
Abstract
Introduction
W
The Potential Use of Cell-Based Therapies for Developmental Lung Injury and Lung Vascular Diseases
Studies of lung injury in mice, including genetic lineage tracing experiments, have revealed the presence of resident lung progenitor cells that are able to proliferate in response to injury and contribute to tissue maintenance and repair. Destructive processes that underlie diseases, such as BPD, affect the global lung architecture, disrupting the functional alveolar–capillary unit, and lead to arrested or dysfunctional lung growth. To optimize physiological lung growth and function, there is a need to regenerate not only the lung epithelium, but the associated capillaries of the vascular network. The feasibility of using endogenous organ progenitor cells to treat injury has been successfully tested for the heart in the SCIPIO trial (17); autologous cells isolated from the hearts of patients with ischemic cardiomyopathy were expanded ex vivo, returned to the patient, and shown to confer improvement in cardiac function. Recently, c-kit+ native lung stem cells were isolated from human lung, clonally expanded in culture, and shown to repair and restore normal lung architecture, including conducting airways and vessels, in a murine cryoinjury model (37). To date, preclinical and clinical studies of lung disease have focused on stem cells harvested from sources outside the lung, such as bone marrow, blood, umbilical cord, and adipose tissue. These stem cells include endothelial progenitor cells (EPCs), a subpopulation of mononuclear cells in the blood that are capable of differentiating into endothelial cells in vitro (2), amnion-derived epithelial cells (AECs) (75), and mesenchymal stem cells (MSCs) (63). The treatment of lung disease with ex vivo expanded EPCs derived from blood or bone marrow has been tested in experimental models of PH, and in adults and children with PH (76, 82). In experimental models of BPD, EPCs or bone marrow myeloid-like angiogenic cells, and AECs have shown therapeutic potential with restoration of the alveolar structure and vascular integrity in the neonatal hyperoxia mouse model (6, 75). AECs were also reported to have regeneration potential in a fetal sheep model of ventilation-induced lung injury (32). In addition, several reports point to the therapeutic potential of MSCs in models of lung disease, which will be reviewed in the subsequent sections. Regardless of the stem cell type, however, significant hurdles remain to be overcome to actualize the potential of cell-based therapies in the clinic. The major among them is the absence of a rigorous characterization of cell lines that can be unequivocally reproduced among experimental groups and the uniformity of culture and expansion procedures. These issues are discussed in more detail below in the context of MSCs.
MSC Characterization
MSCs, originally referred to as bone marrow stromal cells, were first described in 1968 as a fibroblast-like population in the bone marrow. They are the most widely studied to date in preclinical models of disease and in human transplantation studies. Because investigators have used different isolation and characterization methods for these cells, in 2006, the International Society of Cellular Therapy (ISCT) defined MSCs by the following three criteria (21): (i) MSCs must be adherent to plastic under standard tissue culture conditions; (ii) MSCs must express certain cell surface markers, such as CD73, CD90, and CD105, and lack expression of other markers, including CD45, CD34, CD14, CD11b, CD79α, CD19, and HLA-DR surface molecules; (iii) MSCs must have the capacity to differentiate into osteoblasts, adipocytes, and chondroblasts under in vitro conditions. Using this definition, MSCs from different types of tissues have been successfully isolated, including adipose tissue, Wharton's jelly, umbilical cord blood, and even adult lung tissue (58, 61). MSCs of bone marrow origin were first proven to have efficacy in models of acute and fibrotic lung injury. Therapeutic properties were subsequently demonstrated in various disease models with MSCs isolated from other tissues, including the human adult lung and human embryonic stem cells (hESC) stimulated to differentiate into MSCs (35, 74). In general, MSCs are easy to isolate in large numbers from different sources. Adipose-derived MSCs, for instance, are available in large quantities from liposuction procedures, and are thus considered as major candidates for future approaches to regenerative medicine (65). Unlike ES cells, MSCs can be easily expanded in vitro under standard culture conditions, while maintaining their undifferentiated state and continuing to express MSC markers for several passages. In addition, they do not carry the teratogenic potential of ES cells after in vivo transplantation (20). MSCs are also thought to be immunoprivileged in that they evade clearance by the recipient immune system (46) due to low expression of the major histocompatibility complexes and to their ability to inhibit proliferation and function of immune cells, such as dendritic cells, NK cells, and T and B lymphocytes (8, 23, 67). MSCs can also be genetically engineered without losing their stem cell properties and their differentiation potential. However, the role of MSCs in the field of regenerative medicine is not well defined and requires further study. Despite their ease of isolation and the existence of phenotypic markers for MSCs, no correlation between their phenotypic characteristics and immunomodulatory activity or lung regeneration capacity has been established. This is further complicated by the many sources of MSCs, the different culture techniques, as well as by species-specific properties. Although MSCs isolated from different sources express widely accepted markers, adhere to plastic, and show baseline differentiation potential to osteoblast, chondroblast, and adipocyte lineage in vitro, there remains a large variation in differentiation capacity and gene transcription programs depending on the tissue source and species of these cells (34, 43, 71). Even if MSCs maintain their differentiation potential and differentiation markers after genetic manipulation or expansion in culture, these properties do not ensure immunomodulatory and regenerative capacity. Moreover, bone marrow-derived MSCs are the best characterized and have been shown to have cytoprotective effects; yet, they have also been reported to accumulate genetic mutations after extensive expansion (9, 28, 81). This genomic instability may account for the unusual in vitro behavior, with early cultures growing rapidly, but eventually giving rise to heterogenous populations with variable properties that are often unable to reproduce the protective MSC effect.
Currently, our knowledge remains incomplete on the signals required to maintain the therapeutic/regenerative MSC phenotype upon expansion in vitro. In addition, we need insights on the cellular markers defining this phenotype, and the ISCT marker panel may not be sufficient in all cases. Further work is necessary to resolve the above ambiguities and develop rigorous standards to ensure reproducibility in preclinical studies. This hurdle is compounded by the fact that preclinical models of lung disease have diverse precipitating triggers, some more and some less relevant to human disease. Although lung inflammation is the most common denominator in these models, the choice of the particular model may be as important to a successful future translation to the clinic as the MSC phenotype used to treat the animals.
Role of MSCs in Lung Disease
Cellular therapies exhibit great potential for the treatment of a diverse number of lung diseases. MSCs have therapeutic applications in multiple clinical disorders, including myocardial infarction, diabetes, sepsis, graft-versus-host disease, and hepatic and acute renal failure (1, 7, 14, 26, 31, 33, 39, 53). Given the complex architecture of the lung, different cell therapy approaches have been implemented, including the use of 3D bioengineered and decellularized scaffolds populated with progenitor cells. Due to the many cell subtypes of the lung parenchyma, and according to the cell type that is mostly affected in different lung injury models, approaches using ES cells, induced pluripotent stem cells (iPS), EPCs, and MSCs have been implemented with varying results and with different operative mechanisms of action. In preclinical models, EPCs, and ES cells or iPS-derived alveolar epithelial cells, can engraft in areas of injury and repopulate the lung, whereas MSCs seem to work in a paracrine manner with very limited engraftment. In the lung, MSC treatment has proven effective in many preclinical models of PH, BPD, acute lung injury, and pulmonary fibrosis (Table 1). Treatment with allogeneic human MSCs or their conditioned media (CM) administered 1 h following endotoxin-induced lung injury reduced extravascular lung water, improved lung endothelial barrier permeability, and restored alveolar fluid clearance (49). In the hyperoxia-induced murine BPD model, systemic or intratracheal transplantation of bone marrow MSCs suppressed lung inflammation, improved survival and exercise tolerance, while attenuating alveolar and lung vascular injury and PH. Although a potential differentiation capacity of MSCs into the alveolar epithelium was proposed, the protective effect was predominantly paracrine mediated (3, 73). Using the same model, MSC-CM were more effective than MSCs in preventing or reversing established BPD (30). In PH, intratracheal or systemic administration of rat MSCs 2 weeks after monocrotaline (MCT) treatment attenuated the rise in pulmonary arterial pressure and pulmonary vascular resistance, restored pulmonary responses to acetylcholine toward values measured in control rats, and decreased the right ventricular hypertrophy (RVH) (4, 52, 79), and overexpression of eNOS increased MSC efficacy (38). CM from MSCs were also able to alter the proliferative response of pulmonary artery smooth muscle cells in vitro, and MSC treatment ameriolated PH and early pulmonary inflammation in a hypoxic mouse model (48). In this model, MSCs overexpressing heme oxygenase-1 (HO-1) reversed established PH when delivered after 5 weeks of hypoxia and inhibited right ventricular thrombus formation and RV failure in the recipient HO-1-deficient mice (50). In a model of ventilator-induced lung injury, MSCs attenuated alveolar tumor necrosis factor α (TNFα) concentrations, while increasing concentrations of interleukin 10 (IL-10), thus decreasing lung inflammation and histological lung injury and restoring the lung structure (15, 19).
BPD, bronchopulmonary dysplasia; IL-10, interleukin 10; MCT, monocrotaline; MSC, mesenchymal stem cell; PH, pulmonary hypertension; TNFα, tumor necrosis factor α; BAL, bronchoalveolar lavage; RV, right ventricular; CM, conditioned media.
Although exogenous administration of MSCs isolated from tissues other than the lung has proven to be beneficial in many lung injury models, the role that resident lung MSCs play in disease remains to be fully characterized. According to Caplan and Correa (12), perivascular MSCs that arise from pericytes serve as drugstores that react to injury by acquiring an anti-inflammatory and regenerative phenotype. In a bleomycin-induced pulmonary fibrosis model, Jun et al. reported a loss of resident MSCs with an associated impaired T-cell response, while administration of exogenous lung MSCs, isolated as the side population, or their CM, conferred a protective effect (36). On the other hand, resident lung MSCs were found to contribute to vascular remodeling in a superoxide dismutase knockout model (16), and increased numbers of MSCs were identified in tracheal aspirates of newborns with BPD (62). In the latter model, Popova et al. reported activation of the β-catenin pathway leading to myofibroblastic differentiation of neonatal resident lung MSCs isolated from tracheal aspirates of premature infants with respiratory distress, which may have contributed to the BPD phenotype. During chronic hypoxia, MSCs may restore lung vascular homeostasis by inhibiting inflammation and vascular remodeling, thereby promoting vascular stability or they may potentially contribute to vessel wall remodeling (16). Significantly, MSCs have been shown to release growth factors when stressed by hypoxia in vitro (51, 60), and as discussed by Foronjy and Majka, understanding the lung microenviroment is important because the endogenous MSC pool may react in opposing ways to different environmental cues (27).
The recurrent theme in the above studies appears to be that the therapeutic action of exogenous MSC treatment on lung injury is predominantly an immunomodulatory paracrine function. The primary observation is the suppression of inflammatory responses and, in the models tested, media conditioned by MSCs appear to be as efficacious or even more efficacious than cell transplantation. On the other hand, based on the current literature, the role of lung resident MSCs in repairing injured tissue or contributing to the injury itself remains unresolved.
Mechanism of MSC Action
Using adult lung injury models, most early studies focused on the idea that transplanted MSCs engraft in the lung and adopt various lung cell phenotypes, from differentiated airway and alveolar epithelial to interstitial and pulmonary vascular cells. This concept was further supported by reports that bone marrow stromal cells, after transplantation, differentiate into alveolar epithelial cells. Using a lacZ/neomycin resistance transgene (lacZ/neo), it was reported that bone marrow-derived cells engraft as type I pneumocytes in a mouse model (42); and, in a radiation pneumonitis female mouse model, male bone marrow transplantation resulted in Y(+) type II pneumocytes as early as 5 days post-transplant (68). Donor MSC markers in the recipient lung were also found to be pronounced in areas of lung injury, suggesting homing. However, whether significant engraftment and differentiation take place in the injured areas of the lung remains controversial. Using confocal microscopy techniques and 3D reconstruction, it was demonstrated that epithelial engraftment is rare, and lung autofluorescence accounts for many false-positive results (13), indicating that original reports grossly overestimated donor cells in the recipient lung. Even in cases of excessive lung injury, engraftment was transient and no stem cells from primary culture were found 7 days after intratracheal delivery (24, 47). It is safe to assume that, in models of lung disease, regeneration of tissue cannot account for the therapeutic efficacy of MSC treatment.
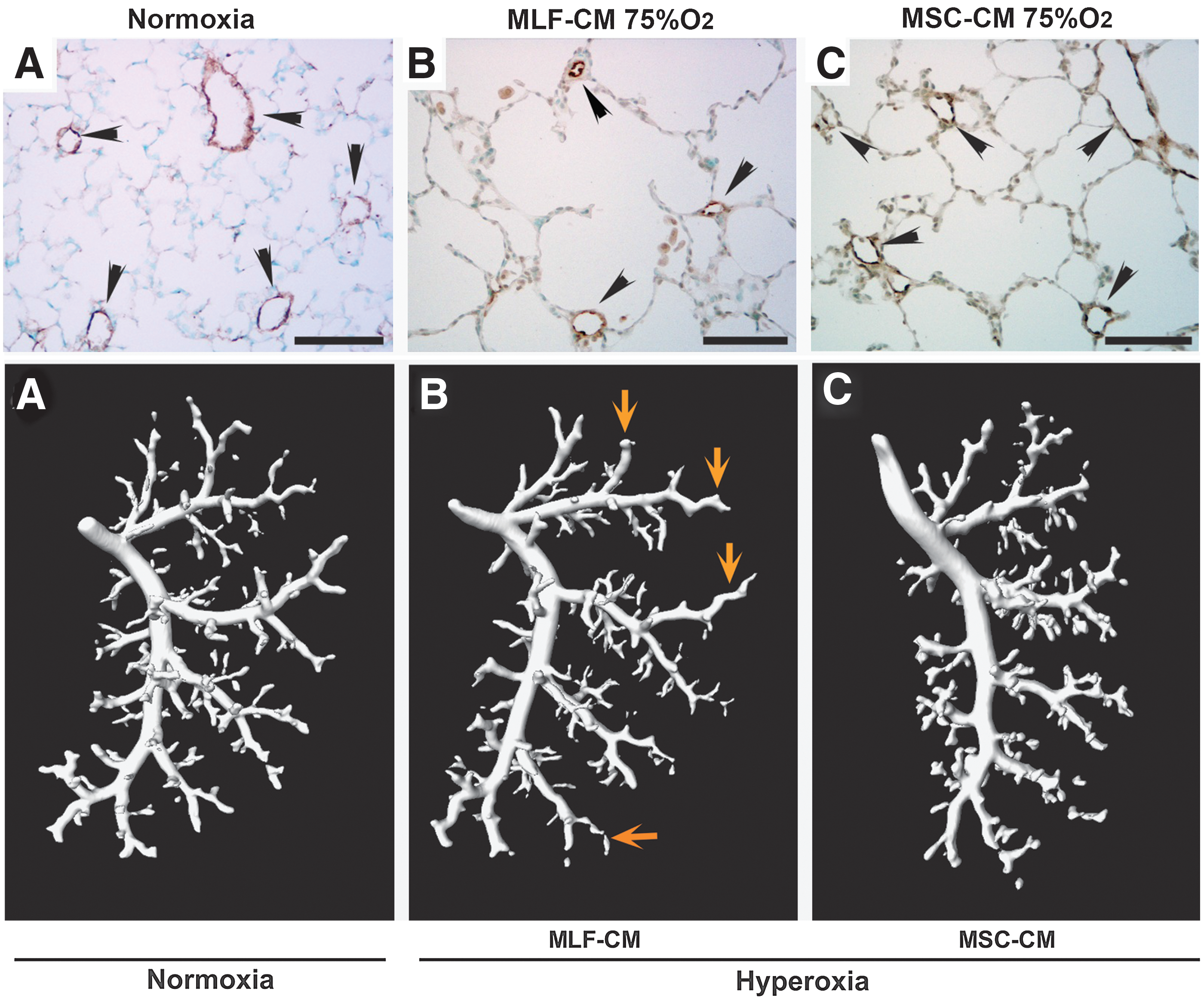
The lack of engraftment of MSCs suggested a cell-autonomous therapeutic process. In 2005, using a myocardial infarction model, Gnecchi et al. first proposed that the effect of genetically engineered MSCs takes place in less than 72 h, before any regeneration can take place, and showed that CM from MSCs cultured in vitro have comparable efficacy to whole cell transplantation in preventing ventricular remodeling (29). In the lung, using an in vitro system, Liang et al. showed that bone marrow MSC-CM can inhibit the proliferation of pulmonary artery smooth muscle cells, an effect not observed with fibroblast-CM (50). This observation demonstrated that, in addition to the immunomodulatory effect of MSCs on lung inflammation, MSCs release factors with an antiproliferative activity on smooth muscle cells, directly inhibiting vascular remodeling. In the hyperoxia-induced lung injury mouse model, bone marrow MSC-CM were more effective than cells in preventing vascular changes associated with PH. A single dose of bone marrow MSC-CM, but not the pulmonary artery smooth muscle medium, was able to suppress RVH, vascular remodeling, and vascular wall thickness in hyperoxia-treated neonatal mice (3). The administration of MSC-CM shortly after hyperoxic exposure abrogated the inflammatory lung infiltration, pointing to the existence of anti-inflammatory mediators in the medium (3). In the same model of established BPD, by delivering MSC-CM, Hansmann et al. were able to reverse the hyperoxia-induced parenchymal fibrosis and peripheral PA devascularization, partially reverse alveolar injury, and normalize lung function (Fig. 1) (30). In addition, MSC-CM fully reversed RVH and attenuated peripheral PA muscularization associated with hyperoxia-induced BPD (30). Further support for the anti-inflammatory mediators of MSC secretome came from Lee et al., who showed that factors secreted by MSCs prevent hypoxia-induced pulmonary inflammation and pulmonary vascular remodeling (48). Pretreatment of hypoxia-exposed mice with bone marrow MSC-CM prevented early lung inflammation with inhibition of macrophage accumulation, suppression of inflammatory cytokines, including monocyte chemoattractant protein-1 (MCP-1) and found in inflammatory zone1/hypoxia inducible mitogenic factor (FIZZ1/HIMF), and prevention of later hypoxic PH.
Extracellular Microvesicles: The Therapeutic Vector of MSCs?
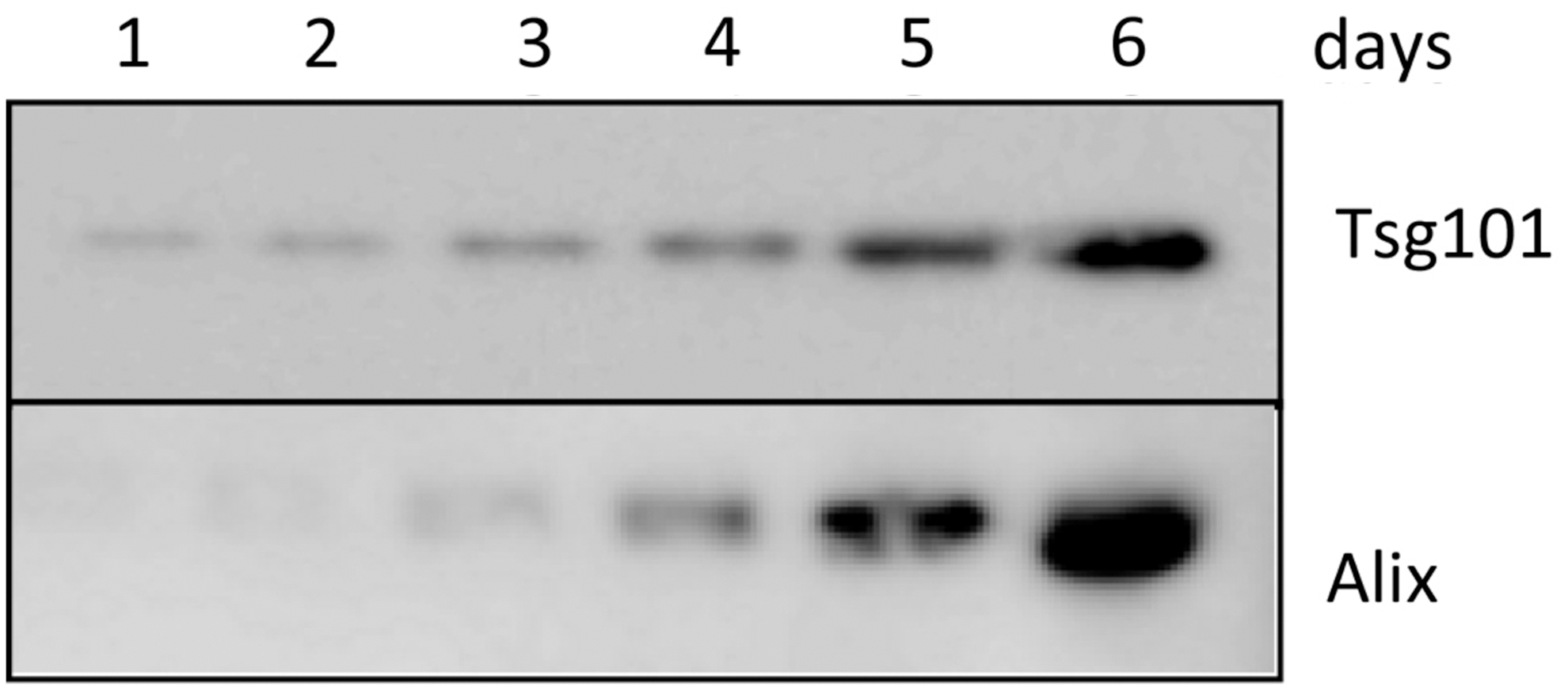
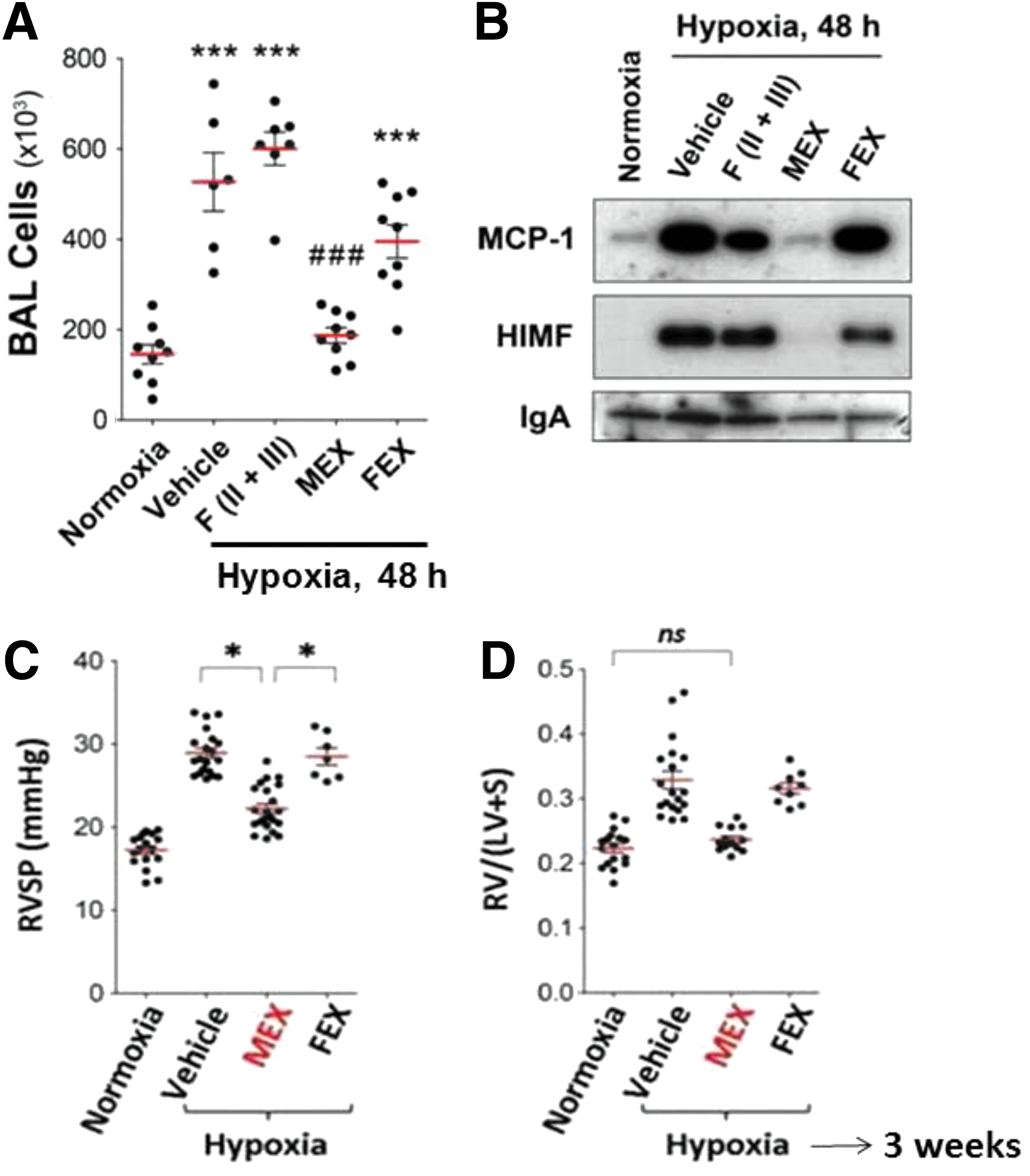
In proteomic analysis of the MSC secretome, many immunomodulators and matrix components were detected, as well as proteins commonly associated with extracellular vesicles derived from different cell types (Fig. 2). Many of these, including CD63, CD81, moesin, Alix, Tsg101, and hsp70, are enriched in exosomes, small microvesicles, 30–100 nm in diameter, which are stored within multivesicular bodies and released into the environment by fusion with the cell membrane (22, 70) (Fig. 3). The horizontal transfer between cells of functional mRNAs and small noncoding RNA species is mediated by exosomes (72), and, as such, they have been identified as an important cell-to-cell communication mechanism. Exosomes are produced by all cells and bear adhesion molecules on their surface, which may serve to target delivery of their cargo into specific cell types (Fig. 4) (54, 55). In the effort to define exosomes, their biogenesis, and their cargo, it has become evident that different subpopulations of microvesicles and exosomes are secreted by different cells. They contain a distinct cargo that not only represents the cell of origin, but may also be differentially enriched in specific nucleic acid or lipid species (18, 66). Because distinct mechanisms account for the uptake of different populations, the mechanism of uptake by the recipient cell, ranging from endocytosis to membrane fusion, has also been debated. The effect of exosomes in vitro has mostly been focused on their interaction with the immune system, including dendritic cell maturation, Treg and B-cell responses (11, 41, 69). Lee et al. demonstrated that exosomes mediate the cytoprotective effect of bone marrow MSCs in hypoxia-induced PH (48). In this model, administration of MSC-derived exosomes, identified through widely accepted exosomal markers and visualized by electron microscopy, protected against the elevation of right ventricular systolic pressure and the development of RVH after 3 weeks of hypoxic exposure, while microvesicle-depleted CM had no effect (Fig. 5). Exosomal treatment was also able to abrogate early hypoxic macrophage influx and downregulate hypoxia-activated inflammatory pathways, thus mediating the anti-inflammatory properties of MSCs. Exosome action was further validated in vitro, as exosomes ameliorated the hypoxic induction of STAT3 in pulmonary artery endothelial cells. This observation was further expanded to MSCs of human origin, using two different sources, bone marrow and umbilical cord Wharton's jelly. Researchers have isolated and characterized exosomes from MSCs originating in almost all sources, including hESC-MSCs, and the MSC exosome has been proposed as the alternative therapeutic vehicle for MSCs in many disease models (5, 59, 64, 78). Such models of disease include a myocardial injury/reperfusion model in which MSC exosomes decreased the infarct size and ameliorated reperfusion injury (44); a cisplatin-induced acute kidney injury model in the rat where adipose tissue MSC exosomes ameliorated oxidative stress and cell apoptosis, promoting cell proliferation in vivo and in vitro (22, 80); and an acute kidney injury model where MSCs activate a proliferative program in tubular cells (10).

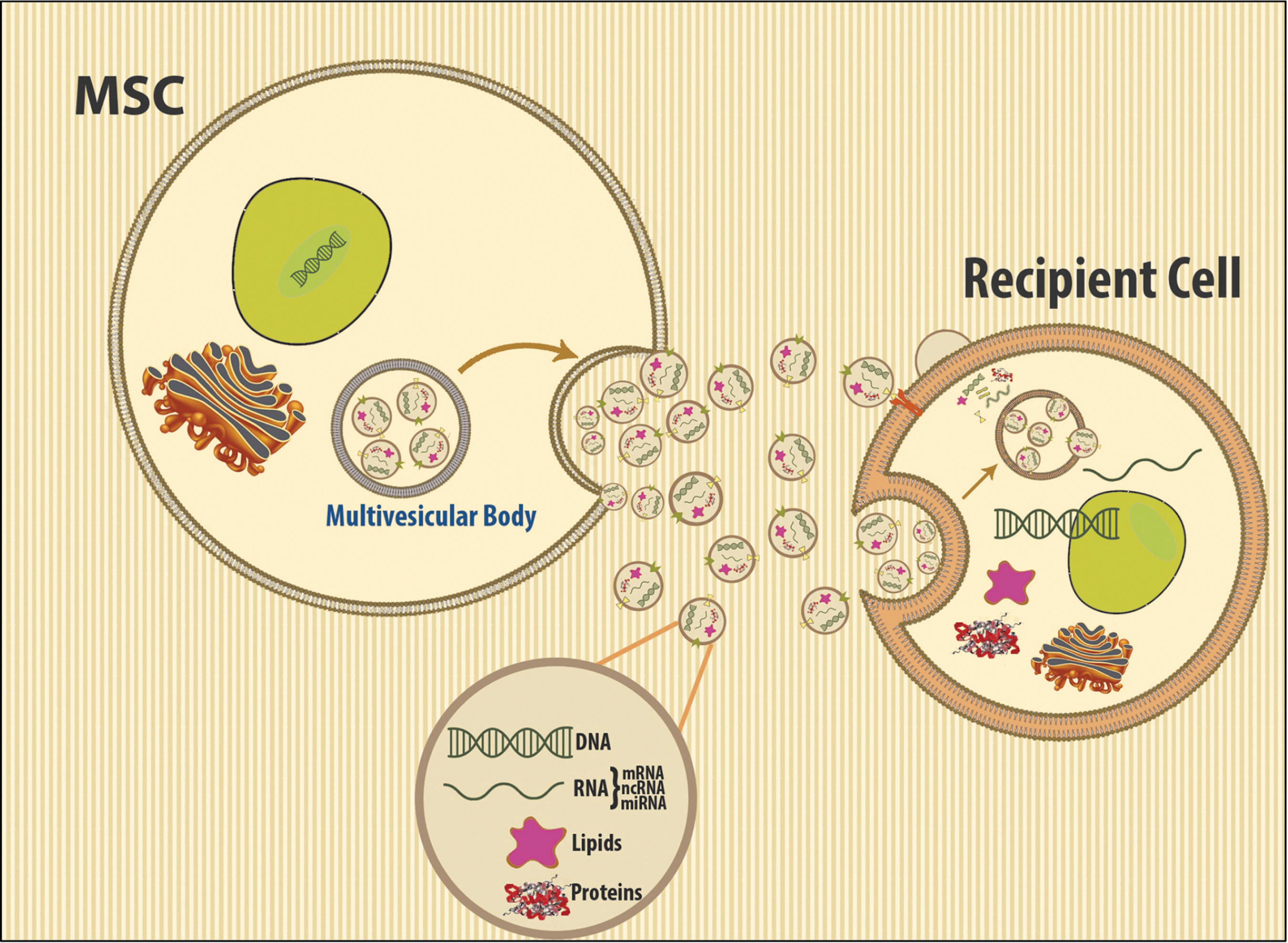
Despite the increasing evidence that microvesicles constitute the active protective moiety of MSCs in models of oxidative injury, no definite mechanism of action is widely accepted although multiple exosomal components are proposed to mediate their protective function. This may reflect their ability to target multiple intracellular processes and to modulate cell injury in different levels, accounting for their efficacy in diverse settings. It is also becoming more evident that MSCs from different sources secrete different populations of exosomes, and that exosomes dynamically reflect the intracellular MSC state with different culture conditions resulting in a totally different exosomal composition. Such variety in the compositions enables a range of capabilities: exosomes are potent carriers of extracellular RNA (exosomal shuttle RNA) and MSC exosomes are shown to transfer functional microRNAs into recipient cells (72) (Fig. 4). All components of the 20S proteasome were identified in MSC exosome preparations, and their action in misfolded protein response was evaluated in vivo (78). Moreover, exosomes may stimulate organ-specific progenitor stem cells to expand, proliferate, and differentiate and, most importantly, to participate in cellular repair to replace injured cells. Phase I trials of microvesicles derived from dendritic cells for immunotherapy of advanced cancer (25, 57) showed that large-scale, clinical-grade production and delivery of MSC exosomes/microvesicles are feasible and safe (45). The unique capacity of the MSC exosome to combine the advantages of cell-based therapies without the potential risks for long-term maldifferentiation of engrafted cells and tumor generation makes their study an active area of investigation that holds great promise for the effective treatment of several inflammatory diseases of the lung.
Conclusion
Cell-based therapies for lung diseases hold tremendous promise. Over the last decade, numerous studies in preclinical models and human clinical trials have demonstrated the ability of EPCs and MSCs to prevent and treat pulmonary vascular diseases, as well as to restore lung architecture in neonatal murine models of BPD. Importantly, cell-free therapies are on the horizon, including MSC-released extracellular vesicles/exosomes. These critical vectors of MSC action have the potential to deliver diverse signals, including proteins, lipids, and nucleic acid material such as mRNA or noncoding RNA, to the recipient lung cells and to alter signaling pathways to restore lung function. The results from preclinical models are very promising, but a number of questions remain: is the production of therapeutic microvesicles a property of all MSCs, independent of their tissue of origin? This is important for future large-scale production and clinical translation, since adipose MSCs, umbilical cord blood, or Wharton's jelly MSCs can be obtained much easier than those from the bone marrow. How do the MSC differentiation potential and passage in culture relate to microvesicle production? This reflects the hurdles associated with MSC characterization and phenotypic markers. A critical issue will be defining the effective therapeutic dose of MSC exosomes/microvesicles in the patient and the need for large-scale MSC cultures, which should be readily produced in bioreactors. In addition, we know very little about the microvesicle mechanism of action. What is the active cargo they contain and to what target cell in the injured lung do they deliver it? Although the predominant effect appears to be immunomodulation, the pathways involved are not defined. In addition, the fact that a single dose of microvesicles has long lasting effects suggests epigenetic regulation of critical signaling pathways of immune function and lung homeostasis. Clearly, more work is required to better characterize the biology of MSC-released vesicles/exosomes, and the molecular and epigenetic mechanisms of their action on inflammatory pathways of injury and repair that underlie lung diseases such as BPD.
Footnotes
Acknowledgments
The authors wish to acknowledge Dr. S. Alex Mitsialis and Ms. Ivy Dodge for editorial review, and Ms. Sarah Gately for expert assistance in preparing the article. This work was supported, in part, by NIH grants RO1 HL055454 and RO1 HL085446 (to S.K.). K.S. is a recipient of a William Randolph Hearst Award.