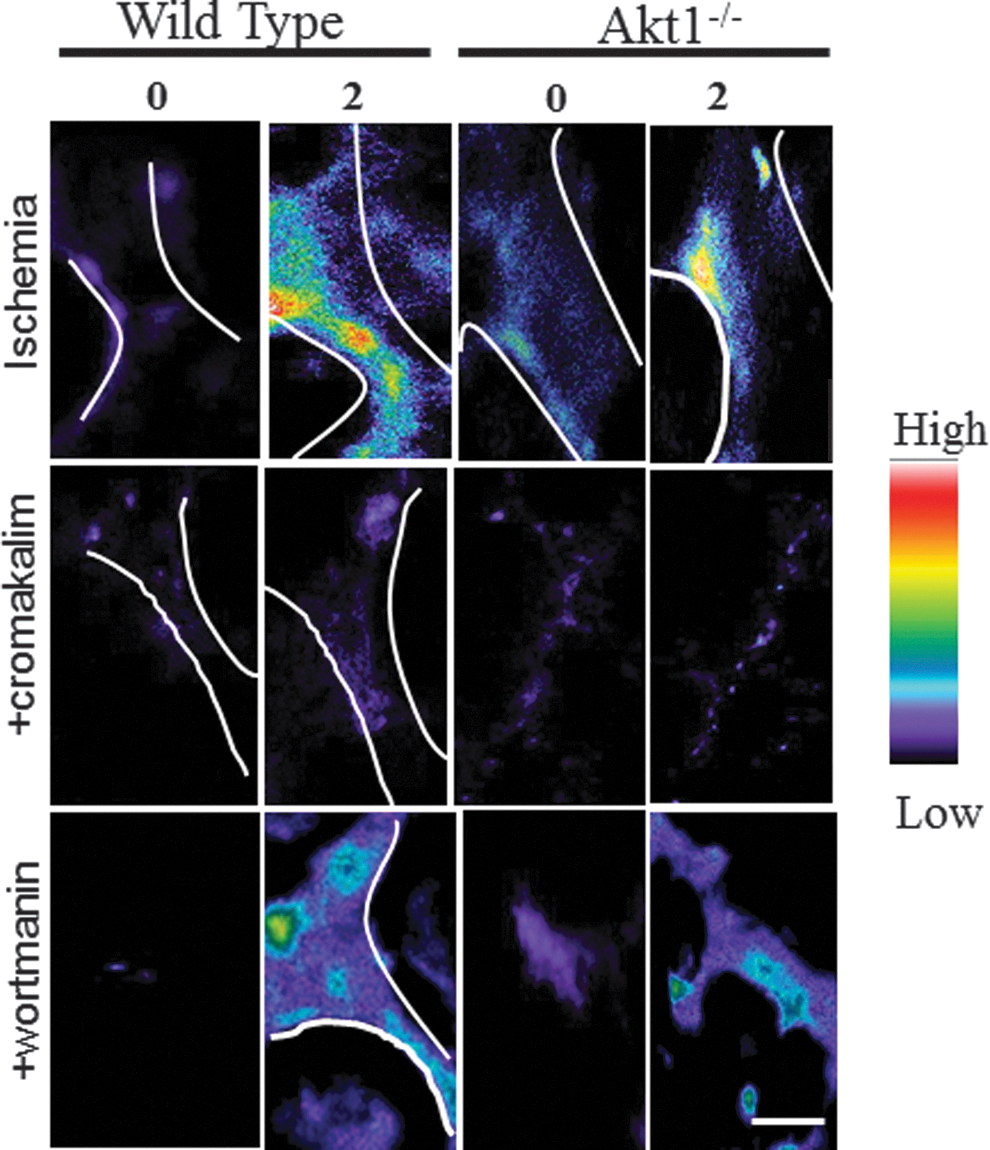
In our studies on stop of flow, we observed that depolarization led to the activation of phosphatidylinositide-3-kinase (PI3K) and protein kinase B (Akt) (25). Akt1, a serine-threonine kinase and a downstream effector of PI3K, was phosphorylated subsequent to depolarization. Depolarization with lung ischemia was not affected by blocking PI3K with wortmannin or by lack of Akt (Akt1 null mice) (25). These results indicate that depolarization is upstream of PI3K/Akt activation (25). The link between the alteration of membrane potential and activation of PI3K is not clear; perhaps voltage-sensitive domains in the p85 subunit of the kinase are sensitive to membrane polarity as it is this unit that gets phosphorylated with altered shear. The change in the concentration of the PI3K substrate, PI(4,5)P 2, may depend on the membrane polarity; PIP2 concentration increased with hyperpolarization and decreased with depolarization (71, 141). However, understanding the mechanism for the voltage sensitivity of PI3K or protein kinase C (PKC), its upstream effector, will require further study.
Increase in intracellular Ca2+
Cessation of flow results in immediate depolarization via KATP channel closure and is followed by calcium influx via T-type Ca2+ channels (92, 129, 134). In studies on stopped flow, the magnitude of cellular depolarization was found to be ∼20 mV. Assuming that ECs have a resting potential of around −60 mV at rest, stop of flow causes the membrane to reach −40 mV. At that potential, T-type Ca2+ channels open resulting in Ca2+ influx into the cell (150). T-type Ca2+ channel activation and Ca2+ influx was blocked by the KATP channel agonist cromakalim, providing evidence that this effect with stopped flow was the result of cell membrane depolarization. In flow-adapted bovine pulmonary artery ECs, the kinetics of [Ca2+]i changes (as monitored by Fura-2) with stopped flow showed resting [Ca2+]i concentrations to be ∼135 nM at ∼30 s after flow cessation in flow adapted and reached a maximum of 270 nM within 10 min (150).
NOX2 activation
As described above, the production of ROS is a prominent feature of the response to stop flow (25, 29, 129) and also has been observed with the onset of shear (87, 158). Studies with NOX2 null mice show essentially complete loss of the ROS response indicating that this NOX isoform is responsible for ROS production in the mouse stop of flow model (Fig. 3) (25, 161). Activation of this oxidase requires the sequential participation of several kinases, including PKC and PI3K (50). Likewise, this signaling pathway is required for NOX2 activation with stop of flow. The elements upstream of NOX2 are KATP and PECAM; indeed, lungs from KATP and PECAM null mice showed a marked reduction in production with stop of flow compared to wild-type NOX2 activation (and ROS generation) (106). Activation of NOX2 was also compromised in Akt1 null lungs with stopped flow compared to wild-type lungs. PI3K-Akt activity is significantly enhanced in systems where depolarization occurs with ischemia (wild-type and NOX2 null cells), but it is not affected in cells that do not depolarize (KIR6.2 null cells) (Fig. 4). Thus, the sequence of events that occurs post flow cessation is membrane depolarization followed by PI3K/Akt activation, which leads to NOX assembly and ROS generation (25). We have recently shown that peroxiredoxin 6 also plays an important role in the NOX2 activation cascade and its PLA2 activity is required for ROS production with ischemia (28, 85). This cytosolic protein is phosphorylated upon the activating signal resulting in its binding to the membrane substrate and generation of the phospholipid hydrolysis products that are necessary to initiate the translocation of cytosolic components of the NOX2 enzyme complex (28).
Nitric Oxide
Increased NO production has been demonstrated with a change of shear, either an increase or a decrease (2, 51, 148) With the abrupt decrease in shear, NO production follows the depolarization event and is blocked by preventing cell membrane depolarization by pretreatment with cromakalim (129, 134). NO production is inhibited by L-NAME indicating that NOS is activated, presumably as a result of the increased cytoplasmic Ca2+. NO production was markedly reduced by the presence of a calmodulin inhibitor. The ∼45 s delay between membrane depolarization and NO generation observed in isolated rat lungs may reflect the time required for the increase in [Ca2+]i and subsequent activation of NOS through [Ca2+]i-mediated and calmodulin-dependent mechanisms. The dependence of NO generation on [Ca2+]i and calmodulin suggests that eNOS is the responsible isoform for the major fraction of NO production (129, 134, 148). Increase of nitrotyrosine was also observed in lungs subjected to ischemia; this was blocked upon the inhibition of NO (70).
Targets of the Mechanosignal
The transmission of the mechanosignal occurs via the production of ROS; indeed, several redox-sensitive pathways are activated with alterations in shear stress. Some of the targets of ROS are also part of the signaling cascade that drives ROS production. Presumably, several of these are part of a feedback mechanism to modulate upstream signaling events.
HIF-1α and VEGF
The accepted paradigm is that the transcription factor HIF-1α and β, which regulates the transcription of >200 genes, is modulated by oxygen tension. In most organs, stop of flow is accompanied by hypoxia that causes the activation of HIF-1α and β. Upon activation, HIF-1 translocates to the nucleus where it mediates the transcription of several genes (126). This does not occur in normal cells as HIF-1 is degraded by prolyhydroxylases (PHD), but under hypoxic conditions, the PHD activity is inhibited. However, studies now suggest that ROS can directly modulate HIF-1α activity via NFκB independent of hypoxia and PHD (15, 58).
HIF-1 is well established to regulate the transcription of VEGF; in our studies on stopped flow in the mouse hind limb ischemia model, we observed HIF-1α activation and increased VEGF expression, both of which were NOX2 dependent. The lack of revascularization with stopped flow in NOX2 mice could be rescued by the delivery of VEGF into the ischemic tissue. We also found that cells subjected to acute stop of flow in vitro showed increased cell proliferation and tube formation in Matrigel (18, 19, 99); this response to loss of shear was both NOX2 and HIF-1α dependent.
Transcription factors
Cessation of flow to pulmonary artery ECs in vitro results in the activation of transcription factors, NFκB and AP-1 (149). Both of these factors are associated with increased cell division. The increased expression of the p50 and p65 subunits of NFκB and the c-jun and c-fos subunits of AP-1 was abolished by pretreatment with ROS scavengers or by blocking NOX2 activation (148, 149). Stopped flow also caused increased DNA synthesis, cellular G0/G1 arrest, and a concomitant increase in the number of cells in S phase (149), an effect that was reversed by inhibitors of ROS generation.
Ion channels
Although ion channel activity is clearly upstream of ROS with altered shear stress, further studies on ion channel regulation have shown that ROS, specifically H2O2, can in turn modulate channel function. ROS can oxidize sulfhydryl groups located on the ion channel proteins, cause peroxidation of membrane phospholipids, or inhibit membrane-bound enzymes that affect channel activity. In the case of the mechanoresponsive KATP channels, oxidant stress results in glutathionylation of thiol groups in the vicinity of the KIR pore protein, thereby preventing opening of the channel pore (156, 157). This important issue is discussed in a separate review in this Forum (155). This is the apparent mechanism for inhibition of KIR channels by H2O2. However, the levels of ROS necessary for this effect are presumably higher than that generated as a signaling mechanism for altered flow. NOX2 and AKT1 null cells and lungs show membrane depolarization with stop of flow but do not generate ROS (25). These results indicate that the inactivation of KATP channels that occurs with stop of flow is not ROS mediated. Additionally, PI3K and Akt have been reported to affect KATP channel function elsewhere, but we observed in our studies that KATP induced depolarization occurred even under conditions where PI3K was blocked (wortmannin) or Akt nulls were used; hence, it does not seem plausible that PI3K drives channel function (25).
Kinases and phosphatases
ROS are known to activate kinases and phosphatases (96). Three mechanisms by which ROS activate these have been proposed. First, ROS may directly activate kinases by altering protein–protein interactions via sulfhydryl groups. Second, protein tyrosine phosphatases that contain a cysteine residue in their activation site may be directly inhibited by ROS, which in turn results in tyrosine phosphorylation of the kinases. Third, ROS may affect regulatory proteins; for instance, ROS-induced MAPK activation often occurs via the modification of the dual-specificity MAPK phosphatases (DS-MKPs), which dephosphorylates MAPK. NOX-derived ROS are reported to modulate DS-MKP and thus cause enhanced MAPK activation under oxidative conditions (40, 41, 65). In our studies with stop of flow, PI3K activation was found to be independent of ROS, as NOX2 null cells showed PI3K activity comparable to that of the wild type.
Physiological Response to Altered Shear
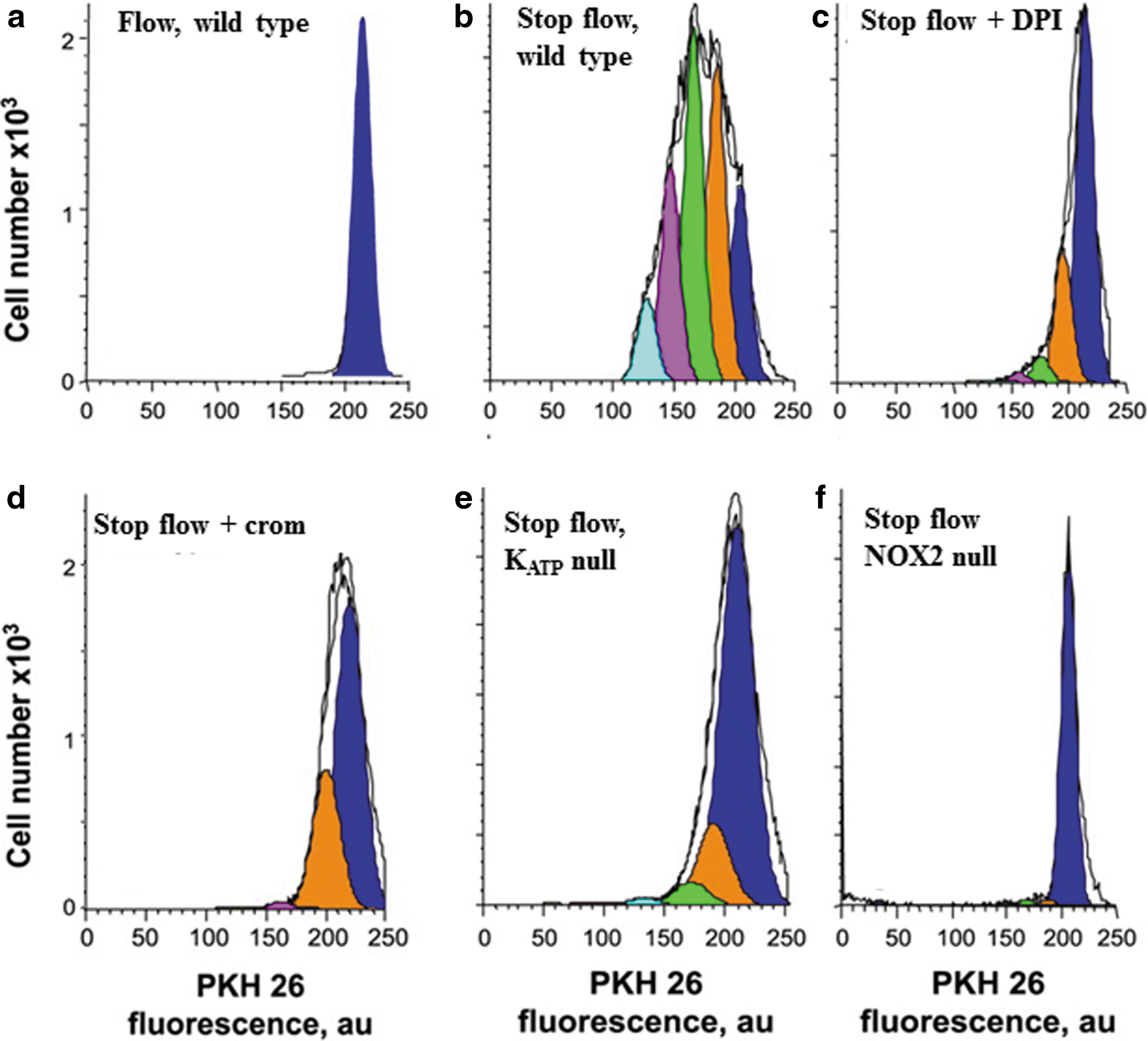
ROS have been shown to result in activation of division in ECs and cell proliferation (98). This physiological response as related to ROS generation with altered shear stress has been confirmed with the in vitro model of stopped flow in flow-adapted ECs (98, 99). These studies showed a 2.5-fold increase in the cellular proliferation index compared to control cells (Fig. 5). Cellular proliferation is abolished in cells pretreated with cromakalim or DPI or in cells with knock-out of NOX2, the KATP channel, or caveolin-1. Each of these manifestations has been shown to abolish ROS generation (98, 99) (Fig. 5). Cessation of flow with ECs in vitro led to tube formation in a Matrigel layer and neovascularization in a subcutaneous Matrigel plug (18). These data provide evidence that cell proliferation is a result of the signaling cascade associated with shear sensing. Thus, cell proliferation is not random but a directed response leading to the formation of new vessels.
Conclusions
There have been significant recent advances in the understanding of the molecular mechanisms by which cells sense physical forces as well as the signal transduction pathway that modulates gene expression and cellular responses. Our laboratory has utilized a stop of flow model in which the activation of NOX2 results in cell signaling medication by ROS that are generated by the activation of NOX2. Additional features of the stop of flow signaling cascade are cell membrane depolarization, NO generation with subsequent vasodilation, and ROS generation with subsequent neovascularization. These responses from a homeostatic standpoint suggest an attempt to restore impeded blood flow (Fig. 6). In contrast to the stop of flow models, most in vitro studies of endothelial mechanotransduction have evaluated the onset of flow with cells grown under static conditions. This experimental model has limited direct physiological relevance since it is known that these cells are not flow adapted unlike ECs in vitro. Despite this caveat, it seems clear that the onset of flow and the stop of flow elicit similar responses, representing a change from a previously flow-adapted state.
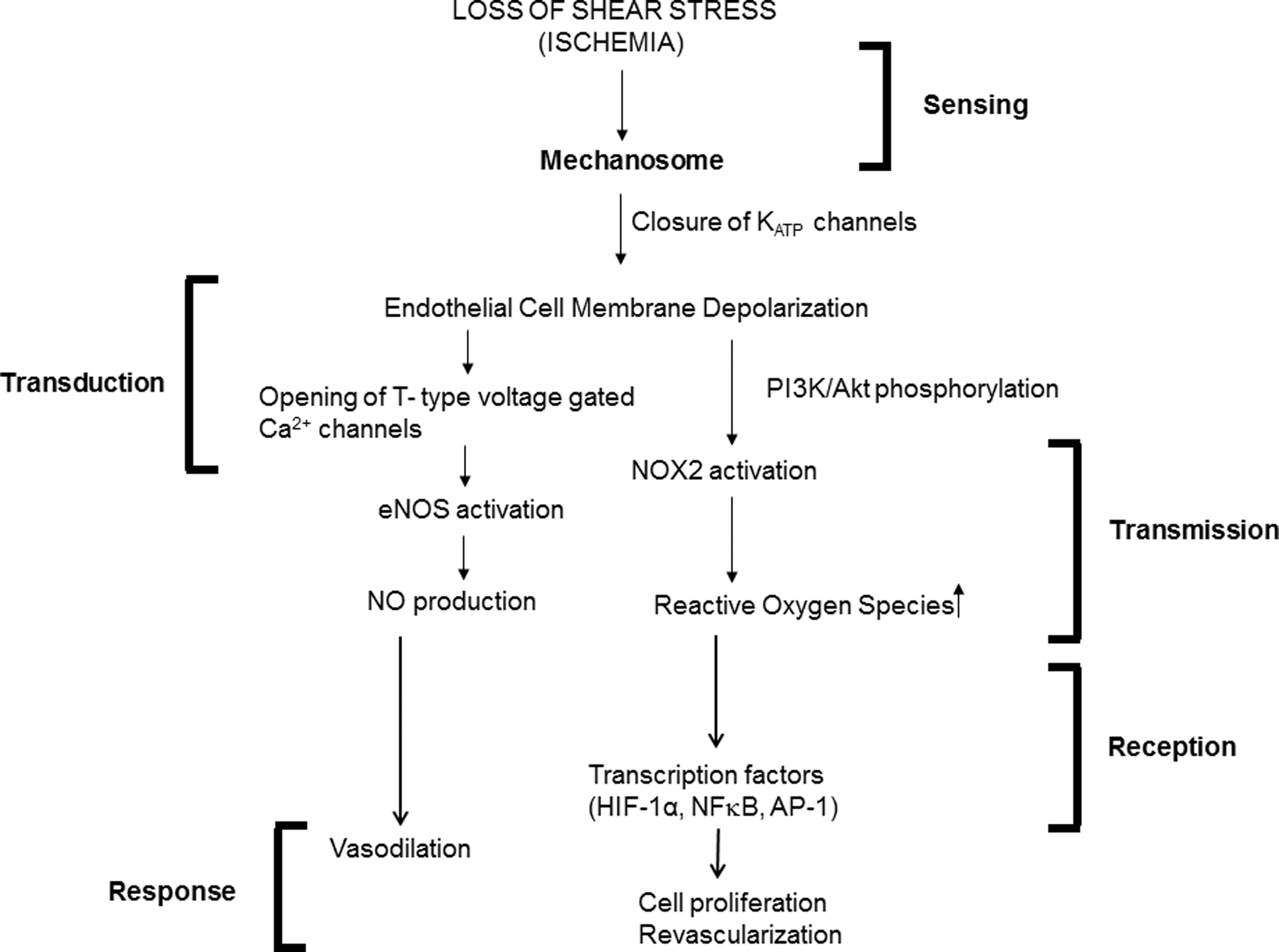
We thus propose a mechanosome hypothesis as the sensor for endothelial mechanotransduction. Under constant laminar flow, the mechanosome network is essentially inactive (OFF mechanosome); alteration of shear forces (ON mechanosome) results in sensing of shear. The concept of the mechanosome has been applied previously to bone loading (13, 14, 111), but here the concept is broadened to include the vascular endothelium and possibly other cell types.
Although the signaling paradigm for mechanotransduction with altered shear stress has been fairly well delineated, there are still aspects that need further inquiry. One of these is the nature of the link between the initial sensing event and transduction of the mechano-pathway. Another is the basis for differential sensing with various shear patterns. Finally, there is a lack of understanding of how the signaling pathway translates into gene expression. Such information is vital for the elucidation of the fundamental mechanisms of mechanotransduction and endothelial function in health and disease.