
Abstract
Aims:
Doxorubicin (DOX) is a chemotherapeutic drug that is used to treat many cancers, but its use is limited by cardiotoxic side effect. Carbonyl reductase 1 (CBR1) is an NADPH-dependent oxidoreductase that reduces DOX to doxorubicinol (DOXOL), a less potent derivative that is responsible for DOX cardiotoxicity. Thus, we aimed to demonstrate that inhibition of CBR1 enhances the chemotherapeutic efficacy of DOX and attenuates cardiotoxicity.
Results:
Pharmacological or genetic inhibition of CBR1 improved the anticancer effects of DOX in preclinical models of breast cancer. RNA interference or chemical inhibition of CBR1 improved the anticancer effect of DOX in breast cancer. Moreover, CBR1 overexpression enabled breast cancer cells to obtain chemotherapeutic resistance to DOX treatment. Intriguingly, inhibition of CBR1 decreased DOX-induced cardiotoxicity in animal model.
Innovation and Conclusions:
Inhibition of CBR1 increases chemotherapeutic efficacy of DOX and reduces cardiotoxicity by blocking DOX reduction to DOXOL. Therefore, we offer preclinical proof-of-concept for a combination strategy to safely leverage the efficacy of doxorubicin by blunting its cardiotoxic effects that limit use of this cytotoxic agent used widely in the oncology clinic. Antioxid. Redox Signal. 26, 70–83.
Introduction
B
Doxorubicin (DOX), an anthracycline antibiotic derived from Streptomyces peucetius var. caesius (3), is one of the most effective chemotherapeutic agents in the primary treatment of breast cancer (19, 21, 47). DOX intercalates between DNA base pairs, resulting in conformational changes in DNA structure. It prevents DNA and RNA synthesis by inhibiting the activity of topoisomerase II (Topo II) (23). DOX also generates reactive oxygen species (ROS), such as superoxide (O2 •−), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH), via the interaction with iron during intracellular metabolism, which plays a critical role in cancer cell death (10, 50). Moreover, DOX is believed to cause dose-dependent inhibition of mitochondrial oxidative phosphorylation (10, 52).
Carbonyl reductase1 (CBR1) is an NADPH-dependent oxidoreductase that reduces doxorubicin (DOX) to doxorubicinol (DOXOL), which has less potent anticancer effects than DOX and leads to chronic cardiotoxicity. Inhibition of CBR1 enhances the chemotherapeutic efficacy of DOX and attenuates cardiotoxicity by blocking DOX reduction to DOXOL. This study offers preclinical proof-of-concept for a combination strategy to safely increase the efficacy of doxorubicin by blunting its cardiotoxicity that limits the use of this agent.
One of the major adverse effects of DOX is cardiotoxicity (29, 46, 51), which limits its use in breast cancer patients. In clinical practice, cumulative doses of DOX can induce irreversible cardiotoxicity, resulting in congestive heart failure (25, 38). Cardiotoxicity is associated with a poor prognosis and high mortality rate. A metabolite of DOX, DOXOL, which is reduced at the side chain C-13 carbonyl moiety of DOX, has been implicated in DOX-induced cardiotoxicity (7, 35). DOXOL has less DNA binding affinity (34) and, thus, exhibits lower chemotherapeutic efficacy than DOX. Several enzymes are involved in the reduction of DOX to DOXOL, including carbonyl reductase 1 (CBR1) (13) and aldo-keto reductase (AKR1C3 and AKR1A1) (13, 32), and may serve as therapeutic targets in ameliorating the cardiotoxicity of DOX.
CBR1 is an NADPH-dependent enzyme of the short-chain dehydrogenases/reductase (SDR) protein family and is widely distributed in human tissues. It can efficiently reduce quinones, prostaglandins, and other carbonyl-containing compounds, including xenobiotics. CBR1 also inactivates lipid aldehydes such as the highly reactive and genotoxic 4-oxonon-2-enal (ONE), 4-hydroxynon-2-enal, and acrolein to less reactive metabolites during oxidative stress (4, 36). Interestingly, CBR1-overexpressing transgenic mice have decreased survival and increased heart damage after DOX treatment (11), which is likely a result of increased DOXOL production. In contrast, conversion from DOX to DOXOL is decreased to an almost undetectable level in CBR1 ± mice, resulting in reduced cardiotoxicity (34). It has also been reported that the activity of CBR1 is relatively high in human breast and lung cancer patient tissues when compared to normal subjects (24).
In this study, pharmacological inhibition of CBR1 is shown to enhance the chemotherapeutic efficacy of DOX in breast cancers and simultaneously ameliorate cardiotoxicity. Therefore, we propose that CBR1 is a valuable target molecule for future drug development in patients with breast cancer.
Results
Combined treatment with the specific inhibitor of CBR1 (OH-PP-Me) and DOX enhances the chemotherapeutic effect of DOX in breast cancer cells
To determine whether CBR1 inhibition enhanced cell death during DOX treatment, we utilized MDA-MB-157, MDA-MB-436, MCF-7, and MCF10A cell lines, which are triple-negative/basal-B mammary carcinoma, ER-positive/PgR-positive luminal mammary carcinoma, and normal breast cell lines, respectively. MDA-MB-157, MDA-MB-436, and MCF-7 cells have higher CBR1 expression levels than MCF10A (Supplementary Fig. S1A; Supplementary Data are available online at

To confirm the synergistic effect of OH-PP-Me and DOX on the cell death, we performed in vitro clonogenic assay. The combination treatment markedly decreased colony formations in MDA-MB-157 and MCF-7 cells, compared to doxorubicin treatment alone (Fig. 1B and Supplementary Fig. S1C). The changes in apoptosis-related proteins were also examined. The expression levels of cleaved PARP and caspase-7 and the expression level of Bax were all significantly increased in combination treatment with DOX and OH-PP-Me in cancer cell lines (Fig. 1C and Supplementary Fig. S1D). Since the MCF-7 cell line has a deficiency in caspase-3 (49), activation of caspase-7 was observed in both cell lines.
To verify the effect of combination treatment on cell death, terminal transferase dUTP nick end-labeling (TUNEL) assays were performed. As expected, TUNEL-positive cells were highly increased in combination treatment with DOX and OH-PP-Me compared with DOX alone (Fig. 1D and Supplementary Fig. S1E). It is well known that DOX induces ROS generation (22, 50), and thus, whether combination treatment affected ROS generation was examined. Combined treatment of DOX with OH-PP-Me increased ROS generation compared with DOX treatment alone (Fig. 1E and Supplementary Fig. S2F, G). Collectively, these results demonstrate that CBR1 inhibition during DOX treatment enhances cell death in breast cancer cells.
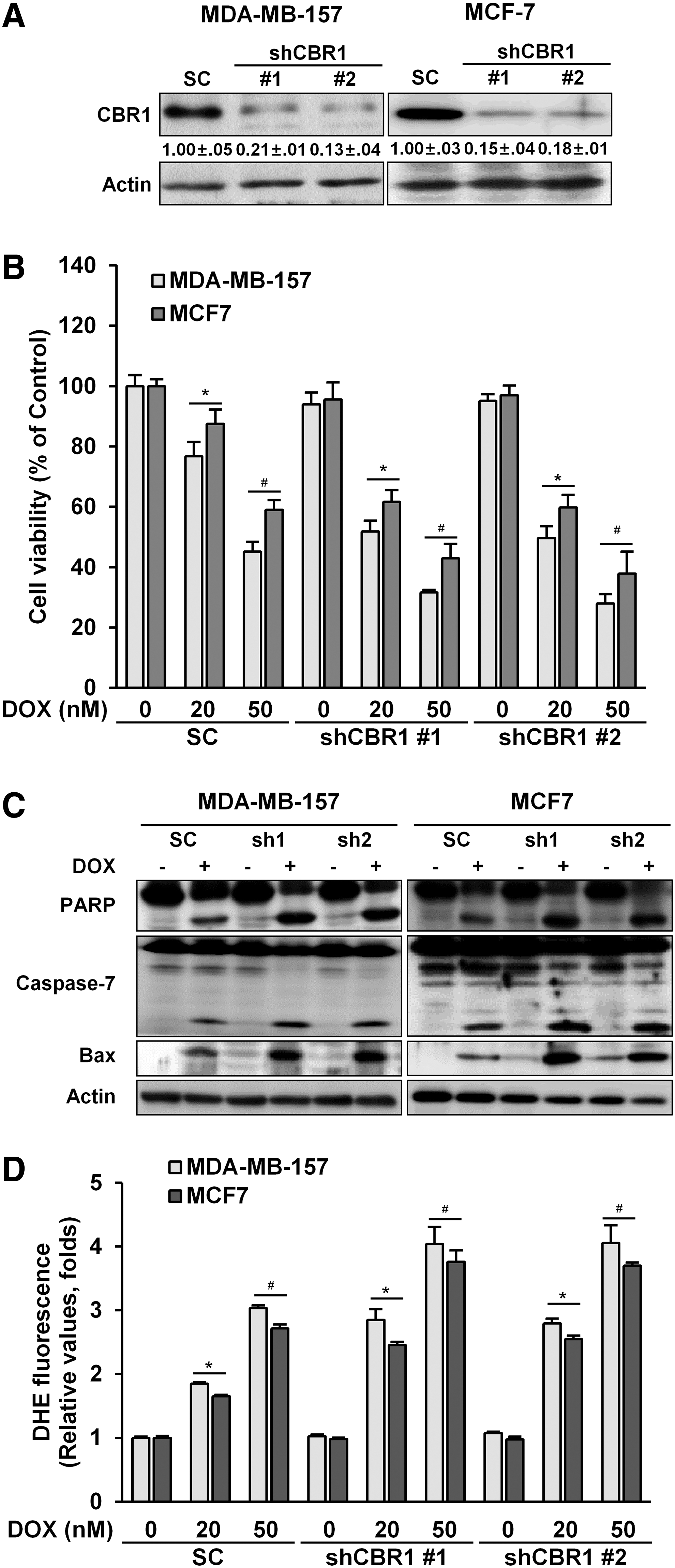
Knockdown of CBR1 increases cell death after DOX treatment in breast cancer cells
To further elucidate the role of CBR1, cells with a stable knockdown of CBR1 were constructed using two independent small hairpin RNA (shCBR1) clones in both MDA-MB-157 and MCF-7 cells. CBR1 expression was strongly and similarly suppressed in two clones of knocked down cancer cells, that is, CBR1 shRNA #1 and shRNA #2, in both cell lines (Fig. 2A). Knockdown of CBR1 increased cancer cell death by DOX treatment, as shown by cell viability (Fig. 2B). The expression levels of cleaved PARP and caspase-7 and the expression level of Bax were higher in shCBR1 transfectants than in cells transfected with the scrambled controls after DOX treatment (Fig. 2C). The increased cell death was associated with an increase in ROS (Fig. 2D and Supplementary Fig. S2A) in shCBR1 transfectants. Together, these data clearly show that knockdown of CBR1 enhances DOX-induced cell death in breast cancer cells.
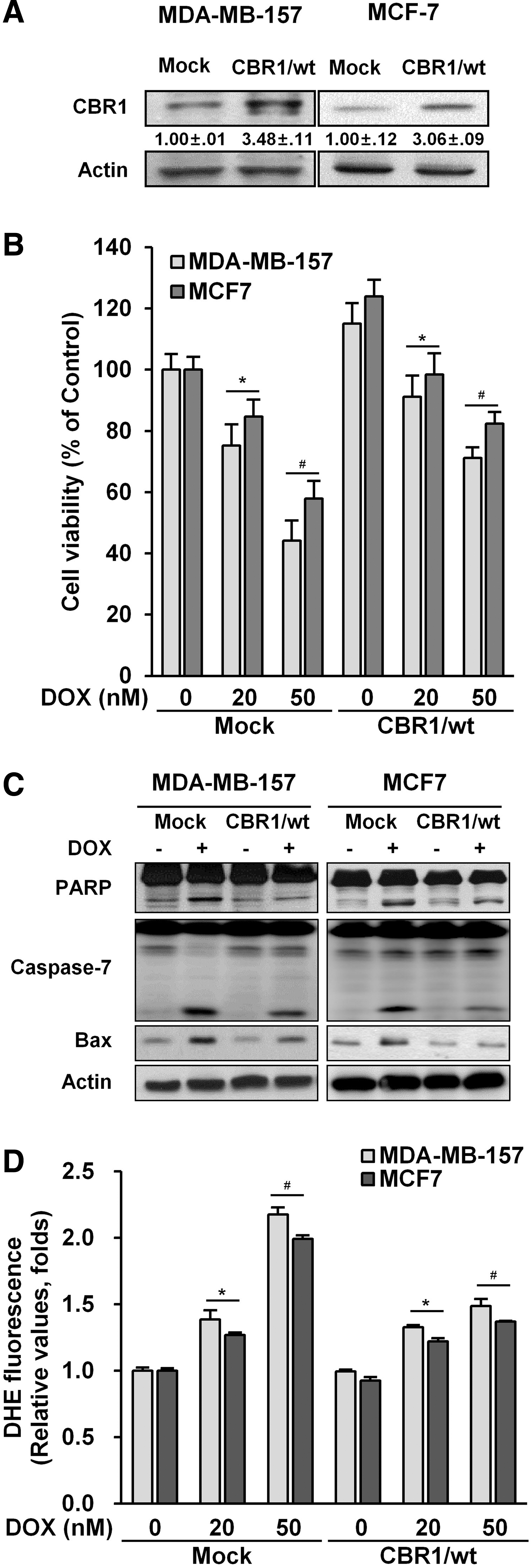
Overexpression of CBR1 protects breast cancer cells against apoptosis during DOX treatment
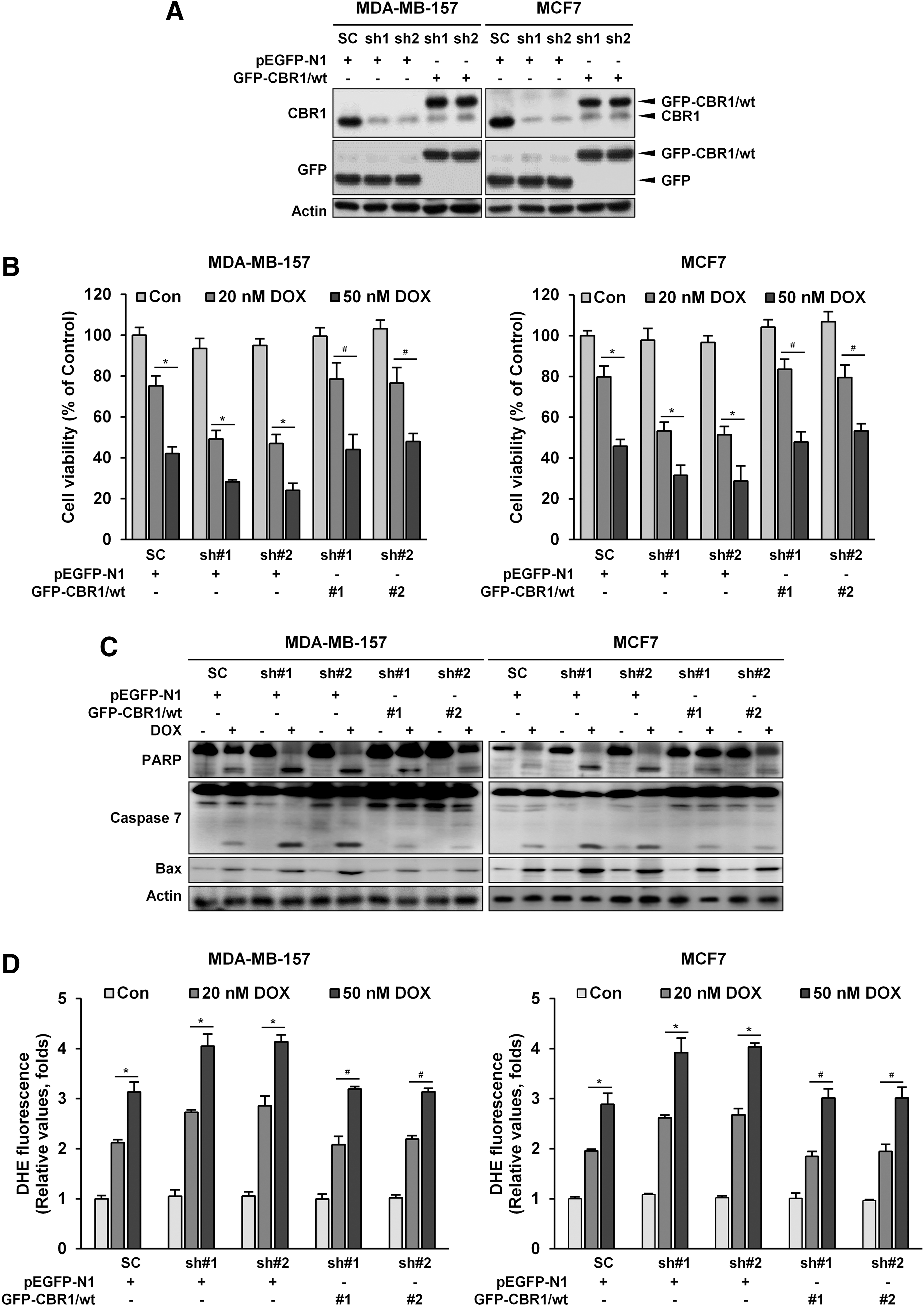
To further support our hypothesis, stable cell lines that overexpressed CBR1 using MDA-MB-157 and MCF-7 cells were made. CBR1 expression was found to be three-fold higher in stably transfected cells than mock transfectants (Fig. 3A). Overexpression of CBR1 decreased DOX-induced cell death (Fig. 3B) compared to mock controls of both cell lines. In the cells stably overexpressing CBR1, expression levels of cleaved PARP and caspase-7, as well as the expression level of Bax, were lower than in the mock controls after DOX treatment (Fig. 3C). In addition, CBR1-overexpressing transfectants exhibited significant reduction of ROS generation (Fig. 3D and Supplementary Fig. S2B). To further confirm the role of CBR1 in DOX-induced cell death, we constructed the two CBR1-GFP plasmids containing a Wobble mutant cDNA encoding mouse CBR1 with synonymous point mutations within the shRNA target sequences, and then re-expressed the functional CBR1 in MDA-MB-157 and MCF7 clones harboring the shCBR1 #1 and shCBR1 #2 (Fig. 4A). The re-expression of CBR1 rescued the cells from cell death (Fig. 4B), apoptosis (Fig. 4C), and ROS generation (Fig. 4D and Supplementary Fig. S2C). Overall, these data indicate that overexpression of CBR1 can induce resistance to apoptotic cell death by DOX in breast cancer cells.
CBR1 attenuates DOX-induced oxidative stress in breast cancer cells
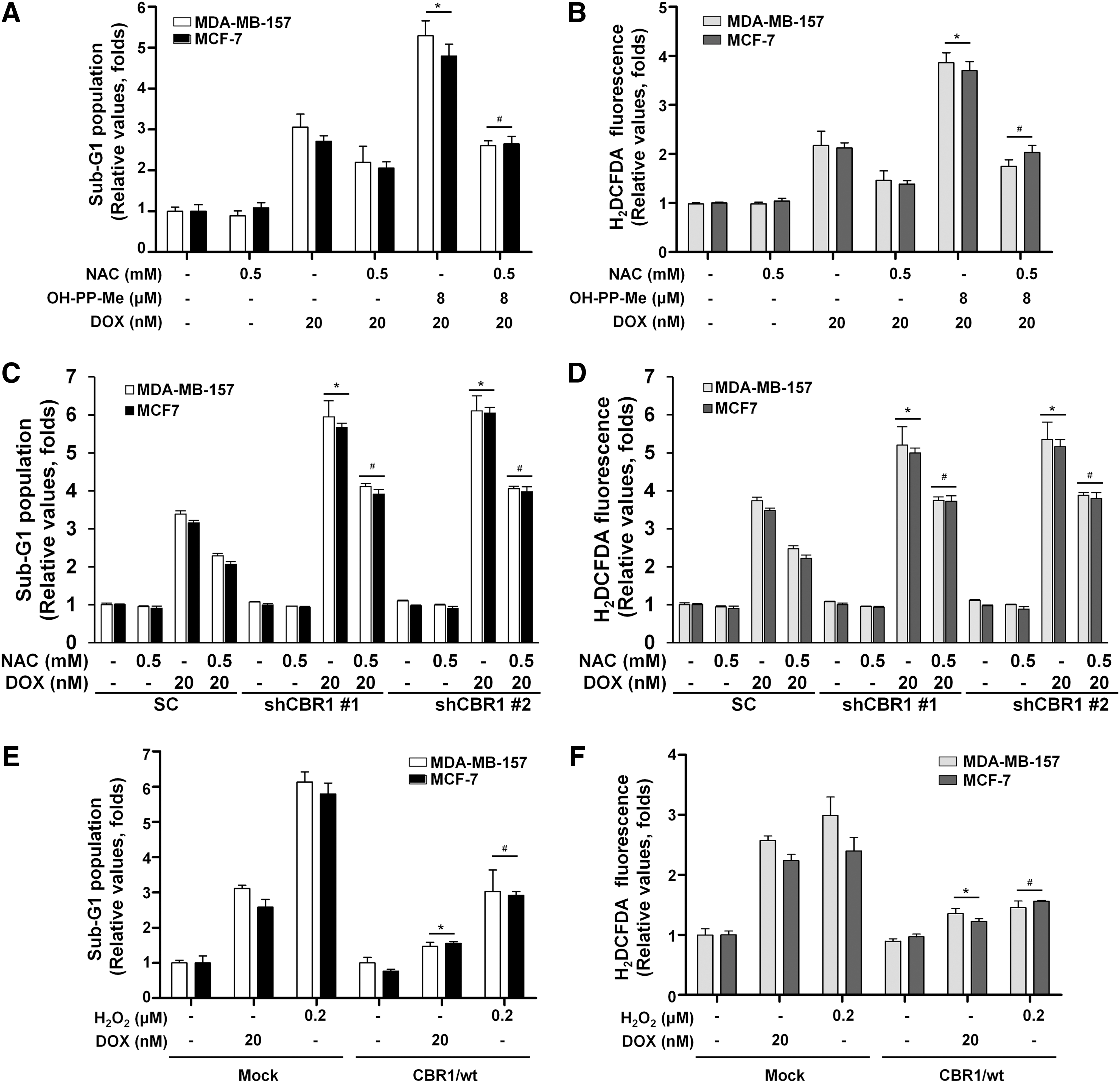
To further explore the effect of CBR1 on oxidative stress, cells were pretreated with the ROS scavenger N-acetyl-l-cysteine (NAC) for 2 h, and then cell cycle was analyzed for cell death and ROS levels were measured after treatment with DOX and OH-PP-Me. As expected, the combination treatment group with NAC had significantly decreased sub-G1 population and ROS generation compared with the combination treatment group without NAC (Fig. 5A, B). In addition, the results showed a marked reduction in sub-G1population and intracellular ROS levels with NAC and DOX treatment in both scrambled and shCBR1 transfectants (Fig. 5C, D). Consistent results were observed with hydrogen peroxide (H2O2) and DOX treatment (Fig. 5E, F). Taken together, these data suggest that CBR1 plays an important role in protecting cells from ROS generation and can cause resistance to DOX treatment in breast cancer cells.
CBR1 inhibitor enhances DOX sensitivity to tumor cells, but prevents cardiotoxicity, in MDA-MB-157 implanted tumor mice
To confirm the effect of combination treatment with OH-PP-Me and DOX on tumor growth in vivo, we first established the implanted tumor mouse using the MDA-MB-157 breast cancer cell line. Since tumor masses were observed 2 weeks after the inoculation of MDA-MB-157 cells, mice were injected intraperitoneally with OH-PP-Me (1.67 mg/kg × 6 times, i.p.), DOX (2.5 mg/kg × 6 times, i.p.), or in combination for 2 weeks. As shown in Figure 6A, the combination treatment of DOX and OH-PP-Me markedly suppressed tumor growth compared with treatment of DOX alone. In addition, hematoxylin and eosin (H&E) staining of dissected tumor sections revealed that tumor cell number was less in the combination-treated mice group than the DOX-treated group (Fig. 6B). The higher DNA fragmentation was also observed in the tumor specimens from mice cotreated with both compounds compared to those from mice with DOX alone (Fig. 6C).

To determine whether OH-PP-Me could reduce DOX-induced cardiotoxicity in these implanted mice, serum creatine phosphokinase (CPK) levels were measured and cardiac histological tests were performed after dissection. Treatment with DOX alone showed explicit signs of cardiac injuries, whereas the combination of DOX and OH-PP-Me distinctly prevented morphological alterations (Fig. 6D). Moreover, treatment with DOX alone resulted in approximately a three-fold increase in serum CPK activity compared with untreated mice. In contrast, the combination treatment of DOX and OH-PP-Me significantly decreased CPK level in the blood (Fig. 6E). In addition, DOX alone led to a further increase in DNA fragmentation in the cardiac tissues, but the combination of DOX and OH-PP-Me rather reduced DNA fragmentation (Fig. 6F).
Finally, we quantified the apoptosis data to compare cell damage between cardiac and tumor tissues. The quantified results showed that cardiac tissues were more vulnerable to doxorubicin than tumor tissues as reported previously (18), and inhibition of CBR1 by OH-PP-ME significantly reduced DNA fragmentation in cardiac tissues while it was enhanced in tumor tissues (Fig. 6G).
We also investigated whether OH-PP-Me could prevent side effects of DOX in heart, blood, and liver using normal mice. Normal mice were treated with the agents in the same manner as the implanted tumor mice, and then H&E staining and complete blood count were performed. In these experiments, DOX induced cardiotoxicity (Supplementary Fig. S3A) and reduced total WBC and platelet numbers (Table 1). To see whether DOX caused the functional and pathological damage in the liver, we performed the blood biochemistry test and H&E staining. The results showed that there was negligible liver damage by DOX as reported previously (20). Furthermore, OH-PP-Me alone or combined treatment did not show any functional and pathological damage in the liver (Table 2 and Supplementary Fig. S3B). Overall, these results indicate that inhibition of CBR1 increases the chemotherapeutic effects of DOX and reduces DOX-induced side effect, including heart damage.
Mean ± SE (range).
Bold, out of normal reference range.
DOX, doxorubicin; RBC, red blood cells; WBC, white blood cell; SE, standard error.
Mean ± SE (range).
GGT, gamma-glutamyl transpeptidase; TP, total proteins; ALB, albumin; ALP, alkaline phosphatase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; T-BIL, total bilirubin; D-BIL, direct bilirubin; T-CHOL, total cholesterol; HDL, high-density lipoproteins; LDL, low-density lipoproteins; LDH, lactate dehydrogenase.
OH-PP-Me attenuates DOX-induced cardiotoxicity in a rat model
To further elucidate the effect of OH-PP-Me on DOX-induced cardiotoxicity, a rat model was utilized. Rats are more optimal than mice as an established model for human cardiovascular diseases, as rats have a heart rate that is similar to humans, while mice have a significantly different heart mass, rate, and cell composition of cardiac muscle (1, 5). Cardiac function after DOX (2.5 mg/kg × 6 times, i.p.) treatment with or without the CBR1 inhibitor OH-PP-Me (1.67 mg/kg × 6 times, i.p.) was observed in rats.
Serum CPK levels were high after DOX treatment, indicating tissue damage, particularly in cardiac muscle. However, DOX combined with OH-PP-Me significantly decreased the levels of CPK compared with DOX alone (Fig. 7A). Next, H&E staining was used to examine the morphological changes associated with drug treatment. Treatment with DOX alone showed overt signs of cardiomyocyte morphological alterations, whereas the combination of DOX and OH-PP-Me markedly rescued morphological alterations (Fig. 7B). In addition, increased DNA fragmentation after exposure to DOX was rescued by OH-PP-Me treatment (Fig. 7C).

Finally, echocardiography results revealed that cardiac function parameters, for example, left ventricular end-diastolic dimension (LVEDD) and left ventricular end-systolic dimension (LVESD), were increased, but ejection fraction (EF) and fractional shortening (FS) were decreased, during DOX treatment (Fig. 7D). In contrast, the combination treatment showed almost no changes in cardiac function parameters. Collectively, these data demonstrate that cardiomyocyte apoptosis and cardiotoxicity after DOX treatment are decreased when combined with CBR1 inhibitor.
Discussion
Breast cancer is one of the most common cancers in women. It is also a major cause of cancer-related death (42), accounting for 15% of all cancer deaths globally in 2012. Breast cancer is a heterogeneous disease influenced by several environmental factors and is susceptible to the development of drug resistance, and thus, the most optimal treatment option still remains undefined (12).
Treatment strategies vary between individuals based on clinical factors such as stage, hormonal status, HER2 overexpression, and comorbidities. Chemotherapy is given as neoadjuvant, adjuvant, or as palliative treatment. The most active agents for breast cancer are anthracyclines and taxanes (27). These drugs are used as single agents or in combination with other agents in various settings. Generally, combination chemotherapy produces better outcomes, although increased toxicities present a major concern (9). Since the targeted agent era, trastuzumab, which targets the HER2 receptor, was introduced for the treatment of HER2-overexpressing breast cancer. Combination of trastuzumab with cytotoxic chemotherapy showed improved outcomes in a prior randomized phase III trial (44).
However, significant cardiotoxicity was a major adverse event. Concomitant use of trastuzumab and DOX is still not recommended because of cardiotoxicity. Furthermore, the combination of DOX with other agents, for example, paclitaxel, also increases cardiotoxicity, despite enhanced treatment efficacy (14, 16). Since cardiotoxicity with active agents, including DOX and trastuzumab, has significantly limited the use of combination therapy against breast cancer, new therapeutic modalities to overcome this toxic effect are urgently needed.
In this study, CBR1 is shown to be a novel molecular target to enhance chemotherapeutic efficacy of DOX in breast cancer cells. It was clearly demonstrated that inhibition of CBR1 by chemical inhibitor, OH-PP-Me, increased anticancer effects of DOX in breast cancer cells. It was also observed that overexpression of CBR1 decreased DOX-induced anticancer effects in breast cancer cells, whereas CBR1 knockdown increased these effects. Our previous studies demonstrated that CBR1 had protective effects against oxidative stress in diverse conditions (17, 45). A recent report has also revealed that CBR1 induces doxorubicin resistance via reducing oxidative stress in diverse gastrointestinal cancer cells (26). Consistent with this concept, the present study found that suppression of CBR1 increased superoxide (O2 •−) levels after DOX treatment. Conversely, overexpression of CBR1 decreased superoxide (O2 •−) levels in breast cancer cells. These data indicate that CBR1 plays a regulatory role in apoptosis and cell survival via oxidative stress.
Due to a lack of specificity, patients undergoing DOX treatment can develop several adverse effects, the most severe of which is cardiomyopathy, leading to heart failure (8). Therefore, DOX-induced cardiomyopathy limits its therapeutic application as an anticancer drug and, thus, continues to present a clinical dilemma in oncology and cardiology practices (41, 43). Recently, DOX was modified to evade these adverse effects. For example, pegylated liposomal DOX (PL-DOX) has shown a better pharmacokinetic profile and less cardiotoxicity than DOX itself (2, 28). However, PL-DOX causes severe skin toxicity due to the unique pharmacokinetics of circulating liposomes (33). Thus, there is still a strong need to improve the anticancer effects and to reduce other adverse effects of PL-DOX.
DOX-induced cardiotoxicity appears to be a multifactorial process, and numerous mechanisms have been proposed (6, 15, 31, 39). DOX generates the DOX-semiquinone that induces DNA damage and lipid peroxidation via ROS formation (30). On the contrary, it has been reported that carbonyl reduction of DOX to the secondary alcohol metabolite, DOXOL, contributes to severe cardiotoxicity (40). Since the metabolism of DOX involves CBR1, the present study hypothesized that a CBR1 inhibitor, for example, OH-PP-Me, might prevent DOX-induced cardiotoxicity. In fact, our results showed this to be the case, that is, inhibition of CBR1 augments chemotherapy efficacy and attenuates cardiotoxicity by blocking DOX reduction to DOXOL.
In summary, the current study demonstrated that inhibition of CBR1 enhances chemotherapeutic efficacy of DOX and attenuates cardiotoxicity by blocking DOX reduction to DOXOL. Therefore, we propose that development of the CBR1 inhibitors that can be administered into cancer patients will pave the new way for better application of DOX in oncology clinics.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), Roswell Park Memorial Institute medium 1640 (RPMI1640), penicillin/streptomycin, and fetal bovine serum (FBS) were purchased from Corning Cellgro (Manassas, VA). The antibody against CBR1 was purchased from Abcam (Cambridge, MA). Antibodies against cleaved caspase-3, caspase-7, and Bax were acquired from Cell Signaling Technology, Inc. (Danvers, MA). PARP and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Doxorubicin, hydrogen peroxide (H2O2), dihydroethidium (DHE), propidium iodide, RNase-A, and Mayer's hematoxylin were acquired from Sigma-Aldrich (St. Louis, MO). Doxorubicin for animal experiments was purchased from Dong-A Pharmaceutical Co. (Seoul, Korea). 2′-7′-5-(and-6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) and trypan blue were acquired from Life Technologies Co. (Carlsbad, CA). The Cell Counting Kit-8 (CCK-8) was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY).
Cell culture
Human breast cancer cells (MDA-MB 157, MDA-MB-436, MCF-7, and MCF10A) were obtained from the American Type Culture Collection (ATCC; Rockville, MD). MDA-MB 157 cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and 100 μg/ml penicillin/streptomycin. MDA-MB436 and MCF-7 cells were maintained in RPMI1640 supplemented with 10% heat-inactivated FBS and 100 μg/ml penicillin/streptomycin. MCF10A cells were maintained in DMEM/F12 with 5% horse serum, 2 mM
Assessment of cell proliferation
To assess the effects of combined treatment on cell proliferation, CCK-8 assays were carried out in 24-well plates, following the manufacturer's instructions. The absorbance was measured at 450 nm using a microplate reader (Bio-Rad Laboratories, Inc., Tokyo, Japan).
Cell viability
Cell viability was detected using Vi-CELL XR (Beckman Coulter, Inc., Brea, CA) after trypan blue staining. Cell survival was expressed as the relative percentage of viable cell numbers.
Cell cycle analysis
Cells were grown to 80% confluence, harvested, rinsed with PBS, and then fixed in 75% ethanol for 1 h at 4°C. The fixed cells were centrifuged and suspended in 0.5 ml PBS containing 0.05 mg/ml propidium iodide and 0.2 mg/ml RNase-A, and then incubated at 37°C for 15 min. Red fluorescence (580–630 nm) of propidium iodide was measured in the FL-2 channel, and 30,000 events were collected for each sample. The data were analyzed with CellQuest software (Beckman Coulter, Inc.).
Clonogenic colony formation assay
Cells were plated at a concentration of 500 cells/well in a 6-well plate and allowed to form colonies for 14 days. The colonies were fixed and stained with crystal violet dye (0.5% in methanol) at room temperature after they were washed in PBS. Cells were washed with water, and plates were photographed with an image scanner. Crystal violet was resolved from colonies by methanol and measured at 540 nm. Based on the absorbance at 540 nm, survival rates were expressed as a percentage relative to DMSO-treated control from three independent experiments.
TUNEL assay for apoptosis
Cells
Breast cancer cells were fixed for 30 min in 4% paraformaldehyde. The fragmented DNA in the cells undergoing apoptosis was detected using the DeadEnd™ Colorimetric TUNEL System (Promega Corp., Madison, WI). The TUNEL-positive nuclei (dark brown) in breast cancer cells were observed using a normal white light microscope (Olympus, Tokyo, Japan).
Tissues
The tumor and heart tissue sections from animal models were used for the TUNEL assay and the FragEL™ DNA Fragmentation Detection Kit (Merck KGaA, Darmstadt, Germany). Staining for the TUNEL assay was performed following the manufacturer's protocol. Incorporated fluorophores were examined with a confocal microscope (Carl Zeiss Microscopy GmbH, Göttingen, Germany) using appropriate excitation wavelengths and filter sets.
Quantification was performed by counting the number of TUNEL-positive cells in at least five random fields. The TUNEL-positive cells are expressed as a percentage of the total number of cells per field.
ROS analysis
Intracellular superoxide (O2•−) levels were measured with DHE using a flow cytometer (Beckman Coulter, Inc.). Cells were incubated with 25 μM DHE at 37°C for 20 min. The mean DHE fluorescence intensity was measured with excitation 488 nm and emission 525 nm.
The H2O2 levels were measured using CM-H2DCFDA. Cells were incubated with 5 μM CM-H2DCFDA at 37°C for 30 min. The mean CM-H2DCFDA fluorescence intensity was measured with excitation 488 nm and emission 525 nm.
Establishment of stable cell lines
MDA-MB-157 and MCF7 cells were transfected with pcDNA3 (Mock) or pcDNA3-CBR1 wild-type (CBR1/WT) plasmid using TransIT-BrCa transfection reagent (Mirus Corp., Madison, WI), according to the manufacturer's instructions. Cells transfected with pcDNA3 plasmid alone without the CBR1 gene (Mock) were used as the control.
Two independent small hairpin RNAs (shRNAs) specific to CBR1 (pLKO-shCBR1) were purchased from Sigma Aldrich; #1 shCBR1, CCGGCAAGCTGAAGTGACGATGAAACT CGAGTTTCATCGTCACTTCAGCTTGTTTTTG; #2 shCBR1, CCGGCCATGGACAATT TGTTTCAGACTCGAGTCTGAAACAA ATTGTCCATGGTTTTTG. Cells transfected with the pLKO plasmid without the CBR1 shRNA (scrambled [SC]) were used as the control. For stable transfection, cells were cultured in selective medium with 600 μg/ml G418 or 20 μg/ml puromycin for 2 weeks. Then, drug-resistant individual clones were isolated and incubated for further amplification in the presence of selective medium. The CBR1 expression levels in these stable cells were confirmed by immunoblotting. To re-express a functional CBR1, two pEGFP-N1-CBR1/wt plasmids were constructed containing a “wobble” mutant cDNA encoding human CBR1 with synonymous point mutations within the two shRNA target sequences (5′- CC
Implantation tumor nude mice
Female athymic nude mice (Foxn1 nu/nu) aged 4 weeks were purchased from Harlan (Udine, Italy) and were allowed to acclimate for 1 week. The animal protocol was approved by the Institutional Animal Care and Use Committee of Kyung Hee University (Seoul, Korea). To establish implantation tumor nude mice of breast cancer, 107 MDA-MB-157 human breast cancer cells in 0.2 ml PBS were injected into the mammary fat pad. Two groups of the experimental mice were established; one were mice that developed tumors and the other were normal healthy mice. The two groups were further divided into different treatment groups as follows: Group 1: Untreated control; Group 2: OH-PP-Me 1.67 mg/kg, 6 times (i.p.); Group 3: DOX 2.5 mg/kg, 6 times (i.p.); Group 4: OH-PP-Me 1.67 mg/kg, and after 1 h, DOX 2.5 mg/kg, 6 times (i.p.).
After 2 weeks of treatment, tumor and heart tissues were collected for histological and immunohistochemical analysis, while blood samples were collected for the CPK activity assay.
Determination of cardiac function in rat
The rats were divided into three treatment groups (DOX alone (2.5 mg/kg × 6 times, i.p.), 3-(7-isopropyl-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidin-5yl)phenol (OH-PP-Me) alone (1.67 mg/kg × 6 times, i.p.), and a combination of OH-PP-Me and DOX), and one group as an untreated control. For the combination treatment group, rats were injected with OH-PP-Me 1 h after the injection of DOX. After 4 weeks, the rats underwent echocardiography, and then blood and heart tissue samples were collected for further analysis. Animal experiments were conducted according to the protocol approved by the Institutional Animal Care and Use Committee of Kyung Hee University (Seoul, Korea). Male Sprague Dawley rats (8–9 weeks old) were purchased from Orient Bio, Inc. (Sungnam, Korea).
Measurement of CPK activity
CPK activities were measured after treatment by using a CPK test kit (Abnova Corp., Taipei, Taiwan). One unit of CPK was defined as the reduction of 1 μM NAD+ to NADH per minute.
Histological analysis of animal models
The tumors and hearts were isolated from the animals of each group. Paraffin-embedded, 5-μm-thick heart tissue sections were used. Routine H&E-stained sections were examined to ensure the structural integrity of the tissues using a normal white light microscope.
Echocardiography
Following anesthetization, rats were shaved and placed in the supine position. Transthoracic echocardiography was performed to obtain two-dimensional M-mode images, using a 13-MHz linear probe (Vivid FiVe; GE Medical Systems, Milwaukee, WI). From M-the mode images, LVEDD and LVESD were determined according to the standard method of the American Society of Echocardiography. All measurements were averaged from values of three cardiac cycles. LV FS was calculated as (LVEDD—LVESD)/LVEDD × 100. EF was calculated using the Teichholz formula.
Western blot analyses
Western blot analyses were performed using whole-cell extracts as previously described (48). Protein concentrations of lysates were measured using a Bio-Rad DC protein assay (Bio-Rad Laboratories, Inc.). For immunoblotting, the proteins were separated on 8–15% SDS-PAGE and transferred onto nitrocellulose membranes (Pall Corporation, Washington, NY). After blocking, primary antibodies were diluted according to the manufacturer's instructions. The blotted proteins were detected with an enhanced chemiluminescence detection system (Santa Cruz Biotechnology, Dallas, TX). Actin was used as a loading control.
Statistical analysis
The results are expressed as the mean ± standard error of the mean of at least three independent experiments. The differences between two means were analyzed for significance by the Student's t-test (SPSS for Windows; version 22.0, SPSS, Inc., Chicago, IL). p < 0.05 were considered significant.
Footnotes
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (No. 2011-0030072 and NRF-2013R1A1A2061214).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.