
Abstract
Introduction
I
Hepatic hepcidin is induced by multiple stimuli, including inflammatory signals caused by infection, autoimmune diseases, and cancer (12, 28). Elevated hepcidin levels in response to inflammation lead to hypoferremia or anemia of inflammation (11, 26). Lipopolysaccharide (LPS) is widely used to induce hepatic hepcidin and inflammatory anemia in experimental conditions (16, 29, 39, 45). It is currently well characterized that interleukin-6 (IL-6) plays the critical role in inflammatory hepcidin expression by activating signal transducer and activator of transcription 3 (STAT3) (9, 53). Binding of IL-6 to its receptor recruits Janus kinase 2 (JAK2) to phosphorylate STAT3. Furthermore, p-STAT3 dimers translocate to the nucleus, bind to the hepcidin promoter, and upregulate hepcidin gene transcription. Hepatic hepcidin fails to respond to inflammatory stimuli or promote anemia in either STAT3 liver-specific or IL-6 knockout mice (21, 42). In general, the IL-6/JAK2/STAT3 pathway is of great importance to inflammation-induced hepcidin expression.
Concerns have been raised regarding the efficiency and safety of conventional management of anemia of inflammation, illustrating an urgent need for alternative therapies. In the study, we demonstrated for the first time that H2S could attenuate inflammatory hepcidin production by both reducing interleukin-6 (IL-6) secretion and promoting sirtuin 1 (SIRT1)-mediated signal transducer and activator of transcription 3 (STAT3) deacetylation. Meanwhile, we established a novel regulatory link between SIRT1 and hepcidin. These findings indicated H2S-releasing compounds as innovative candidates for the treatment of inflammatory anemia.
Hydrogen sulfide (H2S), which has long been known as a noxious gas, is the third gasotransmitter after nitric oxide (NO) and carbon monoxide (CO) (49). Biosynthesis of H2S has been identified in a variety of mammalian tissues. In the liver, H2S is endogenously produced by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) with
Considering the anti-inflammatory effects of H2S, in addition to the dominant role the liver plays in hepcidin secretion and H2S metabolism, we assessed whether H2S could regulate hepcidin expression during inflammation. In the present study, we discovered for the first time that H2S suppressed inflammatory hepcidin through both reducing IL-6 secretion and promoting sirtuin 1 (SIRT1)-mediated STAT3 deacetylation. Our findings elucidated the effects and potential mechanisms of H2S on inflammation-induced hepcidin expression and might throw light on new therapeutic approaches for anemia of inflammation.
Results
H2S attenuates LPS-induced hepcidin expression and regulates iron homeostasis in mice
To investigate our hypothesis that H2S regulates inflammatory hepatic hepcidin, C57BL/6 and C3H mice were pretreated with sodium hydrosulfide (NaHS, i.p., 6 mg/kg/day) for 3 days, followed by LPS challenge (i.p., 0.5 mg/kg), as stated in the Materials and Methods section. C57BL/6 mice were sacrificed for mRNA, protein, and serum analysis 6 h after LPS stimulation. Tissue iron measurements and Perl's Prussian blue staining were conducted in C3H mice at 24 h. As shown in Figure 1A and B, hepatic hepcidin mRNA (Hamp) and serum hepcidin were evoked by LPS, which was significantly suppressed by NaHS. Moreover, immunoblot assays indicated that ferroportin, the membrane iron exporter that is degraded upon binding with hepcidin, was restored by NaHS treatment (Fig. 1C). Due to its enrichment of macrophages, the spleen has a dominant role in iron deposition during inflammation. We next analyzed serum iron levels and iron accumulation in the spleen. LPS lowered serum iron content in C57BL/6 mice by ∼30% after 6 h, which was improved by NaHS (Fig. 1D). As demonstrated in Figure 1E and F, iron accumulation in splenic macrophages of C3H mice was also significantly ameliorated by NaHS treatment.
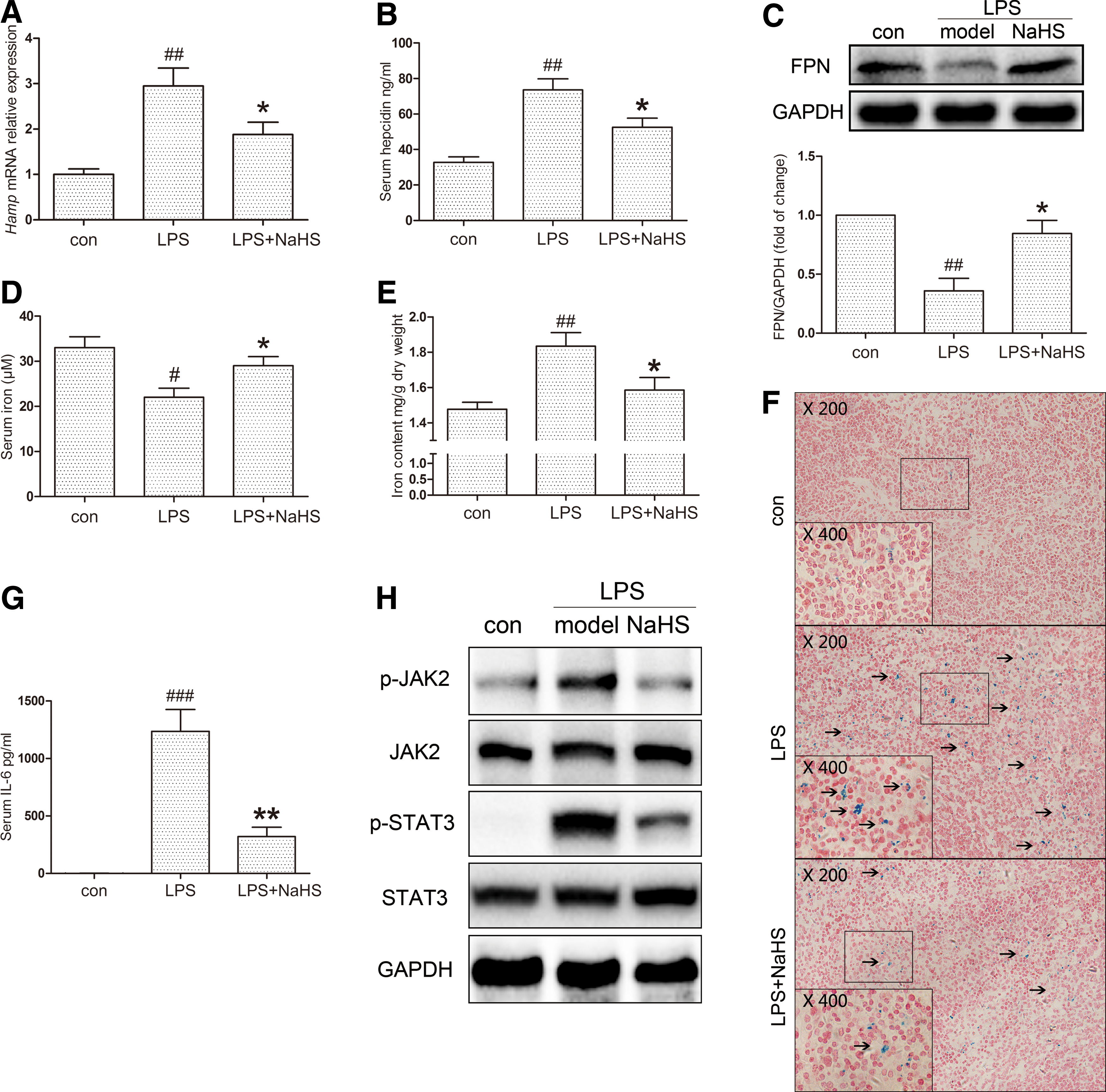
As STAT3, which is activated by IL-6, is the most crucial transcription factor for hepcidin induction during inflammatory conditions, the effect of H2S on the IL-6-mediated JAK2/STAT3 pathway was then investigated. NaHS tremendously lowered serum IL-6 levels (Fig. 1G) and hepatic Il-6 mRNA expression induced by LPS (Supplementary Fig. S1A; Supplementary Data are available online at
H2S inhibits hepcidin expression through decreasing IL-6 secretion in vitro
To evaluate the effects of H2S in vitro, we used a conditioned medium (CM) model consisting of THP-1-derived macrophages and Huh7 hepatoma cells to mimic the pathophysiological conditions in vivo. A time course evaluation suggested 6 h as the peak point of hepcidin mRNA (HAMP) induction in Huh7 cells (Supplementary Fig. S2A). NaHS was first applied to THP-1-derived macrophages 1 h before LPS stimulation to produce different CMs, which was defined as the pretreatment (Fig. 2A). The dose range (50–300 μM) was chosen by reference to previous studies (46, 47). HAMP expression induced by LPS-CM was markedly suppressed by NaHS pretreatment in a concentration-dependent manner (Fig. 2B). Similar results were obtained with
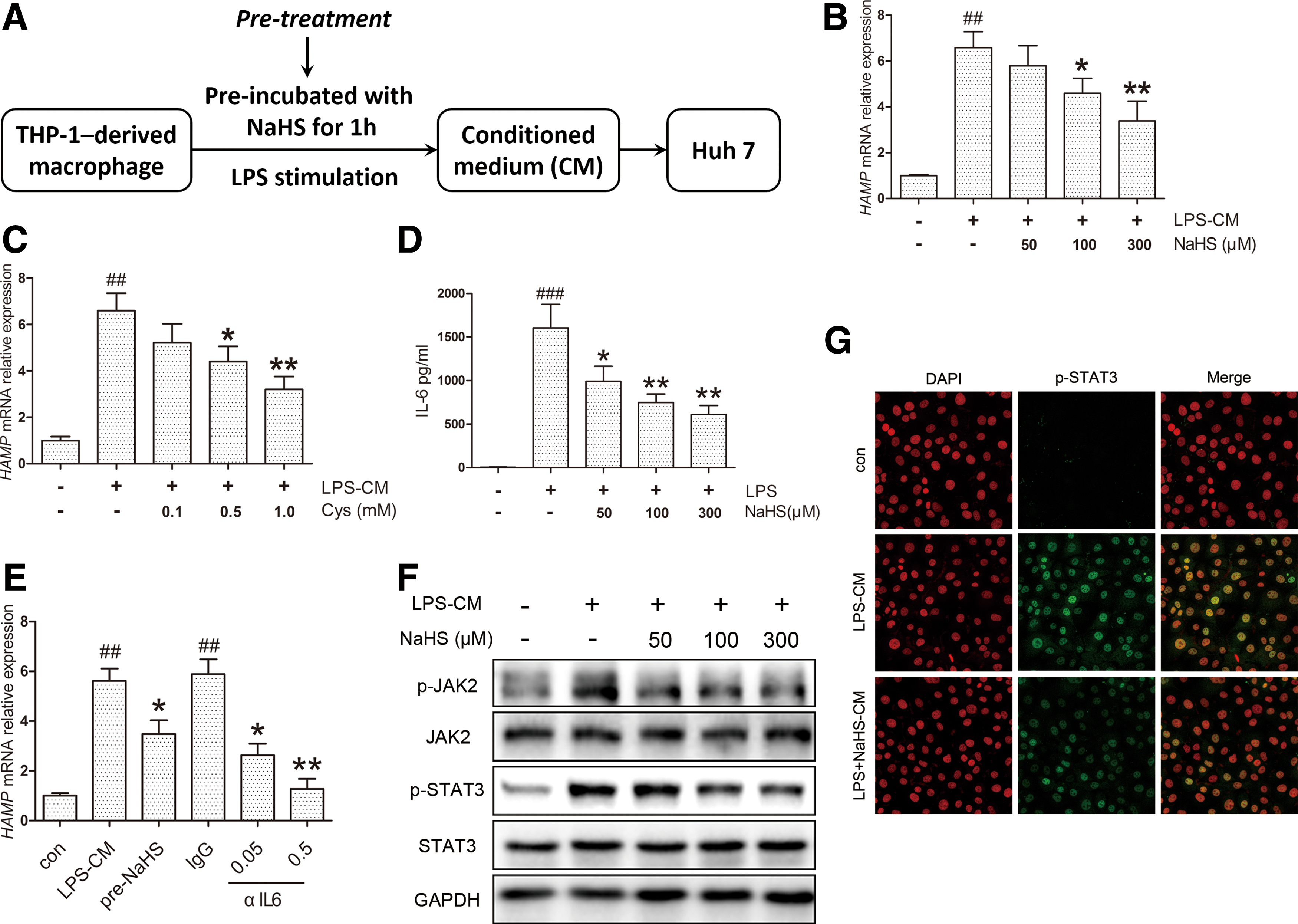
Due to its essential role in inflammatory hepcidin induction, the IL-6 content in CM was quantified by ELISA. NaHS significantly ameliorated IL-6 secretion evoked by LPS (Fig. 2D). Consistent results were observed with IL6 mRNA levels in THP-1 cells, whereas NaHS elicited no significant effect in the absence of LPS (Supplementary Fig. S2B). To confirm the connection between the IL-6 reduction in CM and HAMP inhibition, anti-human IL-6-neutralizing antibody (αIL6, 0.05, and 0.5 μg/ml) was applied to THP-1 macrophages exposed to LPS. As illustrated in Figure 2E, HAMP induction by LPS-CM was suppressed and completely abolished by αIL6 at a higher concentration. In agreement with the decrease in IL-6 levels, JAK2 and STAT3 phosphorylation was inhibited by NaHS (Fig. 2F, Supplementary Fig. S2C, D) along with nuclear translocation of p-STAT3 (Fig. 2G). These results suggest that H2S could inhibit LPS-CM-induced HAMP expression via decreasing IL-6 and JAK2/STAT3 activation.
H2S ameliorates hepcidin expression by inhibiting STAT3 activation beyond reducing IL-6
Instead of treating macrophages with NaHS, we next applied NaHS directly to Huh7 cells exposed to the same CM, which was defined as the post-treatment here (Fig. 3A). Surprisingly, NaHS in the post-treatment model suppressed HAMP expression as well (Fig. 3B). Considering the critical role of IL-6 in LPS-CM, we then used recombinant human IL-6 for HAMP induction in Huh7 cells and similar results as observed with NaHS treatment (Fig. 3C). The effect of NaHS on IL-6-induced HAMP promoter activity was also evaluated by dual-luciferase reporter assay. As shown in Figure 3D, NaHS decreased HAMP promoter activity evoked by IL-6, whereas no significant change was observed in the NaHS-only group. Moreover, NaHS ameliorated Hamp induction by recombinant murine IL-6 in mouse primary hepatocytes (Fig. 3E). To rule out the possibility of off-target effects, Huh7 cells were preincubated with propargylglycine (PAG) or aminooxyacetic acid (AOAA), which inhibits CSE and CBS, respectively. After preincubation with
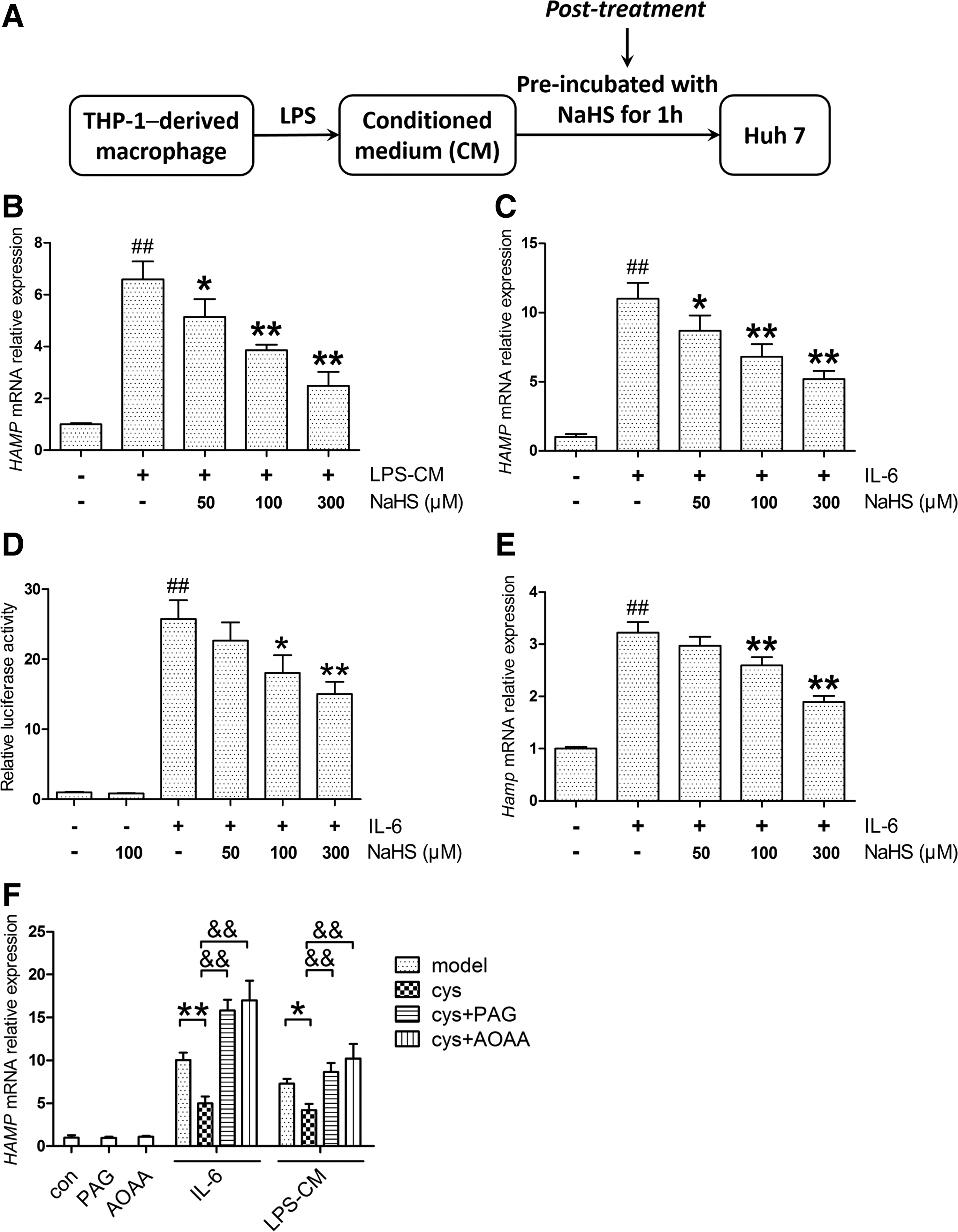
In the post-treatment model, Huh7 cells were treated with either equal amounts of IL-6 or the same CM. As expected, no significant change was observed in p-JAK2 levels (Fig. 4A, Supplementary Fig. S3B). However, NaHS attenuated STAT3 phosphorylation evoked by IL-6 (Fig. 4A, Supplementary Fig. S3C), indicating that an additional pathway is involved. To further evaluate the modulation of STAT3 activation, we analyzed p-STAT3 dimerization, translocation, and transcriptional activity. As indicated in Figure 4B and C and Supplementary Figure S3D, NaHS significantly suppressed p-STAT3 dimerization induced by IL-6, accompanied by decreased nuclear translocation. The transcriptional activity of STAT3 was measured using a luciferase reporter containing five copies of the sis-inducible element (SIE) in the promoter region of the luciferase gene. IL-6 stimulation dramatically increased STAT3 transcriptional activity, which was attenuated by NaHS treatment (Fig. 4D). A similar result was demonstrated in the mRNA level of SOCS3, a target gene of STAT3 (Fig. 4E).
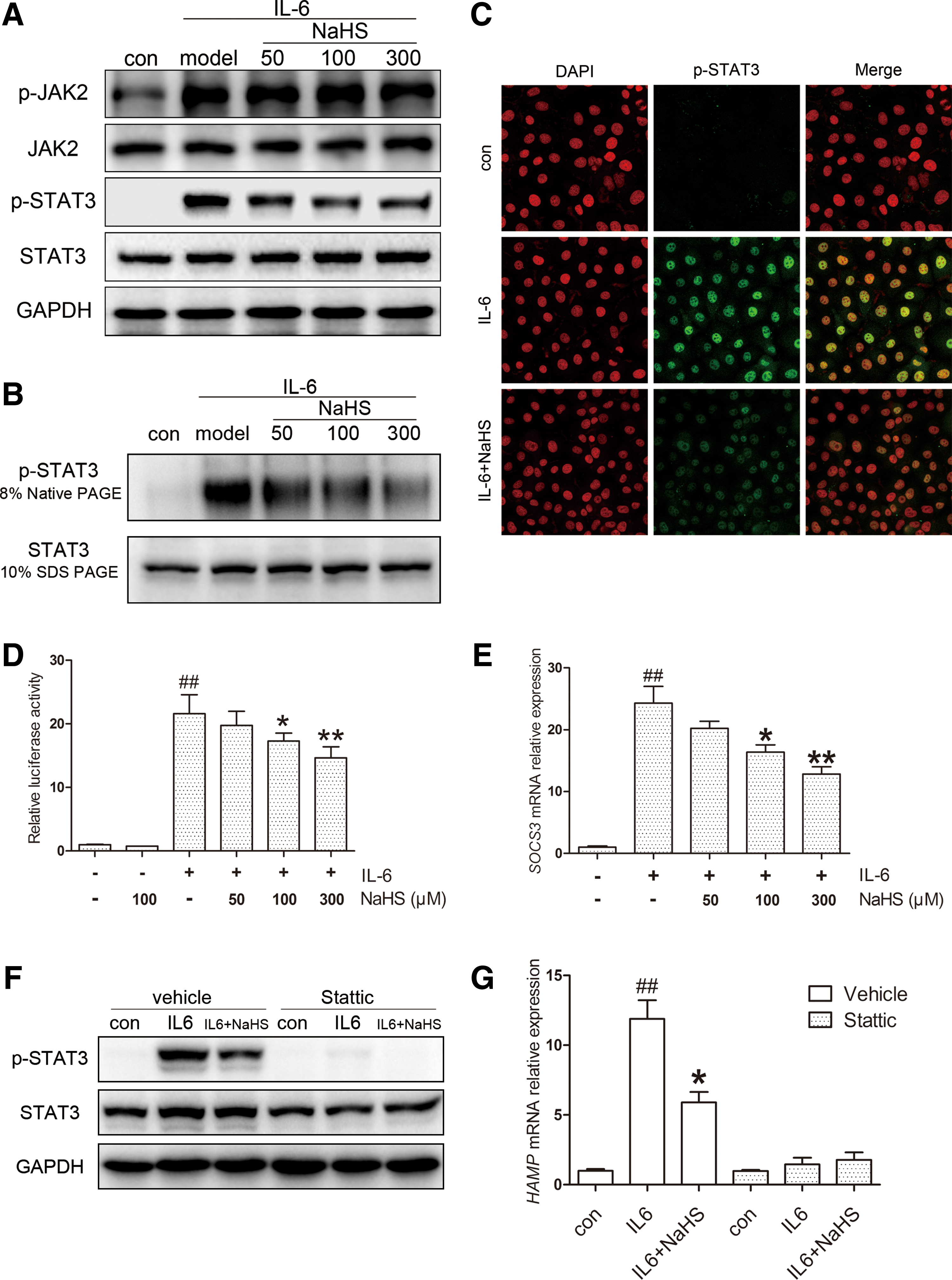
To confirm the involvement of STAT3 in HAMP expression, we treated Huh7 cells with stattic, a potent inhibitor of STAT3 activation. As presented in Figure 4F, stattic effectively blocked STAT3 phosphorylation induced by IL-6. As expected, HAMP displayed no response, and no significant change was observed with NaHS treatment (Fig. 4G).
Taken together, these results suggest that H2S inhibits STAT3 and hepcidin activation through targeting additional pathways other than IL-6/JAK2 signaling.
H2S attenuates IL-6-induced hepcidin expression through promoting SIRT1-mediated STAT3 deacetylation
Since additional mechanisms appeared to be involved in the post-treatment model, we turned our attention to factors regulating STAT3 activation. It was reported that inhibition of STAT3 acetylation (ac-STAT3) impairs its phosphorylation and transcriptional function (54). Thus, ac-STAT3 was investigated in the post-treatment model. As indicated in Figure 5A, NaHS ameliorated the ac-STAT3 levels induced by IL-6. To confirm the regulatory role of ac-STAT3 in self-phosphorylation and HAMP induction, we treated cells with sodium butyrate (NaBu), a histone deacetylase (HDAC) inhibitor, which raised ac-STAT3 levels in previous studies (43). NaBu markedly increased STAT3 acetylation in Huh7 cells, in parallel with the induction of p-STAT3 and HAMP expression, which could be abolished by stattic (Fig. 5B, Supplementary Fig. S4A). These results suggest that STAT3 acetylation regulates HAMP expression through self-phosphorylation.
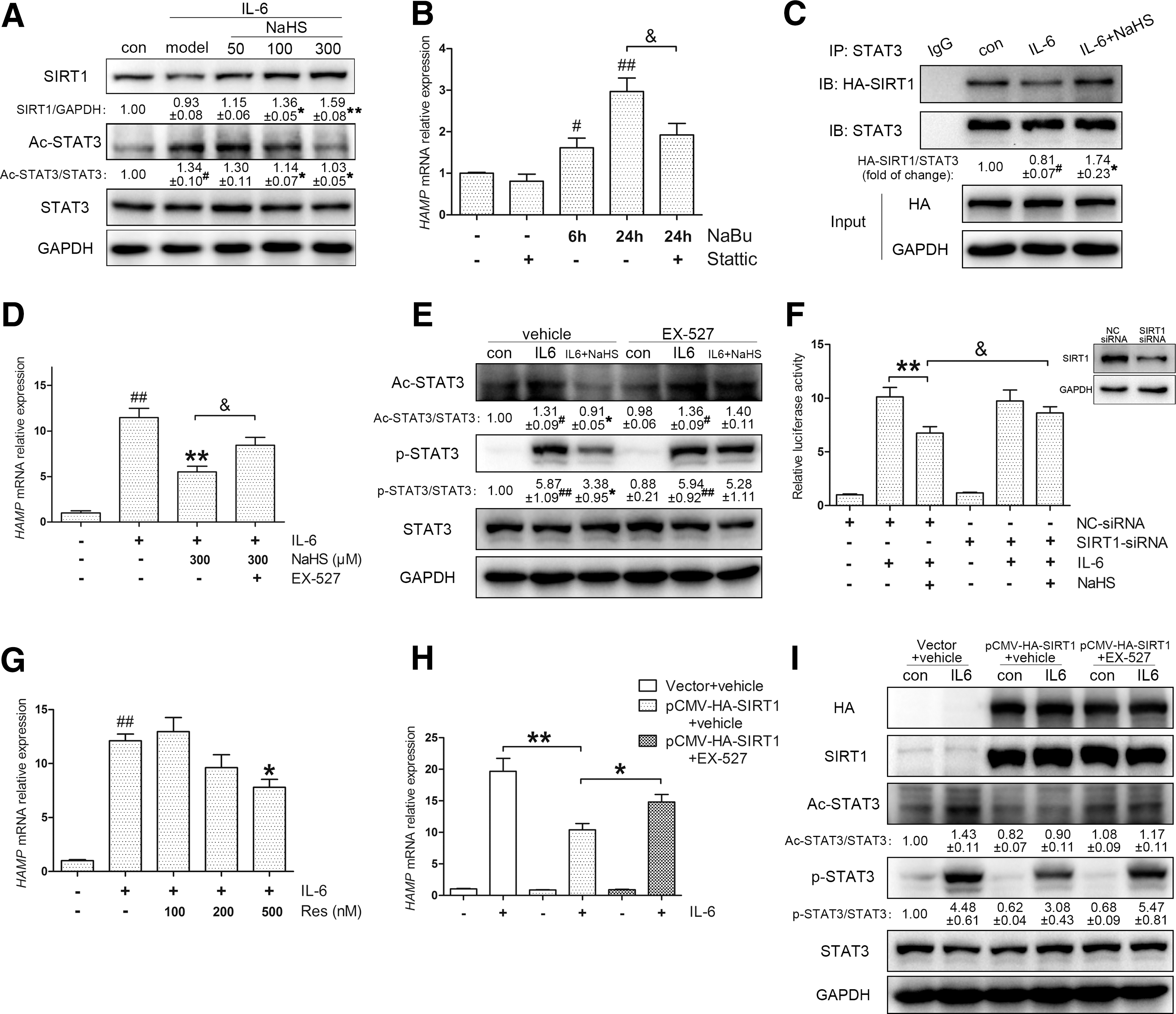
STAT3 acetylation is precisely regulated by histone acetyltransferases and HDACs, including SIRT1 as a class III HDAC (32). Thus, we sought to access the role of SIRT1 in the post-treatment model. As expected, NaHS increased SIRT1 protein levels in Huh7 cells (Fig. 5A) and mouse primary hepatocytes (Supplementary Fig. S4B). Next, we performed coimmunoprecipitation (co-IP) to examine the effect of H2S on the physical interaction between exogenous SIRT1 and STAT3. As shown in Figure 5C, IL-6 stimulation promoted dissociation of the SIRT1-STAT3 complex, which could be effectively stabilized by NaHS.
Then, we used EX-527, a highly selective SIRT1 inhibitor, to block SIRT1 activity. EX-527 abolished the H2S-mediated suppression of HAMP expression (Fig. 5D) in accordance with the STAT3 transcriptional function assessed by SIE reporter (Supplementary Fig. S4C). Immunoblot analysis indicated that EX-527 reversed the inhibition of STAT3 acetylation and phosphorylation by H2S (Fig. 5E). The involvement of SIRT1 was further confirmed by gene silencing. By transient transfection of SIRT1-specific or control siRNA, HAMP promoter activity was evaluated, and a coincident result is presented in Figure 5F.
Afterward, the effects of SIRT1 activation and overexpression were examined. Previous studies suggest that resveratrol improves physiological function by activating SIRT1 (19); hence, we introduced resveratrol as an SIRT1 activator. As was observed with NaHS, preincubation with resveratrol ameliorated HAMP induction (Fig. 5G) and STAT3 activation (Supplementary Fig. S4D). Moreover, SIRT1 overexpression suppressed IL-6-evoked HAMP expression as well as p-STAT3 and ac-STAT3 levels, which could be abrogated by EX-527 (Fig. 5H–I). Altogether, we conclude that H2S inhibits IL-6-evoked STAT3 activation and hepcidin expression by promoting SIRT1-mediated STAT3 deacetylation.
The effect of H2S on inflammatory hepcidin is diminished by SIRT1 inhibition and CSE knockout in vivo
We next investigated whether the regulation of SIRT1 and ac-STAT3 could be confirmed in vivo. C57BL/6 mice were pretreated with either resveratrol (10 mg/kg) only or EX-527 (10 mg/kg), followed by NaHS (6 mg/kg), before LPS (0.5 mg/kg) stimulation. As demonstrated in Figure 6A, EX-527 partially abolished the effect of NaHS. Moreover, resveratrol also inhibited hepatic Hamp expression. Corresponding results were also observed for serum iron content and IL-6 levels, in addition to hepatic SIRT1 and STAT3 activation (Fig. 6B–D). To better understand the H2S-SIRT1-hepcidin interaction in vivo, we also examined mice injected with recombinant murine IL-6. As manifested in Supplementary Figure S5, NaHS application improved hepatic hepcidin mRNA and serum iron levels, which were reversed by EX-527. The involvement of SIRT1 was further supported by immunoblots of STAT3 activation. These results demonstrate that H2S promotes SIRT1-mediated STAT3 deacetylation in vivo.
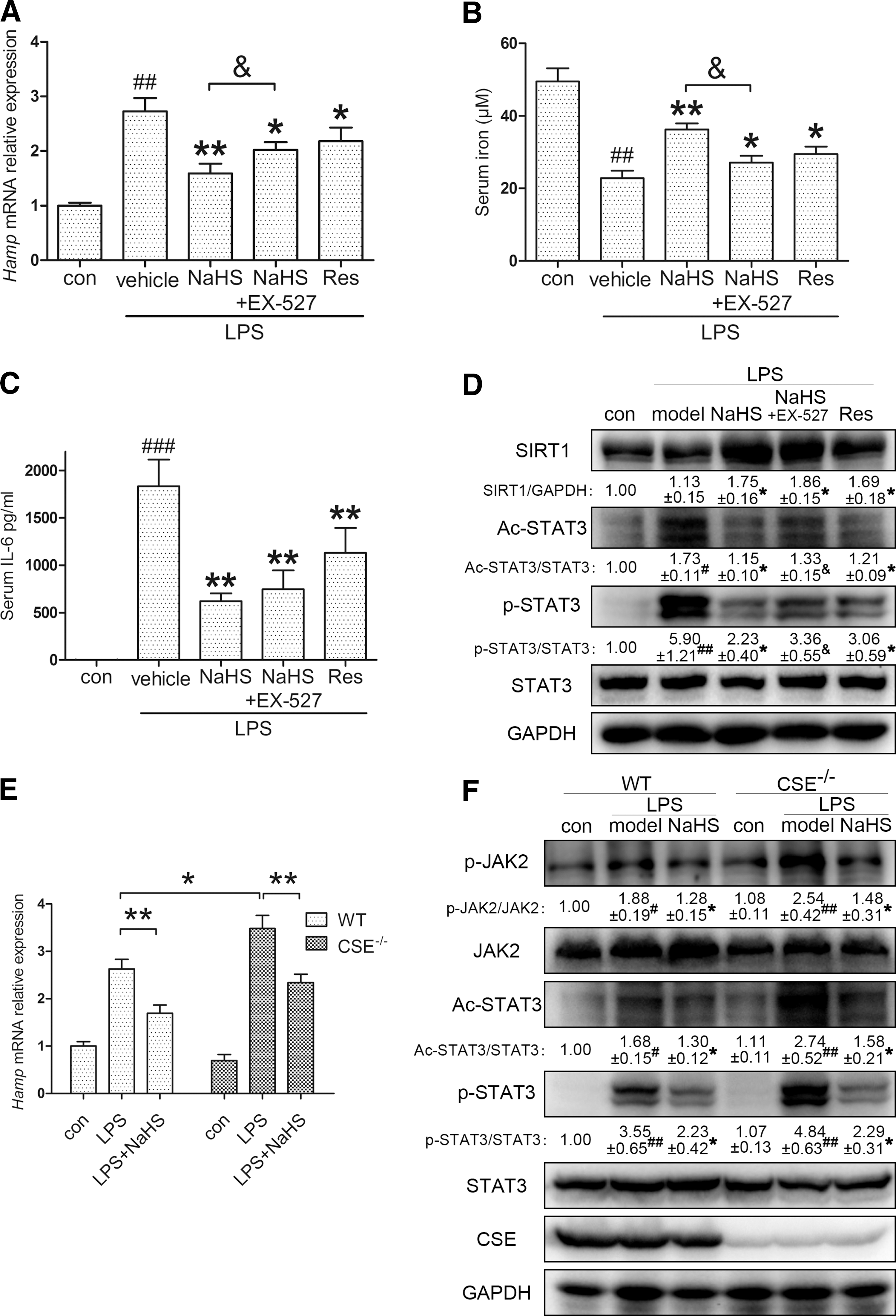
Furthermore, our hypothesis was also tested in CSE knockout (CSE−/−) C57BL/6 mice. As one of the endogenous H2S synthases, CSE is critical for H2S homeostasis. Previous research by our group and other groups demonstrated that plasma H2S levels in CSE−/− mice decreased by ∼50% compared with those in wild-type (WT) mice (17, 44). Thus, we compared the hepatic Hamp response between WT and CSE−/− mice. As manifested in Figure 6E, no significant change was observed in the two control groups. However, CSE−/− mice were more susceptible to LPS (0.5 mg/kg) challenge, indicating the significance of endogenous H2S homeostasis for inflammatory hepcidin production. Moreover, the intense Hamp induction was successfully reversed by NaHS (6 mg/kg) application. Immunoblot analysis confirmed that CSE deficiency exacerbated the LPS-induced JAK2/STAT3 activation, which was ameliorated by NaHS (Fig. 6F). These results provide strong evidence that H2S is critical for hepcidin induction and JAK2/STAT3 activation during inflammation.
Discussion
In the present study, we discovered that H2S suppressed inflammation-induced hepatic hepcidin production both in vivo and in vitro. H2S significantly decreased IL-6 secretion and JAK2/STAT3 activation, which was the dominant pathway for hepcidin induction. In addition, H2S attenuated hepcidin in Huh7 cells and mouse primary hepatocytes in an SIRT1-dependent manner. By promoting SIRT1 expression and stabilizing SIRT1-STAT3 interactions, H2S ameliorated STAT3 acetylation irritated by IL-6. Reduced acetylation suppressed STAT3 phosphorylation and transcriptional function, as well as hepcidin induction. Inhibition or silencing of SIRT1 diminished H2S suppression of hepcidin, whereas SIRT1 activation and overexpression blocked hepcidin induction. Consistent results were obtained in vivo. Furthermore, knockout of CSE, one of the endogenous H2S synthases, exaggerated inflammatory hepcidin expression in mice. Taken together, our results bring forward SIRT1 as a new modulator of hepcidin and H2S as a novel potential therapeutic modality for inflammatory anemia. The schematic diagram is summarized in Figure 7.
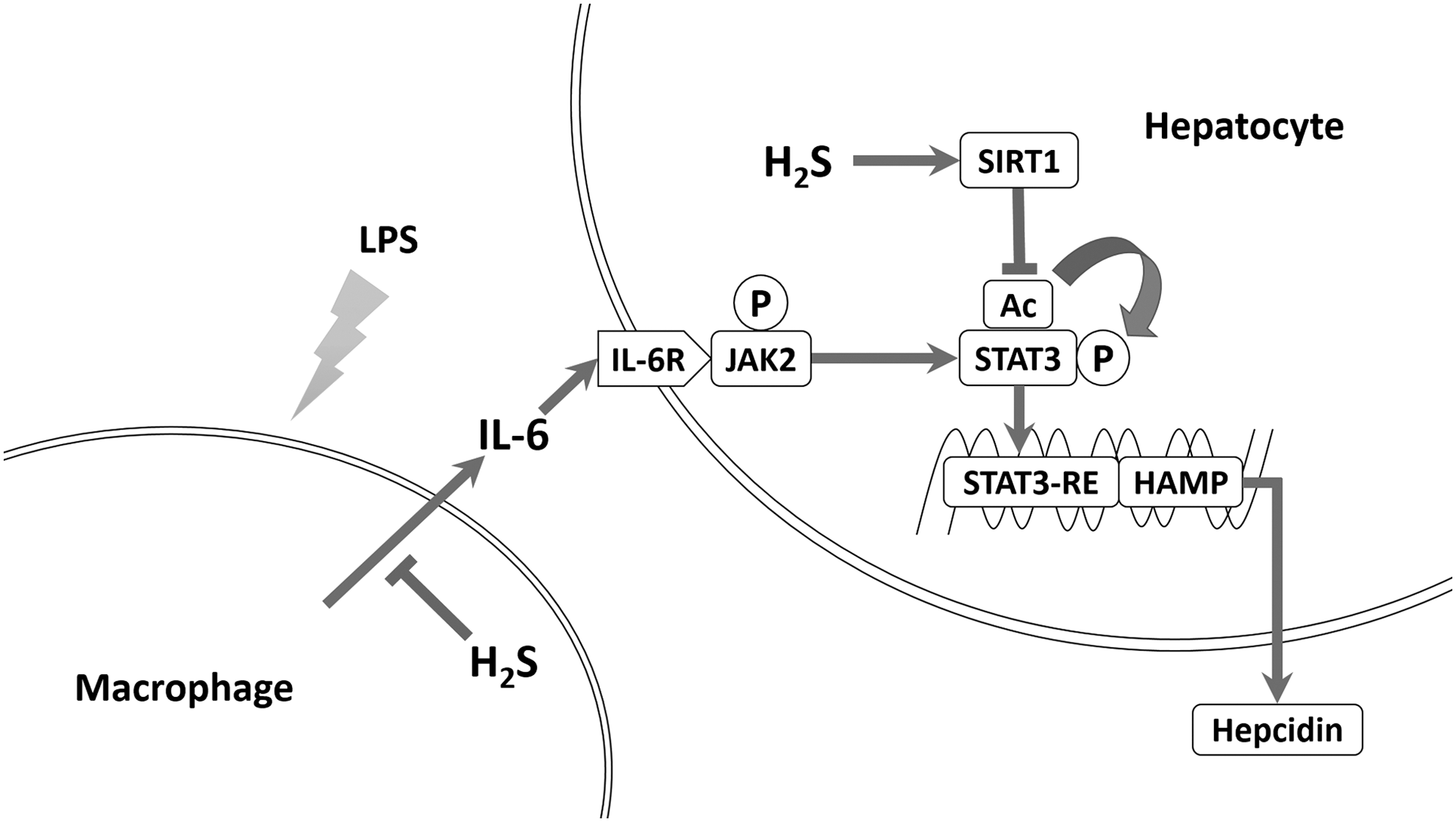
Hepcidin, initially named for its antimicrobial property, is induced during infection or inflammation. This response is regarded as a host defense mechanism against microbial proliferation, which relies on serum iron. Previous studies confirmed that IL-6, secreted from monocytes and macrophages, is responsible for inflammatory hepcidin induction through activating STAT3 (29, 41). The CM model is well accepted in relevant studies, as opposed to exposing hepatocytes to LPS directly (9). In fact, Huh7 cells and mouse primary hepatocytes barely responded to direct LPS challenge with respect to the p-STAT3 level and hepcidin expression (unpublished observations). As a result, we adopted LPS-mediated CM and recombinant IL-6 for hepcidin induction in vitro.
As the third gasotransmitter, H2S plays a paramount role in the regulation of signal transduction. Mounting evidence from independent groups illustrates that H2S is tightly involved in the pathophysiological progression of diseases, such as sepsis, heart failure, and atherosclerosis (50). Since its initial discovery, however, there have been controversies over the role of H2S in inflammation and inflammatory diseases. Currently, it appears that H2S can have both pro- and anti-inflammatory activities depending on the dose and sampling time (14). In this work, we observed that H2S elicited anti-inflammatory effects and suppressed IL-6 secretion in both THP-1-derived macrophages and mice, leading to decreased JAK2/STAT3 activation and hepcidin expression. Previous research indicated that H2S inhibits NF-κB activation under inflammatory conditions, which is the most crucial pathway in the induction of proinflammatory cytokines (34, 48, 52). Thus, the suppression of NF-κB activation by H2S is a potential mechanism involved in IL-6 suppression.
In pharmacological studies, NaHS is widely accepted as an exogenous H2S donor. Due to its chemical property, NaHS releases H2S in a robust manner, but for a relatively short duration after dissolution. Previous work by Whiteman et al. suggested that H2S is completely released from NaHS in 1–2 h (52). To guarantee accuracy and repeatability, NaHS was freshly dissolved immediately before use. In addition, H2S in the CM of the pretreatment model escapes completely after 24 h of incubation and thus has no overlap with that in the post-treatment model.
STAT3 is a member of the STAT family with important roles in cellular transformation, proliferation, inflammation, and metastasis of cancer (1, 6). It is well established that phosphorylation on Tyr705 of STAT3 is essential for its activation upon stimulation with the IL-6 superfamily. On the contrary, increasing studies indicate that STAT3 is also acetylated on some lysine residues. Yuan et al. first reported that STAT3 acetylation on Lys685 by p300 regulates its dimerization and DNA binding ability (54). Recently, Dasgupta et al. suggested that the integrity of Lys685 is required for most unphosphorylated STAT3-dependent genes (7). Furthermore, Nie et al. found that mutations of four key lysines of STAT3 impair its ability of phosphorylation and transactivation functions (32).
Consistently, our results suggest that the deacetylation promoted by H2S suppresses STAT3 phosphorylation and transcriptional function under IL-6 stimulation. In addition, for the first time, we established a link between STAT3 acetylation and hepcidin expression, indicating an alternative approach for hepcidin modulation. However, the specific mechanism by which STAT3 acetylation regulates self-phosphorylation remains unclear. This regulation may be attributed to the change in STAT3 conformational transformation following acetylation since Lys685 locates to the end of the β-sheet region of the SH2 domain (3). It is also noteworthy that acetylation may compete with and negatively regulate protein ubiquitination and stabilization in some circumstances (23, 25).
In line with our observations, other groups have also reported STAT3 acetylation by IL-6 (35), although the mechanisms involved remain unclear. In this work, we found that IL-6 promoted the dissociation of the STAT3-SIRT1 complex. As a deacetylase, SIRT1 plays an important role in STAT3 modification. Thus, dissociation of the complex perhaps accounts for the STAT3 acetylation induced by IL-6. In support of this notion, we observed that stabilization of the STAT3-SIRT1 complex by NaHS reduced STAT3 acetylation levels, which could be abolished by the SIRT1 inhibitor, EX-527. Moreover, consistent results were also demonstrated upon exposure to the SIRT1 activator, resveratrol, as well as after SIRT1 overexpression. It is worth noting that acetylation of STAT3 is also regulated by histone acetyltransferases such as the CREB-binding protein/p300 family (51). Therefore, we conclude that the induction of STAT3 acetylation by IL-6 is, at least partially, attributable to the dissociation of the STAT3-SIRT1 interactions.
SIRT1, a mammalian ortholog of Sir2, is an NAD+-dependent deacetylase involved in a variety of physiological process. While this article was in preparation, other independent groups also reported SIRT1 activation by H2S (24, 56). The mechanisms in H2S-induced SIRT1 activation remain unidentified, but increasing evidence suggests the involvement of energetic status. Concerning the STAT3-SIRT1 interaction, previous studies on hepatic gluconeogenesis have revealed that the inhibitory effect of STAT3 is regulated by the nutritional status through SIRT1-mediated deacetylation (32). In diabetic db/db mice, deletion of SIRT1 increases STAT3 acetylation and exacerbates symptoms of diabetic nephropathy (13).
Unlike basal STAT3 phosphorylation, however, the regulation of SIRT1 on IL-6-induced robust STAT3 activation and downstream hepcidin induction remains unknown. In this work, we present strong evidence that SIRT1 promotion by H2S decreases STAT3 activation and hepcidin expression during inflammation. After all, SIRT1 plays a key role in metabolic regulation and adaptation (4, 5). Recent studies demonstrated the relationship between innate immunity and energy metabolism through antagonistic cross talk between NF-κB and SIRT1 signaling pathways (15), and hepcidin is sensed by innate immune and infectious stimuli (2). Additionally, an RNAi screen study identified that some nutrient metabolism genes might regulate hepcidin expression and iron homeostasis (27). Thus, these results suggested that SIRT1 might be a critical energy sensor connecting cellular metabolism and hepcidin expression. Further confirmatory work is necessary.
In summary, we demonstrated for the first time that H2S suppressed inflammatory hepatic hepcidin by reducing IL-6-induced JAK2/STAT3 activation and promoting SIRT1-mediated STAT3 deacetylation. Our work provides novel insights into SIRT1-regulated hepcidin expression and clarifies the anti-inflammatory property of H2S. Furthermore, these findings highlight H2S-releasing compounds as innovative candidates for the treatment of inflammatory anemia.
Materials and Methods
Reagents
Sodium hydrosulfide (NaHS),
Recombinant human and murine IL-6, goat anti-human IL-6 antibody, and goat normal IgG were obtained from Peprotech. Antibodies to total and phospho-JAK2, total and phospho-STAT3 (Tyr705), acetyl-STAT3 (Lys685), and HA were purchased from Cell Signaling Technology. Antibodies to SIRT1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were acquired from Bioworld Technology. In addition, antibodies to ferroportin and CSE were obtained from Santa Cruz Biotechnology.
Animals
All animal care and experimental protocols complied with the Animal Management Rules of the Ministry of Health of the People's Republic of China and were approved by the Animal Care Committee of Fudan University.
Eight-week-old male C57BL/6 and C3H mice were purchased from the Sippr-bk Experimental Animal Center. The CSE knockout C57BL/6 mice were a kind gift from Shanghai Research Center of Model Organisms, Shanghai, China. Mice were housed under specific pathogen-free conditions at 25°C and maintained under a 12-h light/12-h dark cycle with ad libitum access to food and water. NaHS was freshly dissolved in sterilized saline and administrated intraperitoneally (i.p., 6 mg/kg/day) to mice for 3 days before LPS challenge. Saline was used as a vehicle control. On the fourth day, mice were i.p. injected with 0.5 mg/kg LPS 2 h after the last NaHS treatment. In experiments with EX-527 and resveratrol, these two reagents were dissolved in 0.5% DMSO and 0.5% Tween 20 (vol/vol) saline solution and i.p. administrated 2 h before NaHS or LPS treatment, both at 10 mg/kg. The same solvent was used as a vehicle control. C57BL/6 mice were sacrificed by cervical dislocation for mRNA, protein, and serum analysis 6 h after LPS stimulation. Tissue iron measurements and Perl's Prussian blue staining were performed in C3H mice at 24 h. All efforts were made to ameliorate animal sufferings during the experiments.
Cell culture
Cell cultures were maintained at 37°C under humidified air with 5% CO2. Human Huh7 hepatoma cells (JCRB, Japanese Collection of Research Bioresources) were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; ExCell Bio) and 1% penicillin and streptomycin (Gibco). THP-1, a human monocytic cell line from ATCC, was maintained in RPMI 1640 medium with 10% FBS and 1% penicillin and streptomycin. THP-1 cells were seeded into six-well plates at 6×106 cells per well and treated with 100 nM PMA overnight to induce the differentiation into macrophage-like cells. After washing with phosphate-buffered saline (PBS), differentiated THP-1 macrophages were cultured in fresh medium for 1 day, followed by 1 μg/ml LPS stimulation for another 24 h. After centrifugation, the cell-free supernatant of the culture medium was collected as CM. Huh7 cells (5×105) were exposed to undiluted CM for 6 h, and then harvested for mRNA or protein analysis. For IL-6 treatment, Huh7 cells were directly incubated with 10 ng/ml recombinant human IL-6 without THP-1-derived CM.
Compound treatments
To investigate the effect of H2S on macrophages and hepatocytes separately, NaHS was applied at two different stages—either to THP-1 or Huh7 cells—in the CM model. In one condition, the compound was applied to THP-1 cells 1 h before LPS challenge to generate different CMs, which was defined as the pretreatment in this article; in the other condition, the compound was applied to Huh7 cells 1 h before exposing to the same CM, which was defined as the post-treatment.
Resveratrol, EX-527, and stattic were prepared as DMSO solutions. When cells were challenged with these compounds, the same volume of DMSO was added in parallel as a vehicle control.
Isolation and culture of mouse primary hepatocytes
Mouse primary hepatocytes were isolated from 8-week-old C57BL/6 mice using a modified two-step collagenase perfusion protocol. In brief, after anesthesia, the liver was perfused with calcium and magnesium-free Hank's balanced salt solution containing 0.5 mM EDTA and 0.05 M HEPES, followed by DMEM containing 5 mM calcium chloride and 0.05% type IV collagenase. The liver was then removed and isolated cells were purified with 40% Percoll (GE Healthcare Bio-Sciences). Hepatocytes were seeded onto six-well plates at a density of 8×105 cells per well. After 3 h, the culture medium was changed, and the cells were exposed to 50 ng/ml recombinant murine IL-6 for 6 h on the following day.
RNA isolation and real-time quantitative reverse transcription polymerase chain reaction
Total RNA from cultured cells and mouse livers was extracted with RNAiso Plus (TAKARA Bio) according to the manufacturer's instructions. Reverse transcription of total RNA was conducted with PrimeScript™ RT Master Mix (Perfect Real Time; TAKARA Bio). Primer sequences are provided in Table 1. Real-time quantitative polymerase chain reaction (PCR) was performed and semiquantified on a Bio-Rad IQ5 Real-Time PCR System with SYBR Green Premix Ex Taq (TAKARA Bio).
Plasmid and siRNA transfection
pCMV-HA vector, pCMV-HA-hSIRT1, and pCMV-FLAG-hSTAT3 were purchased from Bioworld. Plasmids were purified using a QIAprep Spin Miniprep Kit (QIAGEN). Huh7 cells were transiently transfected with 2 μg of plasmid per well for six-well plates, using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Six hours later, the cells were exposed to fresh medium and cultured for another 48 h until IL-6 treatment.
For silencing experiments, Huh7 cells in 12-well plates were transfected with 100 nM negative control siRNA or specific siRNA against human SIRT1 (Sigma-Aldrich) using Lipofectamine 2000. The medium was replaced at 6 h post-transfection, and silencing efficiency was determined by Western blotting 72 h after transfection.
Dual-luciferase reporter assay
The human hepcidin promoter (−1000/ +71) was constructed by PCR amplification and inserted into the pGL3 basic vector (Promega Biotech) using MluI and XhoI restriction enzyme sites (Obio Tech). The pGL4.47 [luc2P/SIE/Hygro] vector was purchased from Promega Biotech as an SIE reporter. Huh7 cells were cotransfected with 240 ng of hHAMP-luc or SIE constructs and 8 ng of CMV Renilla control plasmid using Lipofectamine 2000. At 48 h after transfection, cells were stimulated with IL-6 for 6 h. For gene silencing experiments, Huh7 cells were cotransfected with reporter plasmids together with SIRT1-specific or control siRNA. Relative luciferase activities were determined using the Dual-Luciferase System (Promega Biotech), according to the manufacturer's instructions. All transfection experiments were performed in quadruplicate.
Immunoprecipitation and immunoblot analysis
For immunoprecipitation (IP), cells were lysed in IP-specific buffer (20 mM Tris–HCl, 150 mM NaCl, 1% Triton X-100, pH 7.5) and incubated with antibody and Protein A/G Plus-Agarose according to the manufacturer's instructions (Santa Cruz Biotechnology). Normal IgG (Santa Cruz Biotechnology) was used as a negative control. Finally, immunoprecipitates were washed four times before boiling with 2×loading buffer.
For SDS-PAGE, livers were homogenized in RIPA lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS, pH 7.4) containing a protease and phosphatase inhibitor cocktail (Sigma-Aldrich). Cell samples were lysed in 1×NuPAGE® LDS Sample Buffer (Invitrogen). Fifty micrograms of protein was subjected to SDS-PAGE gels for each sample.
For native PAGE of p-STAT3 dimers, native protein extracts were prepared in the same lysis buffer used for IP. After 30 min of lysis, cell extracts were centrifuged at 12,000×g for 10 min, and the supernatant was loaded onto 8% SDS-free PAGE gels.
After electrophoresis, protein samples were transferred to PVDF membranes (Millipore), followed by blocking with 5% skim milk and incubation with primary antibodies overnight at 4°C. The blots were then developed with horseradish peroxidase-conjugated antibodies at room temperature. Immunoreactive proteins were visualized using ECL (Millipore) and quantified by densitometry using a Bio-Rad Image Laboratory system. GAPDH served as the loading control.
Immunofluorescence staining
Huh7 cells were fixed with 4% formaldehyde and treated with ice-cold methanol for 10 min at −20°C. After blocking in PBS with 5% BSA and 0.3% Triton X-100 for 1 h at room temperature, the cells were incubated with phospho-STAT3 (Tyr705) rabbit mAb diluted in PBS with 1% BSA and 0.3% Triton X-100 at 4°C overnight. Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen) was used as the secondary antibody and was diluted in PBS with 0.1% Tween 20 and incubated for 1 h in the dark, followed by staining with DAPI for another 10 min. The slides were observed by confocal laser scanning microscopy (Zeiss LSM 710).
Quantification of H2S concentration
H2S determination was performed using the methylene blue method as described previously (44).
Serum iron, hepcidin, and IL-6 analysis
Mouse blood samples were collected in nonheparinized tubes and centrifuged at 3000 rpm for 10 min to separate serum. Serum iron content was measured using a commercial kit according to protocols described by Roche Diagnostics.
Murine serum hepcidin and IL-6, together with human IL-6 in CM, were quantified by ELISA following protocols described by the manufacturers (hepcidin kit from Uscn, IL-6 kit from Boatman).
Tissue nonheme iron analysis
Mouse spleen samples were first dried at 65°C for 48 h, and then digested in acid solution (3 M hydrochloric acid and 10% trichloroacetic acid) at 65°C for 24 h after weighing. Afterward, the acid extracts were centrifuged, and the supernatant was collected. 1, 10-Phenanthroline monohydrate, a heterocyclic compound widely used for photometric determination of Fe (II), was mixed with the extract at a volume ratio of 3:1 and incubated for 20 min at room temperature. The absorbance at 510 nm was measured using a spectrophotometer. A serial dilution of 45 mM ferrous ammonium sulfate (Sigma-Aldrich) was used to construct a calibration curve.
Perl's Prussian blue staining of paraffin-embedded sections
To evaluate iron deposition in mouse spleens, tissues were fixed in 4% formalin PBS solution, embedded in paraffin wax, sectioned, and stained with Perl's Prussian blue solution for 30 min at room temperature. A neutral red counterstain was then applied to provide a contrasting background. Images were captured using the Zeiss Axio Scope A1 system.
Statistical analysis
Data are expressed as the mean±SEM. Statistical analysis was performed using one-way ANOVA, followed by Student's t-tests. Values of p<0.05 were considered statistically significant.
Footnotes
Acknowledgments
This work was supported by grants from the National Major Scientific and Technological Special Project (No. 2012ZX09501001-001-003; 2012ZX09103101-064), National Natural Science Foundation of China (No. 81330080; 81173054), Shanghai Committee of Science and Technology of China (No. 14JC1401100), a key laboratory program of the Education Commission of Shanghai Municipality (No. ZDSYS14005), and the college students' innovation project of Fudan University (Wangdao, No.13024).
Authorship Contributions
The authors who participated in the research design were H.X. and M.W. M.W., H.X., W.T., Z.S., L.M., W.W., C.L., and X.X. conducted experiments. H.X, M.W., and X.W. performed the data analysis, and M.W., H.X., and Y.Z.Z wrote or contributed to the writing of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.