
Abstract
This Forum addresses the role of mitochondrial dysfunction in the multifactorial nature of diabetic cardiomyopathy (DCM) from multiple angles. Contributors deliver a diverse and in-depth view of the state-of-the-art in DCM, from bench to bedside. What emerges is a picture of mitochondrial dysfunction as a central upstream defect, inflicted on the heart by diabetes. Collectively, the authors pinpoint high-value knowledge gaps, propose new conceptual frameworks, and highlight understudied, but promising, research themes. Antioxid. Redox Signal. 22, 1499–1501.
T
Cardiovascular complications account for most of the morbidity and mortality in the population affected by metabolic syndrome. In particular, the link between diabetes and heart failure (HF) has gained increased attention. Epidemiological studies have confirmed that people with diabetes are more than twice as likely to develop HF compared with people without diabetes. For example, the absolute risk of cardiovascular death is three times higher for men with diabetes than nondiabetic ones after adjustment for several risk factors, including age, race, and income (4).
Chronic diabetes can affect the myocardium directly, resulting in progressive deterioration of cardiac function, a condition termed “diabetic cardiomyopathy” (DCM), as first coined by Rubler in the early 1970s (3). DCM develops independently from other risk factors, including coronary heart disease. In its early stages, DCM presents left ventricular (LV) hypertrophy and diastolic dysfunction, with systolic dysfunction occurring later and progressing to decompensated HF. Although early treatment and screening of DCM would be optimal to prevent a further decline in cardiac performance, specific diagnostic criteria do not exist as the most common clinical features associated with DCM—LV hypertrophy and diastolic dysfunction—also characterize nondiabetic forms of HF (see Schilling in this Forum).
Mitochondrial function plays a crucial role in DCM. In the human body, heart mitochondria process the highest amount of oxygen to harness the essential redox and phosphorylation energy required for organ function (see Aon et al. in this Forum). Thus, the heart faces the highest degree of exposure to possible oxidative damage. To a lesser extent, although equally significant, this also holds true for all major organs of the body. However, in a chronic disease like diabetes, the significance is highest for oxidative and postmitotic organs such as the heart and skeletal muscle.
Overview
In this Forum, eight contributions address the role of mitochondrial dysfunction in the multifactorial nature of DCM from multiple angles. Together, the authors provide comprehensive, timely, and up-to-date perspectives on the key questions driving the field. What emerges is a picture of mitochondrial dysfunction as a central upstream defect, inflicted on the heart by diabetes. Many of the pathologic hallmarks of DCM, including lipid and glucose excess, oxidative stress, and inflammation that ultimately lead to diastolic and systolic dysfunction, can be traced back to impaired mitochondrial function. The pathologic abnormalities elicited by diabetes arise either directly or indirectly from defects in the redox and energetic capacity of mitochondria, provoked by deficits in their natural turnover and alterations in their morphological dynamics driven by fusion–fission processes. As a result of these perturbations, defective mitochondria accumulate, causing oxidative stress, impaired energetics, and altered calcium cycling, leading first to diastolic and ultimately to systolic heart dysfunction over the progression of DCM.
This Forum delivers a diverse and in-depth view of the state-of-the-art in DCM, from bench to bedside. Collectively, the authors have pinpointed high-value knowledge gaps, proposed new conceptual frameworks, and highlighted understudied, but promising, research themes that can be summarized as follows: 1. Mitochondrial fission–fusion dynamics have direct consequences for mitochondrial energetics and the progression of diabetes. A pathological transition between at least two distinct mitochondrial stages characterized by altered morphology and energetic behavior correlates with the disease progression (see Galloway and Yoon in this Forum). 2. The impairment of autophagy during diabetes leads to defective mitochondrial turnover (mitophagy) and attendant dysfunction. Kubli and Gustaffson propose that kick-starting mitophagy may help combat DCM by culling defective mitochondria and enriching healthy ones (see Kubli and Gustaffson in this Forum). 3. Protective mechanisms of mitochondrial function are present in the diabetic heart, favoring the preservation of the myocardial redox environment essential for optimal contractile performance. Understanding the nature and timing of these mechanisms may help to properly harness and stimulate them to generate novel therapeutic approaches (see Aon et al. in this Forum). 4. Early detection of DCM is particularly important, as is the accurate assessment of the stage of progression, from compensated through transitional to decompensated, if we are to refine therapeutic strategies. Moreover, understanding the interplay between diabetes and other cardiac stressors such as hypertension, ischemia, and valvular disease is critical (see Schilling in this Forum). 5. Three contributions (see Hafstad et al., Sung et al., and Schrauwen-Hinderling et al. in this Forum) frame the important debate about substrate selection in cardiac and skeletal muscles, and the links with oxidative stress and insulin resistance. The salutary role of exercise and the potential use of resveratrol for the management and treatment of DCM are thoroughly analyzed. 6. The fact that DCM and HF share similar outcomes, in terms of oxidative stress, but from contrasting metabolic phenotypes, suggests that the two may benefit from distinct therapeutic strategies. Moreover, it may spur a critical evaluation of whether the pharmacological treatment of patients with DCM should be similar to other forms of HF (see Roul and Recchia in this Forum).
Over the course of assembling this review series, our interest was sparked by a recent report by Picard et al. (2) who showed that reprogramming of nuclear gene expression can be elicited by high levels of mitochondrial DNA heteroplasmy (i.e., an intracellular mixture of mutated and normal mtDNAs; 50%–90%), via retrograde signaling, with dramatic phenotypic effects. Moreover, even moderate heteroplasmy (20%–30%) is sufficient to elicit changes in oxidative phosphorylation. The results are particularly germane to this review series, which examines the relationship between diet, exercise, redox balance, mitochondrial fission/fusion dynamics, and the resultant effects on mitochondrial DNA integrity. On the one hand, substrate excess promotes mitochondrial fragmentation favoring mtDNA modification. On the other hand, caloric restriction or exercise would trigger mitochondrial fusion, bolstering cell survival while maintaining mtDNA integrity. Finally, the mechanisms mediated by mtDNA damage may be relevant for DCM because they connect basic mitochondrial fusion/fission (see Galloway and Yoon in this Forum) that impinges on apoptosis, which in turn is closely integrated with the mitophagy quality control pathway (see Kubli and Gustaffson in this Forum). This is a novel and crucial research area with potentially very important and novel therapeutic implications.
Future Directions, New Questions
The scope and context of this thematic series, on the state of mitochondrial research as it pertains to DCM, are summarized in the accompanying figure (Fig. 1). It emphasizes the manner by which lifestyle (nutrition, physical activity, exercise) may have an impact on the health of the mitochondrial network. We submit that a proper balance of energetic supply and demand favors a healthier mitochondrial pool (center, green arrows), whereas a chronic imbalance, as when energy supply exceeds demand (red arrows at left), may lead to a dysfunctional mitochondrial ensemble. As for the mechanism, mitochondrial fusion–fission dynamics and mitophagy play a central role in concert with the mitochondria energy/redox behavior.
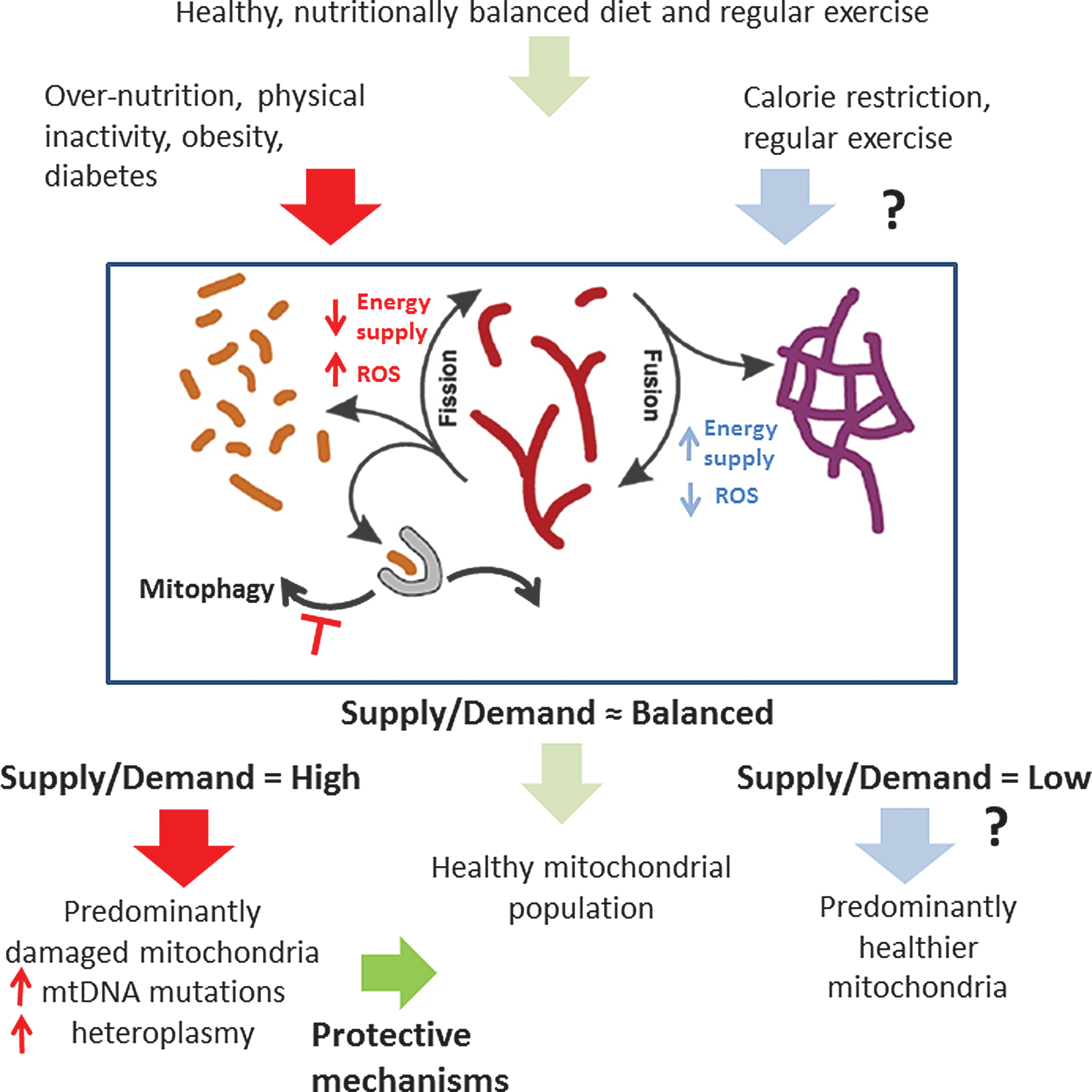
Compelling and extensive experimental evidence supports the view that the performance of the mitochondrial population from diabetic patients or animal models exhibits three main deficits from an energetic standpoint: lower mitochondrial respiration, reduced activity of individual respiratory complexes, and impaired OxPhos coupling (see Aon et al. in this Forum). These deficits likely stem from the accumulation of damaged mitochondria due to altered fission/fusion dynamics and impaired mitophagy.
Yet questions remain. What if the ratio of energy supply over demand is low, as in the case of caloric restriction (blue arrows at right)? Are the effects of exercise on mitochondrial function the same in the setting of caloric restriction as they are in normocaloric individuals? Does a predominantly fused mitochondrial network remain functionally competent in the long term? How can contractile efficiency be maximized in the diabetic heart? Finally, what is the role of heteroplasmy? Do increased mtDNA mutations activate retrograde signaling to the nucleus of the diabetic heart? If so, is the gene reprogramming protective, adaptive, or maladaptive?
Answering these questions will, no doubt, foreshadow the answer to the bigger question. Ultimately, can we harness the knowledge of these relationships to intervene and preserve mitochondrial health and, in so doing, forestall the onset and curb the progression of DCM in patients?