
Abstract
Introduction
T
It is impossible to investigate the result of vitamin C deficiency in humans because it is fatal. Gulo−/− mice cannot synthesize vitamin C like humans, but show highly similar physiological responses upon vitamin C deficiency. Our experiment was done with Gulo−/− mice with the control in their vitamin C uptake. This is the first report regarding the direct in vivo effects of vitamin C in the development of the neonatal brain and in the already developed brain. Our data provide new insights into the crucial role of vitamin C in the normal development of the brain and maintenance of motor functions.
Vitamin C is regarded as one of the important nutrients in the development of the brain (21, 31). In the neonatal brain, the extensive growth during development increases the demand for antioxidants to prevent oxidative damage (35). Lower vitamin C levels are accompanied by increased malondialdehyde (MDA), F(2)-isoprostanes, and total glutathione in the brain, and vitamin C transport during pregnancy protects embryonic and postnatal pups from oxidative stress (18). Experiments with sodium-dependent vitamin C transporter (SVCT)-2 knockout mice have shown that this transporter is essential for perinatal survival and intraparenchymal brain hemorrhages resembling complications reported in premature infants and associated with learning and cognitive disabilities in later life (32, 61, 67). It indicates that vitamin C is crucial for early brain development, which can affect brain function in adults. Moreover, vitamin C deficiency leads to persistent impairment of hippocampal development in guinea pigs (66, 68). Prenatal vitamin C deficiency during pregnancy resulted in a significant reduction in postnatal hippocampal volume and migration of newborn cells into the granular layer of the hippocampal dentate gyrus (68). Postnatal vitamin C deficiency caused impairment of hippocampal memory function accompanied by a loss of hippocampal neurons (66). Therefore, pre- or postnatal malnutrition of vitamin C causes serious consequences in the development and functional maintenance of the brain (36, 67). To examine the effect of vitamin C on fetal and neonatal brains, it is important to control the in vivo vitamin C concentration through supplementation during gestation. However, the effect of vitamin C deficiency in the developing brain has not been fully investigated due to the limitation of vitamin C-controlling animal models.
Vitamin C also plays an important role as an antioxidant in the already developed adult brain (20, 52 –54). Several reports discussed oxidative stress in relation to the therapeutic function of vitamin C in the development of neurodegenerative diseases (e.g., Alzheimer's disease, Parkinson's disease, and ischemic stroke). In fact, vitamin C decreases β-amyloid generation and acetylcholinesterase activity and prevents endothelial dysfunction by regulating nitric oxide in Alzheimer's disease (20). Pretreatment with vitamin C decreased the expression of caspase-3 and prevented the deleterious effects of seizure and neuronal degeneration in a PTZ-induced parkinsonism model (43). However, the specific changes in the brain depending on the absence or presence of vitamin C have not been fully investigated in a nondisease state without neurodegenerative diseases. In addition, the potential role of vitamin C in its own right as a preventive or therapeutic agent for brain diseases is largely unknown.
In humans, there is a mutation in the gene encoding L-gulono-γ-lactone oxidase (gulo), which is responsible for the conversion of L-gulono-y-lactone, a metabolite of glucose, into vitamin C; however, most animals used in studies published elsewhere can synthesize vitamin C endogenously (5, 44, 45). Therefore, it was impossible to investigate the in vivo effect of vitamin C and its related mechanisms due to the ability of most experimental animals' ability to synthesize vitamin C. Gulo−/− mice were generated by the deletion of the gulo gene to examine its preventive effect on the development of atherosclerosis by regulating vitamin C supplementation (38), which is the relevant animal model for the investigation of in vivo vitamin C effects in humans. In our previous study, we found that vitamin C preferentially accumulated in the liver, heart, and brain of Gulo−/− mice (26). In the present study, we investigated the role of vitamin C in preventing abnormalities in the developing and already developed brain by controlling vitamin C supplementation in Gulo−/− mice.
Results
Increase in stillbirths and brain hemorrhages in neonates from vitamin C-deficient Gulo−/− mice
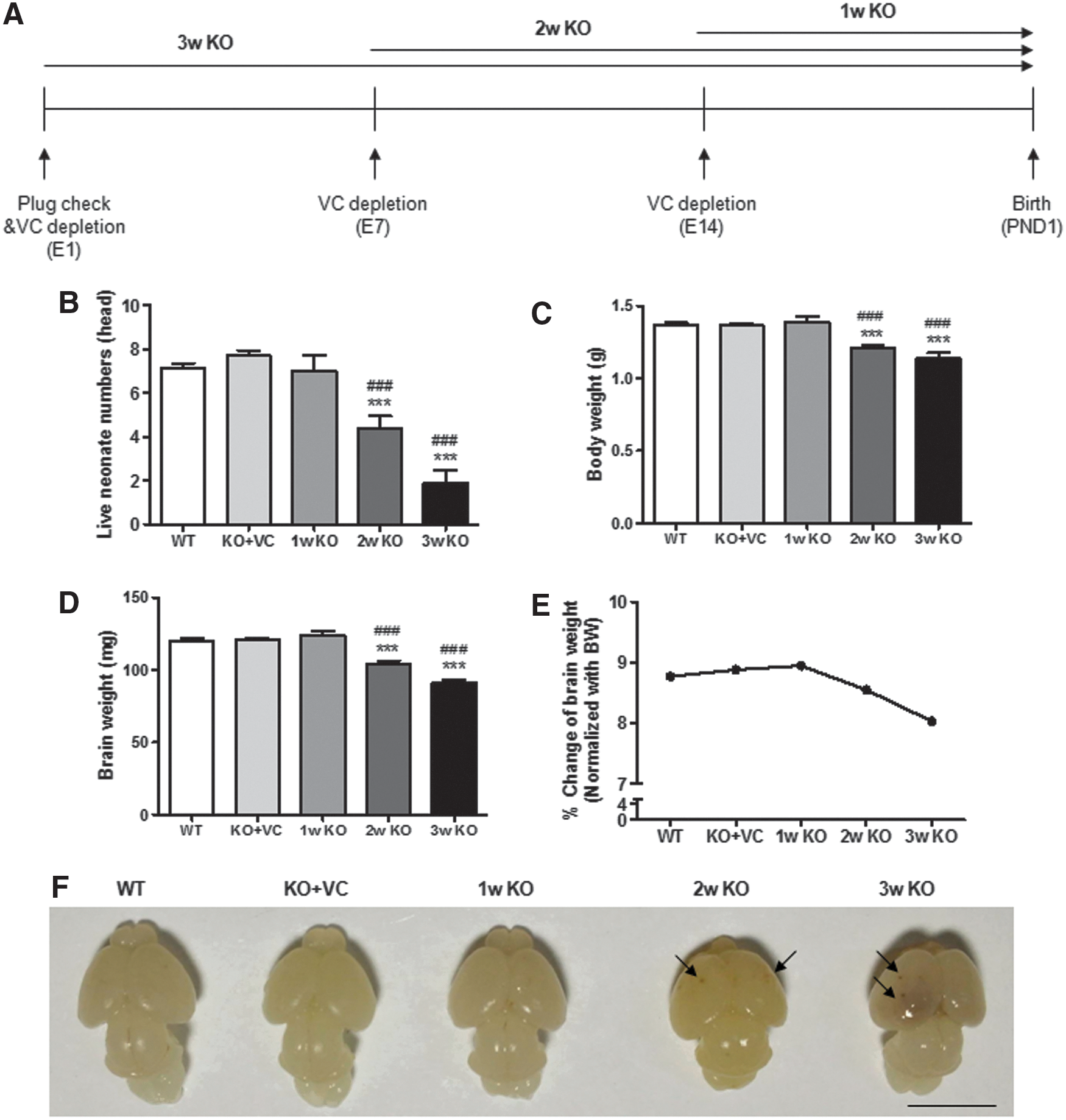
To examine the effect of vitamin C deficiency in the brain during gestation, vitamin C supplementation was withdrawn for 1, 2, and 3 weeks until delivery (Fig. 1A). Six to seven neonates were born from wild-type (WT) and vitamin C-sufficient Gulo−/− mice (KO+VC), but the number of live neonates was definitely decreased in the vitamin C-deficient Gulo−/− mice (KO) with a vitamin C deficiency of over 2 weeks (Fig. 1B). In addition, neonates from the 2w KO and 3w KO mice were underweight compared with the WT and the KO+VC neonates (Fig. 1C). This result means that there is a defect in the growth of neonates in vitamin C-deficient Gulo−/− mice, although they survived in a vitamin C-deficient condition. We also confirmed the decrease in brain weight upon vitamin C deficiency based on the % ratio of the brain weight to the body weight. The values were 8.766% (WT), 8.879% (KO+VC), 8.943% (1w KO), 8.545% (2w KO), and 8.026% (3w KO) (Fig. 1E). Interestingly, we found remarkable brain hemorrhages in the brains of the neonates from the KO mice accompanied by the loss of brain weight (Fig. 1D, F). Therefore, it seems that vitamin C deficiency causes an increase in stillbirths and severe defects in neonatal brain development.
Decrease in vitamin C levels and an increase in oxidative stress in neonatal brains from vitamin C-deficient Gulo−/− mice
Hemorrhages were more distinctively increased in the brains of neonates from Gulo−/− mice with vitamin C supplementation ceased for 3 weeks and they looked more severe in the 3w KO samples than in the 1w KO and 2w KO samples (Fig. 1F). About 18% of the neonates with vitamin C supplementation ceased for 1 week had brain hemorrhages and such hemorrhages were seen in approximately 78% of the neonatal brains when vitamin C supplementation was ceased for 2 weeks (data not shown). For this reason, we did all further analyses by stopping vitamin C supplementation for 2 weeks. First, we confirmed that the vitamin C level in the blood from the KO mother and neonates was lower than that of the WT and KO+VC mice (Fig. 2A, B). Interestingly, the vitamin C level in the blood of the neonates was much higher than that in the blood of the mothers. This result suggests that vitamin C is highly needed during neonatal development. In addition, the concentration of vitamin C in neonatal brain tissue lysates was decreased in the KO mice compared with the WT and KO+VC mice (Fig. 2C).
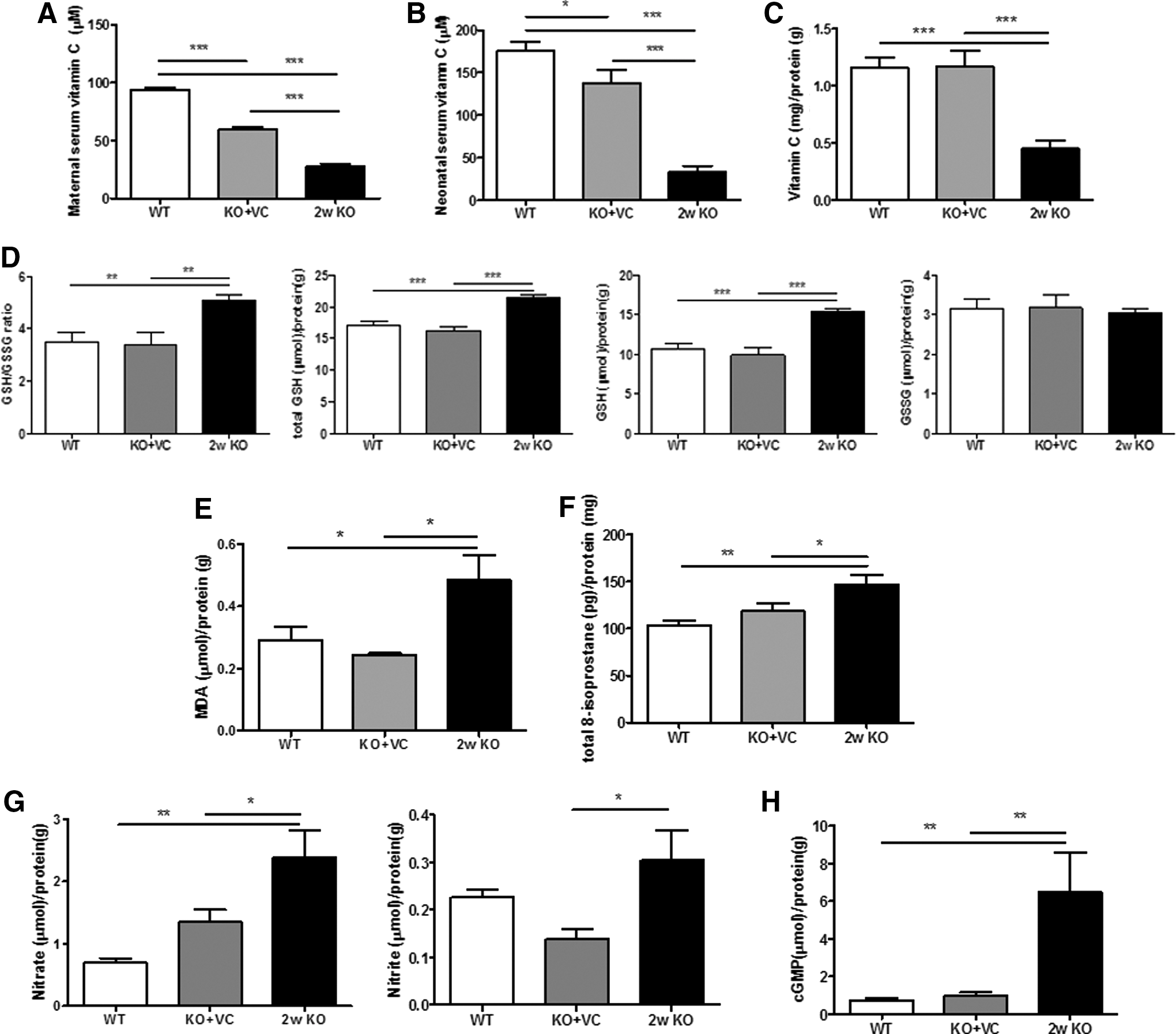
Vitamin C plays an important role in antioxidant network maintenance to protect against damage in the brain from ROS/RNS (33). It is involved in the reduction and oxidation of glutathione (GSH), the most potent endogenous antioxidant. GSH is the reduced form and GSSG is the oxidized form of glutathione (10, 46). Because vitamin C levels in the KO mice were decreased in our results, we determined the oxidative stress upon vitamin C deficiency by measuring the concentrations of GSH and GSSG. In addition, MDA and total 8-isoprostane, products of lipid peroxidation, were also measured. The ratio of GSH/GSSG was increased due to an increase in GSH (Fig. 2D), and MDA and 8-isoprostane were also increased (Fig. 2E, F). This result means that vitamin C deficiency is correlated with the increase in oxidative stress due to a defect in glutathione recycling in the neonatal brain.
Nitric Oxide (NO) plays a role of a neurotransmitter, but it is highly toxic in oxidative stress-related brain diseases, such as Alzheimer's disease and Parkinson's disease (9, 15). We measured the levels of nitrite and nitrate produced by the oxidation of NO and cyclic guanosine monophosphate (cGMP) in the brain. Interestingly, NO and cGMP in the brain of the KO mice were definitely higher than the levels in the WT or KO+VC mice (Fig. 2G, H). This suggests that the brain damage in the KO mice is caused, at least in part, by an increase in the levels of NO.
Abnormal foliation in the neonatal cerebellum by a decrease in brain-derived neurotrophic factor and glial cell-derived neurotrophic factor expression upon vitamin C deficiency
Based on the data shown in Figure 2, it seems that the decreasing vitamin C and increasing oxidative stress are closely related with the changes to the brain in the KO neonates. Next, we examined the morphological changes in the brain of the KO mice. Intraparenchymal hemorrhages were observed in the brain of the KO mice when ceasing vitamin C supplementation for 2 weeks and they were randomly found in several regions of the brain (Fig. 3A).
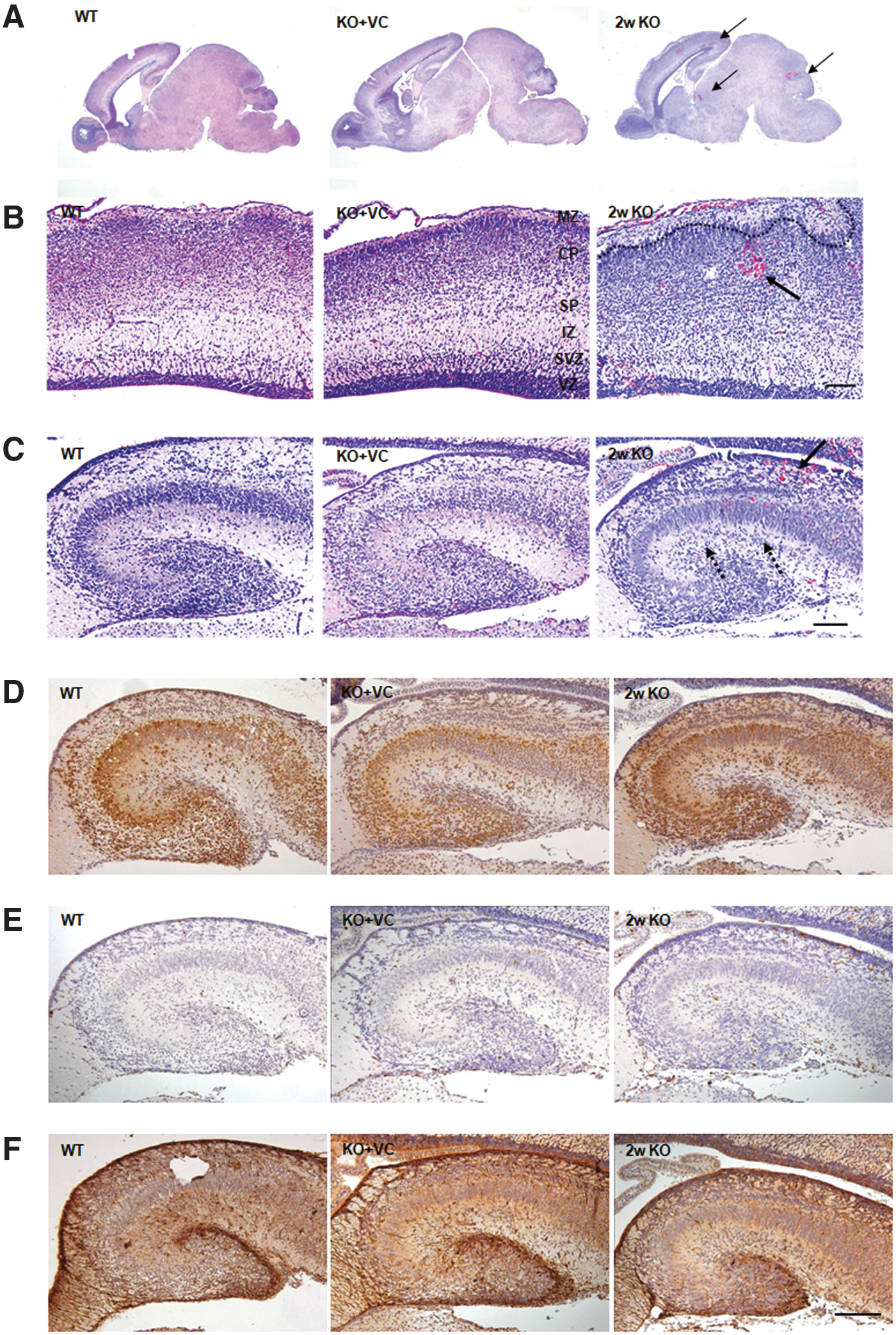
In the cerebral cortex layer, the boundary between the marginal zone and the cortical plate was irregular (dashed line), and the division of the cortical plate, subplate, and intermediate zone was not clear (Fig. 3B). Many cells were observed in the striatum radiatum, striatum lacunosum, and white matter in the hippocampus of the KO mice (Fig. 3C). To identify what types of cells were increased in the hippocampus of the PND 1 neonates from the KO mice, the brain tissues were stained with antibodies against brain cell-specific markers. Antibodies against NeuN, Iba-1, and GFAP were used for neurons, microglia, and astrocytes, respectively. As shown in Figure 3D–F, most of the cells in the striatum radiatum, striatum lacunosum, and white matter in the hippocampus were stained with anti-NeuN antibody, but not with anti-Iba-1 and GFAP antibodies. Therefore, the increased cells in the hippocampus of the PND 1 neonates from the KO mice are neurons.
At postnatal day (PND) 1, neonates from the WT and KO+VC mice showed normal cerebellar foliations with preculminate, primary, secondary, and posterolateral fissures, but abnormal foliation and fissure formation were seen in the cerebellum of the neonates from the KO mice (Fig. 4A). In addition, the layer formation of the cerebellar cortex was not clear (Fig. 4B). Because the layer in the cerebellar cortex consists of Purkinje and granule cells, we examined whether vitamin C deficiency affects the cellular integrity of the Purkinje cells. As shown in Figure 4C and D, we found smaller cell bodies of Purkinje cells and less dendrites in the KO mice than in the WT and KO+VC mice in the neonatal brain at PND 1 based on calbindin-28K expression (Fig. 4D). Additionally, these defects are the reason for the abnormal formation of cerebellar fissures in the cerebellum of the KO mice. Therefore, decreasing vitamin C induces the changes in the brain of KO neonates, especially in the cerebellum.
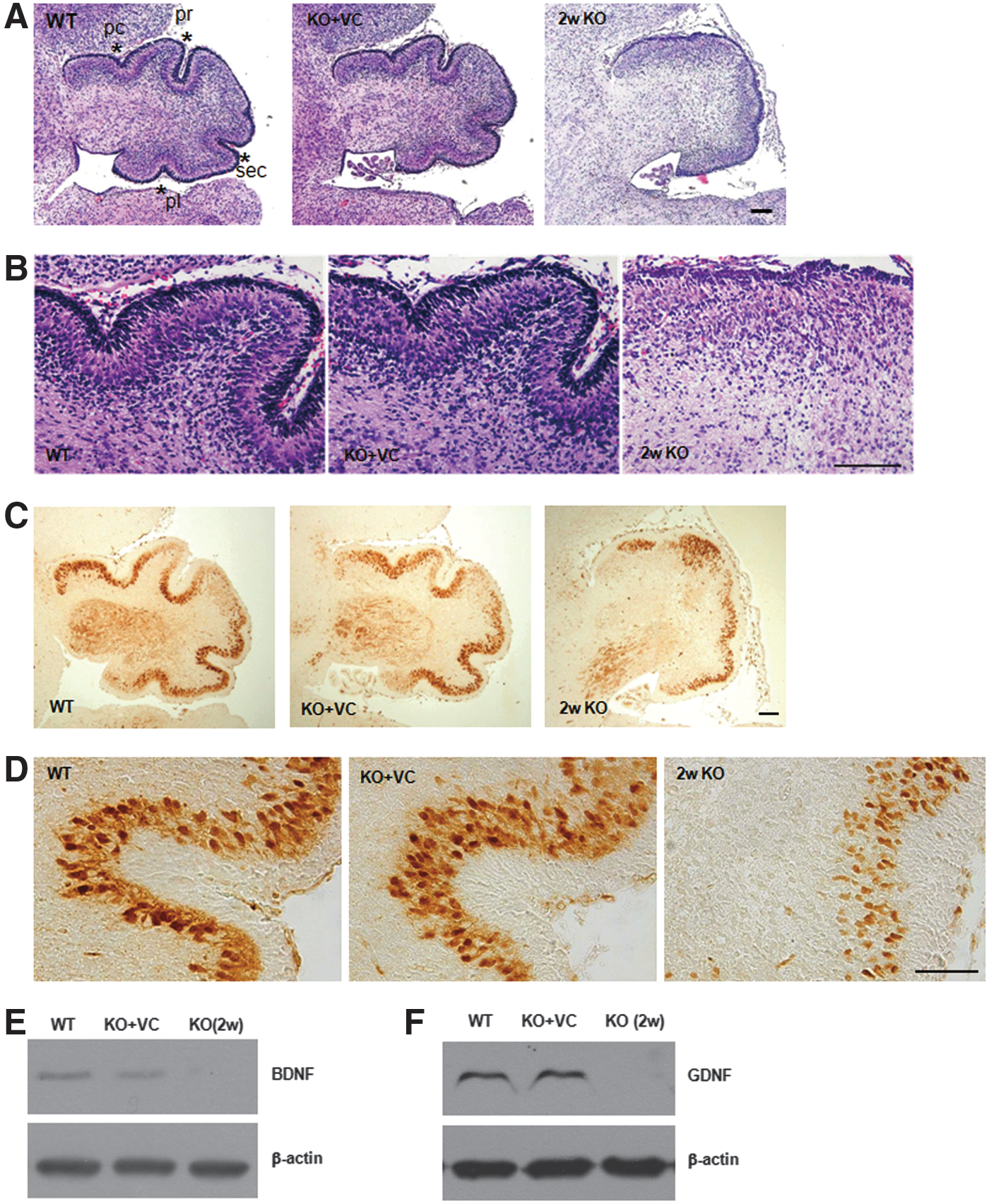
Because abnormal cerebellar development and foliation were revealed in brain-derived neurotrophic factor (BDNF) knockout mice (58), BDNF is one of the candidates responsible for abnormal foliation in Gulo−/− mice by vitamin C deficiency. As shown in Figure 4E, BDNF expression was remarkably decreased by vitamin C deficiency. The expression of glial cell-derived neurotrophic factor (GDNF) was also decreased in the neonatal brains of the KO mice (Fig. 4F). It was reported that BDNF expression is induced by GDNF in dopaminergic neurons (48). Therefore, vitamin C is an important factor for the development and structural maturation of the cerebellum through the regulation of GDNF expression and the subsequent BDNF expression.
Increase in TNF-α and IL-6 production and oxidative stress in the adult brains of vitamin C-deficient Gulo−/− mice
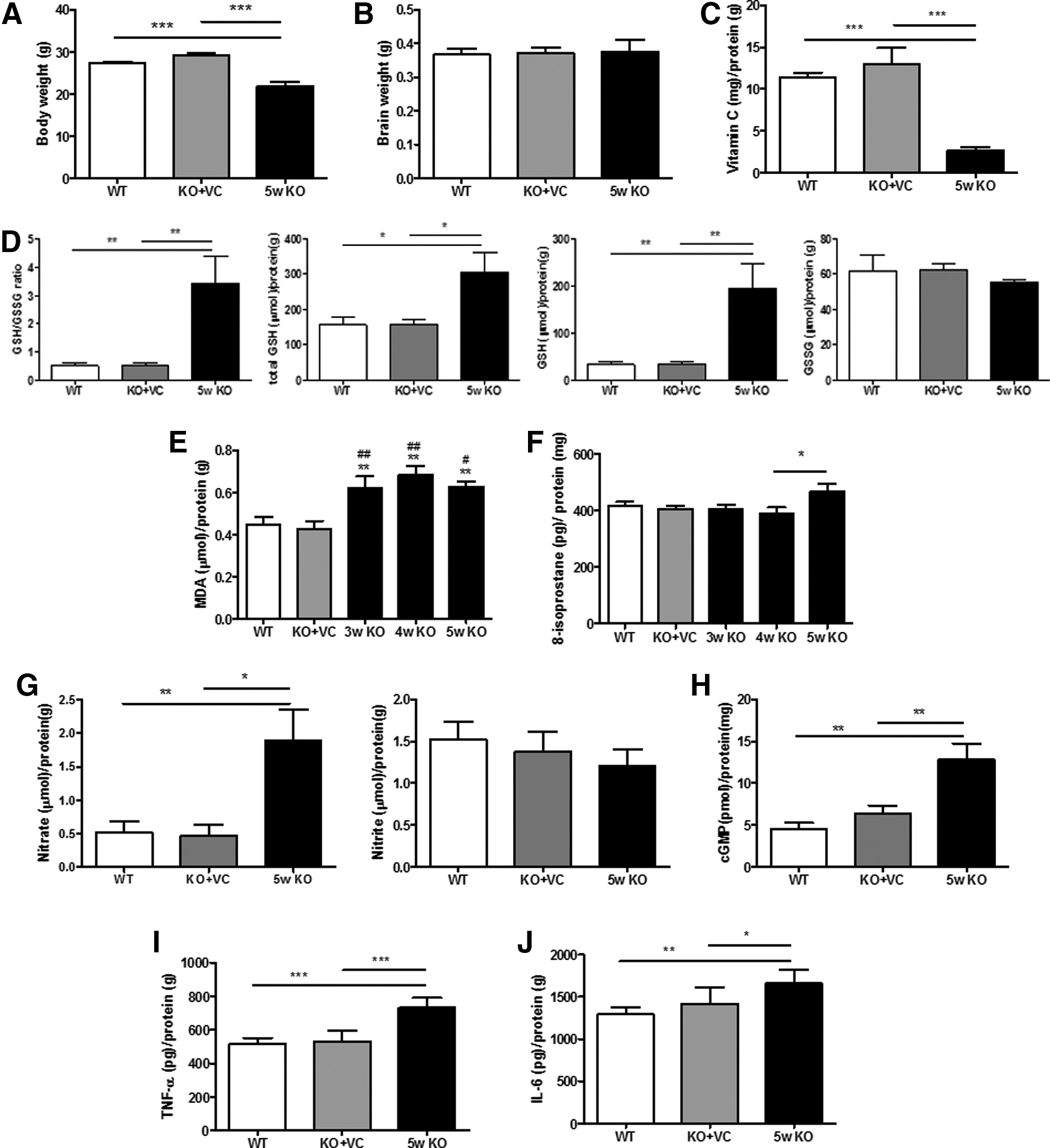
Next, we examined the changes in the adult brains of Gulo−/− mice upon vitamin C deficiency. Because we have already observed that vitamin C is preferentially stored in the brains of KO mice for 5 weeks (26), the brains were examined at 5 weeks after ceasing vitamin C supplementation. The body weight and vitamin concentration of the KO mice were decreased, but there were no differences in the adult brain weights (Fig. 5A–C). Like in the neonatal brains, the GSH/GSSG ratio was increased in the adult brain of the KO mice (Fig. 5D). The production of MDA, 8-isoprostane, NO, and cGMP was also increased (Fig. 5E–H). This result implies that oxidative stress in the adult brain of KO mice is increased due to a defect in the maintenance of the antioxidant network. It is also known that oxidative stress is closely related with an excessive inflammatory response accompanied by the secretion of high levels of proinflammatory cytokines (12, 57). In addition, we observed a significant increase in the production of tumor necrosis factor (TNF)-α and interleukin (IL)-6, which are the representative proinflammatory cytokines in the brains of KO mice (Fig. 5I, J). These results suggest that vitamin C deficiency induces changes in the adult brain of Gulo−/− mice through an imbalance in the antioxidant system and through an increase in proinflammatory cytokine production.
Increase in the changes of cerebellum-related molecules upon vitamin C deficiency
In contrast to the morphological changes in the neonatal brains, there were no significant morphological changes in the adult brains of the KO mice without vitamin C supplementation for 5 weeks (Supplementary Fig. S1; Supplementary Data are available online at
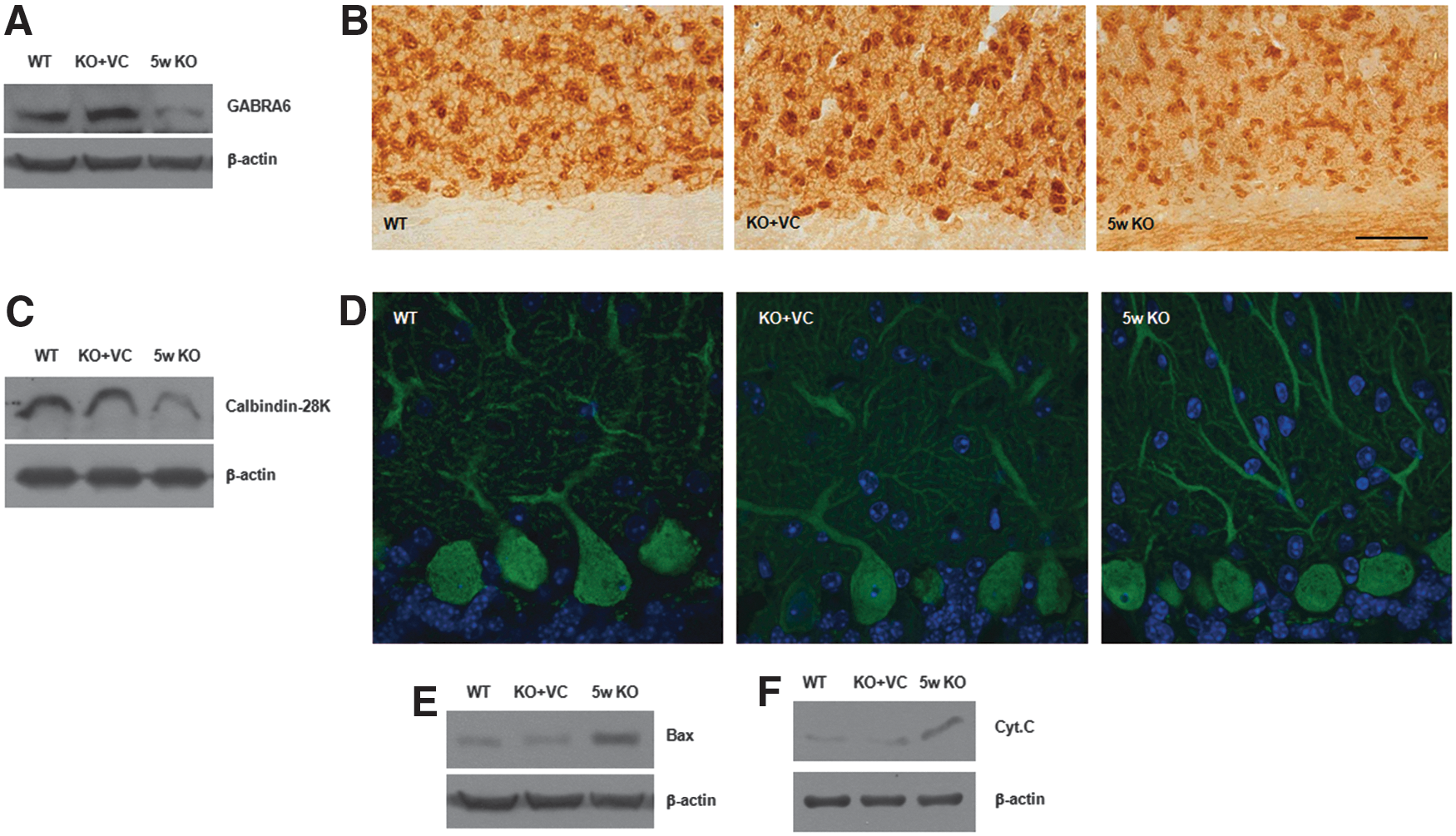
The most downregulated gene upon vitamin C deficiency is the GABAA receptor alpha6 (GABRA6), a marker of mature granule cells in the cerebellum (24). The decrease of GABRA6 in the brain tissues of vitamin C-deficient Gulo−/− mice was confirmed by immunoblotting and immunohistochemistry (Fig. 6A, B). Moreover, cells expressing GABRA6 in the KO mice were more shrunken compared with the WT and KO+VC mice (Fig. 6B). In addition, the expression of calbindkin-28k, a marker of Purkinje cells, was definitely decreased in the KO mice (Fig. 6C, D). Especially, it was remarkable that the dendritic arborization of the Purkinje cells was decreased and the axons were shrunken. Next, we examined whether the shrinkage of the cells in the KO mice was caused by apoptosis. We have previously reported that Bax and cytochrome c are key molecules in vitamin C-induced apoptosis (22, 28). Therefore, we examined the expression of Bax and cytochrome c as proapoptotic molecules. As we expected, Bax and cytochrome c expression was increased upon vitamin C deficiency, but they were inhibited by vitamin C supplementation (Fig. 6E, F). Thus, it seems that vitamin C deficiency in the adult brains of the KO mice causes atrophic changes in the granule and Purkinje cells in the cerebellum.
Defects in motor function upon vitamin C deficiency
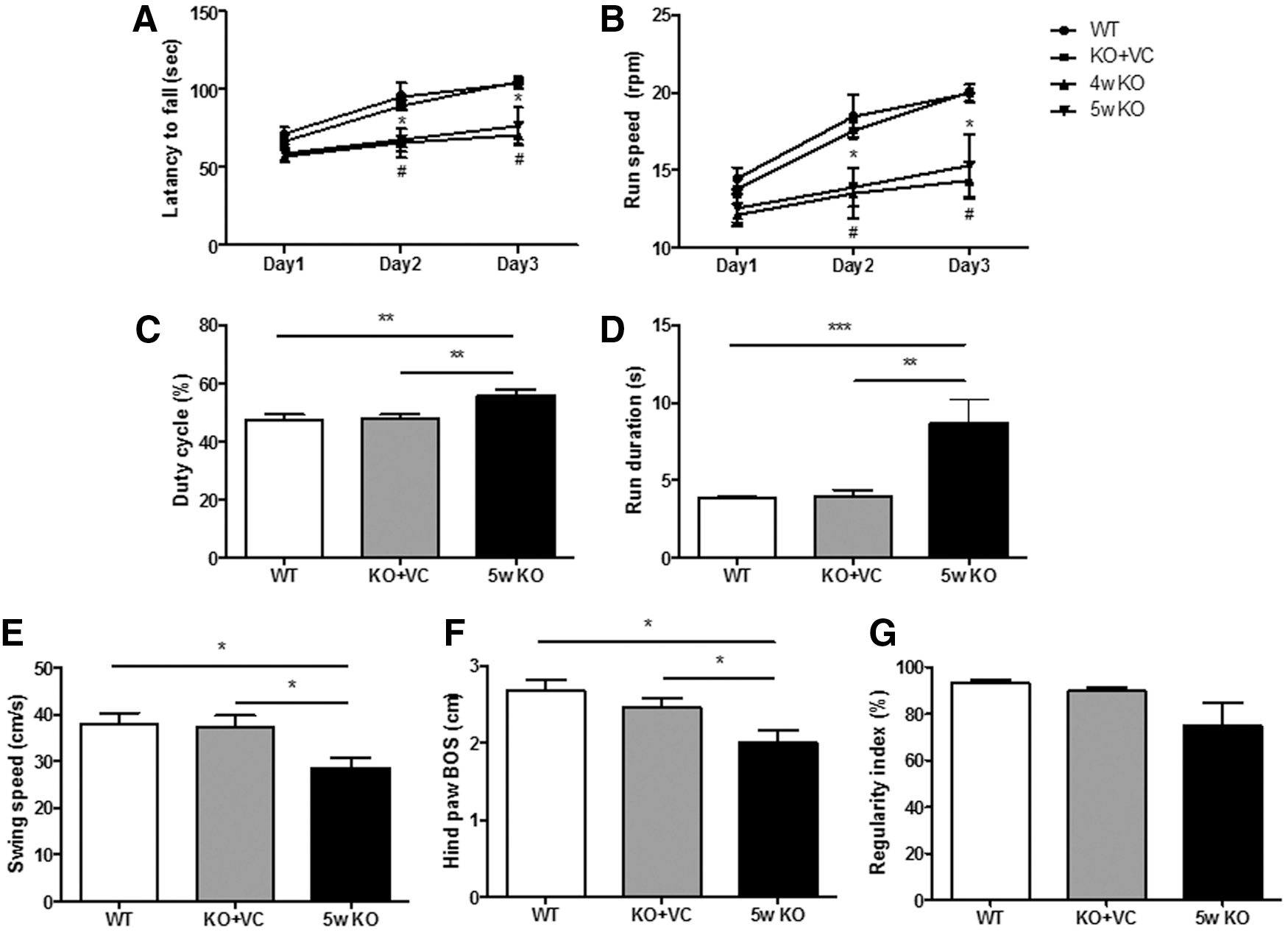
We have shown abnormal changes in the cerebellum, and the cerebellum is the part of the brain for motor coordination. Behavior tests, such as the rotarod test, horizontal bar, static rods, parallel bars, and foot-fault test (CatWalk), are usually used to evaluate motor coordination, which is the function of the cerebellum, especially by Purkinje cells (7, 55, 64). Therefore, changes in motor function upon vitamin C deficiency were examined by the rotarod and CatWalk tests. Before beginning the behavior tests, we compared the integrity of the femoral muscle fibers in the WT, KO+VC, and KO mice. We confirmed that there were no noticeable differences among the experimental groups (Supplementary Figs S2 and S3). The rotarod test is usually used to assess sensorimotor coordination and overall motor function (19). Coinciding with the changes in the cerebellum of the KO mice, the KO mice exhibited a lower latency to fall from a rotating rod and running speed than those of the WT and KO+VC mice (Fig. 7A, B). The CatWalk test is commonly used for gait analysis, which analyzes neurodegeneration and behavioral measures for neurological diseases (e.g., Parkinson's disease) (37, 70). As shown in Figure 7C and D, the values for the duty cycle and run duration were increased. The results represent the stance duration as a percentage of the step cycle duration and the duration and average speed of a run. In contrast, the swing speed and hind paw base of support (BOS) were decreased (Fig. 7E and F). These results show the speed of the paw during the swing and the average width between the hind paws, respectively. These findings strongly imply that vitamin C causes defects in the motor behaviors of the KO mice through alterations in the brain, especially in the cerebellum.
Discussion
The in vivo effects of vitamin C as an anti-inflammatory, antiviral, and antitumor agent and its related mechanisms are still not clearly understood because the effect of endogenous vitamin C synthesized from glucose could not be excluded. Thus, Gulo−/− mice are useful to investigate the in vivo effect of vitamin C because they are unable to synthesize vitamin C and exhibit physiological responses that resemble those in vitamin C-deficient humans. We have recently reported on the regulatory role of vitamin C in the inflammation of the liver and heart, as well as on the viral infection of vitamin C-deficient Gulo−/− mice (2, 25, 29). We have also found a preferential accumulation and maintenance of vitamin C in the brain. It has already been reported that a high concentration of vitamin C is in the brain and cerebrospinal fluids (CSFs) (33). We assume that vitamin C has an important role in developing the brain and maintaining its function by protecting the brain against damages caused by ROS/RNS, but the precise mechanisms of vitamin C in the brain still remain to be elucidated. Therefore, this is the first report that presents the concrete effect of vitamin C and its related mechanisms on brain development by controlling vitamin C supplementation in vivo.
Prenatal or postnatal malnutrition, including vitamin C deficiency, results in serious consequences in the developing brain as well as in fetal growth (36, 67). It has been reported that neonates from mice with a defect in the expression of the SVCT-2, a specific channel protein for vitamin C uptake, have severe brain hemorrhages and an abnormal cellular composition. The alternative ways for vitamin C uptake through facilitative glucose transporters (GLUTs) and SVCT1 are intact in SVCT2−/− mice (56).The expression of SVCT-1 and -2 is different in each organ. SVCT-2 is highly expressed in the brain (65). Figure 1F shows that severe brain hemorrhages were observed in Gulo−/− mice with vitamin C deficiency. Because vitamin C is not endogenously synthesized and SVCT2 is normally expressed in Gulo−/− mice, this is direct evidence regarding the effect of vitamin C on the prevention of brain hemorrhages. However, further experiments on the role of SVCT-1/-2 and GLUT in preventing hemorrhages in the brain of KO mice are also needed.
It is interesting that vitamin C deficiency also caused hemorrhages in the liver, spleen, and other organs of the neonates from the Gulo−/− mice when ceasing vitamin C supplementation for 2 or 3 weeks (Supplementary Figs S4 and S5), although hemorrhages in the SVCT2−/− mice were restricted to the brain only (61). According to a report by Polidori et al., decreased vitamin C levels in plasma are correlated with brain damage in patients with intracranial hemorrhage or head trauma (50).
It has been reported that vitamin C maintains the antioxidant network by reducing other oxidized antioxidants, for example, glutathione disulfide (GSSG) to glutathione (GSH) and tocopheroxyl radical to tocopherol (10, 33, 46). In addition, vitamin C levels and antioxidant enzyme activities (e.g., superoxide dismutase and glutathione peroxidase) in plasma are modestly decreased in stroke patients with the worst outcome at the early stage of the disease (5). We showed a decrease in vitamin C levels in the plasma and brain of the KO mice (Figs. 2C and 5C). Thus, we hypothesized that GSH will be increased to compensate for the lower vitamin C levels in vitamin C-deficient Gulo(−/−) mice. In fact, there are reports that GSH synthesis increases under conditions of oxidative stress with low ascorbic acid, presumably to regulate the antioxidant capacity (18, 40). As shown in Figures 2D and 5D, GSH synthesis was increased, but there was no change in the GSSG level. This result suggests that GSH is more rapidly synthesized than the conversion of GSH to GSSG. In addition, it seems that there might be a defect in the recycling between GSH and GSSG, especially in the conversion of GSH to GSSG, in vitamin C-deficient Gulo(−/−) mice because vitamin C has an important role in the sparing or recycling of GSH (41). This is the reason why GSH/GSSG is increased in our results. Harrison et al. reported that total glutathione (GSH+GSSG) in the brain of Gulo(−/−) mice was increased, but GSH/GSSG ratios did not differ. They measured on postneonatal day (PND) 18, but we measured on PND 1 because PND 1 more closely reflects the effect of vitamin C during pregnancy than the measurement on PND 18. It is also probable that the antioxidant defense system is relatively less organized in the fetus. This might be another reason for the increased GSH/GSSG ratio in our results. In addition, the changes in glutathione upon vitamin C deficiency were measured in the Gulo(−/−) mice without vitamin C supplementation for 5 weeks. Based on our previous results, we found that there are damages to most of the physiological systems, including the cardiovascular system, immune system, and antioxidant defense system, in the Gulo(−/−) mice without vitamin C supplementation for 5 weeks (2, 25 –27). Because GSH is the most abundant and effective antioxidant in mammalian cells, it might be increased first to compensate for the damaged antioxidant network in Gulo(−/−) mice upon vitamin C deficiency. Therefore, the activities of glutathione peroxidase and glutathione reductase in the fetuses and adults of Gulo(−/−) mice upon vitamin C deficiency require further investigation to clarify the reason why GSSG did not increase and the ratio of GSH/GSSG did increase, although GSH was increased.
NO is produced from arginine by nitric oxide synthetase. It is oxidized to nitrate and nitrite (34). Additionally, it increases cGMP production through the activation of soluble guanylate cyclase (62). NO is a signaling molecule with broad physiological actions across many organs. In the brain, NO dose-dependently modulates synaptic transmission and mediates activity-dependent changes in postsynaptic excitability. NO is highly toxic in oxidative stress-related brain diseases, such as Alzheimer's disease and Parkinson's disease (3, 9, 15, 62). NO activates soluble guanylate cyclase to increase cGMP. Moreover, we also found an increase of cGMP in the brain upon vitamin C deficiency (Figs. 2H and 5H). According to a report by Montoliu et al., neuronal death by NO and glutamate is prevented by the inhibition of cGMP formation (42). Because the role of cGMP in the brain is still controversial, the involvement of NO and cGMP in the brain damage of the KO mice should be further examined.
There are two important findings in the present study. One is that vitamin C deficiency changes the cerebellum in the brains from both the neonates and adult KO mice. In the cerebellum of the neonates from the KO mice, abnormal foliation and fissure formation were found (Fig. 4A). Additionally, the layer formation of the cerebellar cortex, consisting of Purkinje and granule cells, was unclear (Fig. 4B). Moreover, there was a definite change in the integrity of the neonatal Purkinje cells in the inward folds from vitamin C deficiency (Fig. 4C). Even though a defect in vitamin C uptake caused damage in the brains of SVCT2−/− mice (61), it was not specifically verified which region of the brain is affected by the vitamin C deficiency. In this report, we showed that the cerebellum is the most fragile region in the brain upon vitamin C deficiency. The second finding is that the axons of the Purkinje cells were shrunken in the cerebellum of the adult brains, followed by abnormal motor behaviors (Figs. 6D and 7). The lower levels of vitamin C are related with the pathogenesis of neurodegenerative diseases caused by defects in motor function, such as Parkinson's disease and Huntington's disease (8, 69). The pathologic findings show that Parkinson's disease and Huntington's disease are caused by changes in the cerebellum. Furthermore, there are several reports that oxidative stress might be involved in their pathogenesis (55). The data regarding the motor behaviors of adult Gulo−/− mice under vitamin C deficiency are meaningful because this might be relevant to patients with Parkinson's and Huntington's disease.
In humans, it has been reported that vitamin C levels decrease as pregnancy progresses and during lactation (39, 51). From the last week of gestation, vitamin C levels in the brain are increased until birth (30), which implies a higher consumption of vitamin C during this period. We showed that the vitamin C concentration in the blood of neonates was much higher than that in the maternal blood in the WT and KO+VC mice (Fig. 2A, B). Furthermore, the vitamin C plasma level was slightly higher in the WT mice than in the KO+VC mice. Nevertheless, it was nearly at the same levels in the brains (Fig. 2C). Inversely proportional to the vitamin C level, the contents of MDA and 8-isoprostane, markers of lipid peroxidation, were high in the neonatal KO brains compared with the brains of the WT and KO+VC mice (Fig. 2E, F). It is known that MDA and especially isoprostanes are considered specific markers for oxidative stress in the cerebellum and brain cortex, particularly at the earlier stages of oxidative damage (19). Therefore, the reason that large amounts of vitamin C are stored in the brain of neonates is to prevent damage to the developing brain, especially the cerebellum.
One of the cytoarchitectonic characteristics in the brain is the formation of a neuronal cell layer. The neonates from the vitamin C-deficient Gulo−/− mice showed abnormal cellular layers in the cortex and hippocampus. It was also definitely observed in the cerebellum. In the cerebral cortex and hippocampus, the distribution of cells and the distinction of layers were less clear (Fig. 3B, C). Additionally, the arrangement of the molecular layer, Purkinje cell layer, and granule cell layer was atypically irregular in the cerebellum (Fig. 4B). Because the cerebellum is a morphologically unique brain structure made up of an elaborate set of folia separated by fissures (63), the most striking findings are the abnormal cerebellar foliation and the formation of fissures in the KO neonates (Fig. 4A). Vitamin C acts as a neuromodulator in dopaminergic neurotransmission (11, 17). Furthermore, it has been reported that BDNF is important in the foliation of the cerebellum, and GDNF induces BDNF expression in dopaminergic neurons (48, 58). Because we showed in the present study a decrease in the expression of BDNF and GDNF (Fig. 4E, F), it appears that vitamin C has an essential role in the foliation of the cerebellum by maintaining the expression of BDNF and GDNF. Although the structural morphology of adult brains, such as neuronal lamination and cerebellar foliation, was not affected by vitamin C deficiency, the cellular morphology of the cerebellum was changed by a lower level of vitamin C in the vitamin C-deficient Gulo−/− mice (Fig. 6). The expression of GABRA6 was mostly decreased by vitamin C deficiency (Fig. 6A, Supplementary Table S1, and Supplementary Fig. S6). GABRA6 is a mature cerebellar granule cell-specific protein and its expression is downregulated in the ataxic mouse, Stargazer (47). Interestingly, BDNF induces the expression of granule neuron terminal differentiation markers, particularly GABRA6 mRNA, to regulate the maturation of developing cerebellar granule neurons (4). As a consequence of the alteration in the cerebellar neurons, a severe defect in motor function in the KO mice might be induced (Fig. 7). Even though there are reports that combined vitamin C and E deficiency induces motor defects in Gulo−/−/SVCT2+/− mice and low levels of vitamin C are responsible for the exacerbated motor deficits in Gulo−/−/mice (19, 49), this is the first report that explains why the vitamin C deficiency exacerbates motor deficit.
The developed adult brain, like the developing neonate brain, is susceptible to oxidative stress. A low level of vitamin C is noted to be associated with elevations in several markers of oxidative stress (18, 19, 36). It is known that oxidative stress regulates the expression of BDNF (23, 71). In addition, GDNF suppresses oxidative stress-mediated damages in the brain (59, 60). Moreover, inflammatory cytokines (e.g., IL-6 and TNF-α) from microglia and astrocytes increase ROS/RNS, which in turn induce neuronal damage through the downregulation of neurotrophins and their receptors (13, 71). In the case of the brains of the KO mice, the production of IL-6 and TNF-α, as well as the expression of apoptosis-related Bax and cytochrome c, was increased (Figs. 5I, J and 6E, F). However, the expression of BDNF and GDNF protein was decreased (Fig. 4E–F). Therefore, vitamin C is necessary for neonatal brain development, especially the cerebellum, and for the functional maintenance of the adult brain by preventing damage to or the death of the brain from oxidative stress and its mediated products, IL-6, TNF-α, Bax, cytochrome c, BDNF, and GDNF.
Vitamin C is not only engaged in modulating glutamatergic neurotransmission but also in the synthesis of noradrenaline and many other neuropeptides to promote myelin formation by Schwann cells (50). As we showed in Supplementary Figure S7A and B, there was a severe defect in dopamine production and tyrosine hydroxylase (TH) expression in the KO mice. Dopamine is produced from L-DOPA by TH (14). In addition, vitamin C is a cofactor of dopamine-β-hydroxylase to produce noradrenaline from dopamine (44). This suggests that there must be a decrease of noradrenaline production in the KO mice. Therefore, functional changes in the brain of the KO mice might also be mediated by a defect in noradrenaline.
Taken together, vitamin C deficiency during the gestation period induces abnormal development of the neonatal brain, especially in the cerebellar lamination and foliation. In addition, its deficiency makes cerebellar granule and Purkinje cells in the adult brain become vulnerable to oxidative stress and proinflammatory/apoptotic mediators. This process is accompanied by defects in the expression of GABRA6, BDNF, GDNF, and TH and the production of noradrenaline. Finally, it causes an increase in the stillbirths of neonates with hemorrhages and severe motor deficits in the Gulo−/− mice.
Materials and Methods
Animals
C57BL/6 wild-type (WT) mice and Gulo−/− mice were housed in a specific pathogen-free condition at the animal facility in the Seoul National University College of Medicine. Gulo−/− mice were supplemented with vitamin C (3.3 g/L) in their drinking water to prevent death by vitamin C deficiency. Fetuses and neonates were obtained by the mating of 6–8-week-old mice, and 10–12-week-old male mice were used for the behavior tests. For the experiments, plug-checked female Gulo−/− mice were maintained with or without vitamin C for 1–3 weeks until delivery. In addition, vitamin C supplementation was continued (KO+VC group) or discontinued (KO group) in adult vitamin C-supplemented Gulo−/− mice. All experiments using animals were reviewed and approved by the Institutional Animal Care and Use Committee of Seoul National University.
Immunoblotting
Brain tissues were homogenized with lysis buffer and quantified with the BCA method. Protein was mixed with 5× SDS sample buffer and loaded into each lane of a 10–15% SDS-PAGE gel. Proteins were separated by electrophoresis and transferred from the gel to a nitrocellulose membrane with an electroblotting apparatus. Nonspecific sites of the membranes were blocked with 5% skim milk for 1 h, and then the membranes were incubated with primary antibodies against GDNF, Bax (Santa Cruz, Palo Alto, CA), GABRA6 (Chemicon, Temecula, CA), BNDF (Abcam, Cambridge, MA) or β-actin, and calbindin-28k (Sigma, St. Louis, MO) at 4°C overnight. Washed with PBS-T (0.05% Tween-20 in PBS), membranes were subsequently incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling, Danvers, MA) and detected with the ECL detection kit (Amersham, Piscataway, NJ).
Immunostaining
Brain tissues were freshly isolated and fixed in 4% PFA at 4°C. Paraffin-embedded tissues were sectioned at 5 μm thickness. After deparaffinization and hydration, tissue sections were stained with hematoxylin and eosin (Sigma), dehydrated, and mounted. Otherwise, antigen epitopes were retrieved by heating with 0.1 M citrate buffer (pH 6.0) under a microwave for immunostaining. Following the blocking of endogenous peroxidase with H2O2 and inhibiting nonspecific signals with 5% goat serum, sections were incubated with primary antibodies against GABRA6, NeuN (Chemicon), Iba-1 (Abcam), GFAP (Dako), or calbindin-28k (Sigma) at 4°C overnight in a humidified chamber. Then, the sections were incubated with a matching biotinylated secondary antibody (Vector laboratories, Burlingame, CA) for 1 h at room temperature. ABC solution (Vector laboratories) was loaded onto the sections for 30 min, and the DAB kit (Vector laboratories) was used for chromogenic detection. Subsequent to dehydration and clearing, the sections were mounted with DPX mountant (Fluka, St. Louis, MO) and observed with a light microscope (Olympus, Center Valley, PA).
Immunoassays
Brain tissues were homogenized with lysis buffer and quantified with the BCA method. The concentrations of TNF-α, IL-6, IL-1β (R&D system, Minneapolis, MN), glutathione (Arbor Assays, Ann Arbor, MI), MDA, 8-isoprostane, nitrite/nitrate, cGMP (Cayman, Ann Arbor, MI), and vitamin C (Immundiagnostik AG, Bensheim, Germany) were measured with assay kits according to the manufacturer's instructions, and the final concentration in the brain was normalized to the amount of total protein in the brain tissue lysates.
Behavior tests
A rotarod machine with automatic timers and falling sensors (Rotamax; Columbus instruments, Columbus, OH) was used. The mouse was placed on a 9-cm-diameter drum rotating at 3 rpm. The surface of the drum was covered with hard chloroethylene, which does not permit gripping on the surface. Before the training sessions, the mice were habituated to stay on the stationary drum for 10 min. For analyzing balance and motor coordination, mice were tested on the accelerating rotarod; the rotational speed increased 1 rpm every 6 s from 3 to 50 rpm over 5 min, and mice were trained for three trials. Gait analysis was performed on walking mice using the CatWalk method. Briefly, when light rays from a fluorescence tube are sent through a glass plate, they are completely reflected internally. As soon as anything is in contact with the glass surface, light is reflected downward. It results in a sharp image of a bright paw print. The whole run is recorded via a camera placed under the glass plate. The following parameters were analyzed: * Duty cycle (%): the ratio between the stance duration and the step cycle duration * Run duration: the time needed to cross the walkways * BOS of the hind paw (cm): the distance between the two hind paws of the mouse * Swing speed (cm/s): the velocity of the moving limb during the swing phase * Regularity index (%): (numbers of normal step sequence patterns ×4/total number of paw placements) ×100
Statistics
Data are expressed as the mean±SEM for each group in individual experiments. For comparison of three or more groups, data were analyzed by the t-test or one-way analysis of variance (ANOVA) following the Newman–Keuls multiple comparison test; p values<0.05 were considered statistically significant. Statistical tests were done with GraphPad InStat (GraphPad Software, San Diego, CA).
Footnotes
Acknowledgment
This work is supported by the grants from the National R&D Program for Translational Research (A110654), Ministry of Health, Welfare, and Family Affairs, to Hyemin Kim.
Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.