
Abstract
Introduction
W
Signaling and signal cascades are essential for inter- and intracellular communication, which regulates vital biochemical pathways. Each signal transduction begins with the binding of a messenger molecule to its target receptor, leading to receptor activation, and further triggering downstream effector molecules that regulate the desired cellular response. Small molecules such as cAMP, cGMP, diglycerides, and inositol triphosphate have been reported for their roles in signal transduction (53, 118, 136). More recently, the role of ROS/RNS in signal transduction and regulating cellular biochemistry has been an emerging area of investigation (27, 44, 49, 131).
A widely accepted mechanism of redox signaling involves the reversible oxidation of redox-sensitive thiol residues (in low-molecular-weight thiols or thioproteins), thereby modulating their function and activity. While the majority of cellular thiols exist in the protonated form (-SH) at physiological pH, a small fraction will remain in the form of thiolate anions (-S−), which can also be favored in a particular local protein microenvironment, increasing the susceptibility to oxidation when compared with the conjugate acid form (-SH) (27, 148). The oxidation of the thiolate anion by hydrogen peroxide (H2O2), for example, results in the generation of sulfenic acid (-SOH), causing allosteric modification of protein structure and function. This sulfenic acid form enters the signal transduction pathway, in which it is reversibly reduced to the thiolate anion by glutaredoxin and thioredoxin, restoring the original structure and function of the protein (28).
While transient increases in the generation of ROS/RNS bring about a reversible oxidation of thiolate anion to sulfenic acid, chronic elevations can result in further irreversible oxidations, generating sulfinic (-SO2H) and sulfonic (-SO3H) residues, rendering the protein structure and function irreparable, and thus resulting in oxidative stress (28).
This reversible signaling process has been observed in many different signal transduction cascades such as metabolism, cell growth, gene expression, differentiation, immunity, senescence, and apoptosis (29, 36, 117, 124, 125, 148). Current understanding of redox signaling in growth, stem cell differentiation, and immune response is briefly summarized below.
ROS/RNS are known to play a role in growth factor signaling pathways, which regulate nutrient uptake signals for cellular growth and mitotic proliferation (108). These processes involve phosphorylation of key tyrosine residues of the epidermal growth factor receptor (EGFR), initiating a chain of several signal transduction pathways such as mitogen-activated protein kinases (MAPKs)–extracellular signal-regulated kinases and phosphatidylinositol 3-kinase, signaling cell survival, growth, and proliferation (116).
Phosphorylation of EGFR is regulated by protein tyrosine phosphatase 1B (PTP1B), mandating therefore that PTP1B be inactivated to enable cell proliferation. The signaling role of H2O2 in this process is through inactivation of PTP1B by reversible oxidation of the thiolate anion of the catalytic cysteine residue to sulfenic acid (8). Furthermore, while transient PTP1B inactivation signals cell proliferation, chronic elevations in ROS/RNS levels can result in permanent deactivation of PTP1B, resulting in uncontrolled cell multiplication and tumorigenesis, thus demonstrating the fine balance between the physiological and pathological effects of ROS/RNS (157).
The role of ROS/RNS in mediating stem cell function has also recently gained a lot of attention. It was initially reported that elevated levels of ROS/RNS activate p38 MAPK and p16INK4a, a cell cycle inhibitor, resulting in cell cycle arrest and apoptosis and thus limiting stem cell renewal (46). Recently, however, it has been understood that ROS/RNS oxidize a key cysteine residue of ataxia telangiectasia-mutated kinase, which is involved in the DNA damage checkpoint and initiates a cascade of antioxidant defenses through the protein BH3 interacting domain (BID), a proapoptotic member of the Bcl-2 family (20, 93). Studies also indicate that stem cell differentiation is an ROS/RNS-dependant process: when stem cells, whether neural (70), hematopoietic (50), mesenchymal (132), or epidermal (40), exhibit reduced ROS/RNS levels, they undergo impaired differentiation. However, it still remains unclear as to what levels of each ROS/RNS impairs stem cell differentiation and self-renewal.
In the past two decades, substantial evidence has emerged highlighting the role of ROS/RNS in innate and adaptive immunity. Proteins such as the Toll-like receptors, which are a component of innate immune system, have been shown to activate inflammatory cytokines by upregulating the production of ROS/RNS when they encounter a pathogen (144). ROS/RNS are also known to initiate a signaling cascade vital for the formation of phagolysosomes and microbicidal activity (133). Similarly, ROS/RNS are involved in stimulation of T- and B-cell receptors, instigating a signaling pathway that results in the proliferation of both T and B cells (52, 145). Again, while elevated ROS/RNS levels can ensure proper immune response, the extent of this elevation and the differential responses of the types of immune cells to ROS/RNS remain unknown.
While some studies identify the role of a particular ROS/RNS (such as H2O2) in a specific signaling pathway, it is highly probable that subsequent publications will suggest the involvement of a different ROS/RNS. Such reports exist for several signaling cascades thus far, including those involved in cell proliferation (48, 104). Furthermore, the role of H2O2 in cellular biochemistry has been far more widely investigated compared with other ROS/RNS; this does not necessarily signify that H2O2 is a more significant signaling molecule, but instead is largely a consequence of the myriad of H2O2 selective fluorescent probes available, as discussed in the following sections, as well as the higher relative abundance and stability of H2O2 compared with other ROS/RNS (39).
The primary ROS/RNS that participate in signaling identified so far include superoxide, hydrogen peroxide, nitric oxide (NO), and peroxynitrite (ONOO−); nevertheless, the potential of other ROS/RNS such as hydroxyl radical, singlet oxygen, and nitrogen dioxide to act as signaling effectors cannot be ruled out.
The Challenge of Imaging Selectivity and Reversibility
To date, as exemplified above, it has been understood that ROS/RNS can act as effective mediators of cellular signaling pathways, most of which are reversibly regulated. However, the kinetic profiles of these signaling processes and the physiological levels that trigger the reversal of the signaling cascades are not well understood. While the roles of ROS/RNS signaling in growth factor signaling, stem cell differentiation, and immunity have been best elucidated, it is likely that ROS/RNS induce the reversible oxidation of other signal transduction pathways. Currently, our understanding of in vivo redox homeostasis, signaling, and pathological changes has been constrained by the lack of appropriate tools and methodology, as well as compromised cross talk between chemists who actually develop fluorescent tools and biologists who utilize them.
From the above discussion, it is clear that there is a lack of understanding about the specificity of signaling pathways to individual ROS/RNS. While some studies have reported a single ROS/RNS for a certain signal transduction pathway, in many cases, contradictory studies highlight the role of multiple ROS/RNS for the same pathway (48, 104). Clear answers to such questions will require selective tools that can unambiguously report on individual ROS/RNS.
Second, current understanding of redox signaling proposes that the concentration and time of exposure to ROS/RNS are critical in determining fate of the pathway: sufficiently low concentrations will cause reversible oxidation and hence effective signaling, while if concentrations are too high or exposure times too long, irreversible oxidation will occur, leading to constitutively active signal cascades, causing toxicity and oxidative stress. To better understand redox signaling, it is essential to distinguish these two events, necessitating tools that detect the dynamics of redox state.
It is apparent then that there are two distinct parameters to be achieved in tools for redox signaling to achieve imaging of the redox dynamics (Fig. 1). Studies must not only be sensitive to a specific ROS/RNS but also be able to identify the reversibility (or otherwise) of the signaling process. Ideally, these two needs may be met in a single probe, but, practically, work to date has focused on achieving only one of these two goals. While there are a large number of selective fluorescent probes, the development of reversible probes has been rather slow, largely because the power of reversible probes was not realized until a few years ago. In this review, we will consider critical design features that allow us to achieve these two aims, highlighting notable examples of probes of each type. We then consider the small subset of probes that exhibit both selectivity and reversibility, before considering possible avenues for future research.
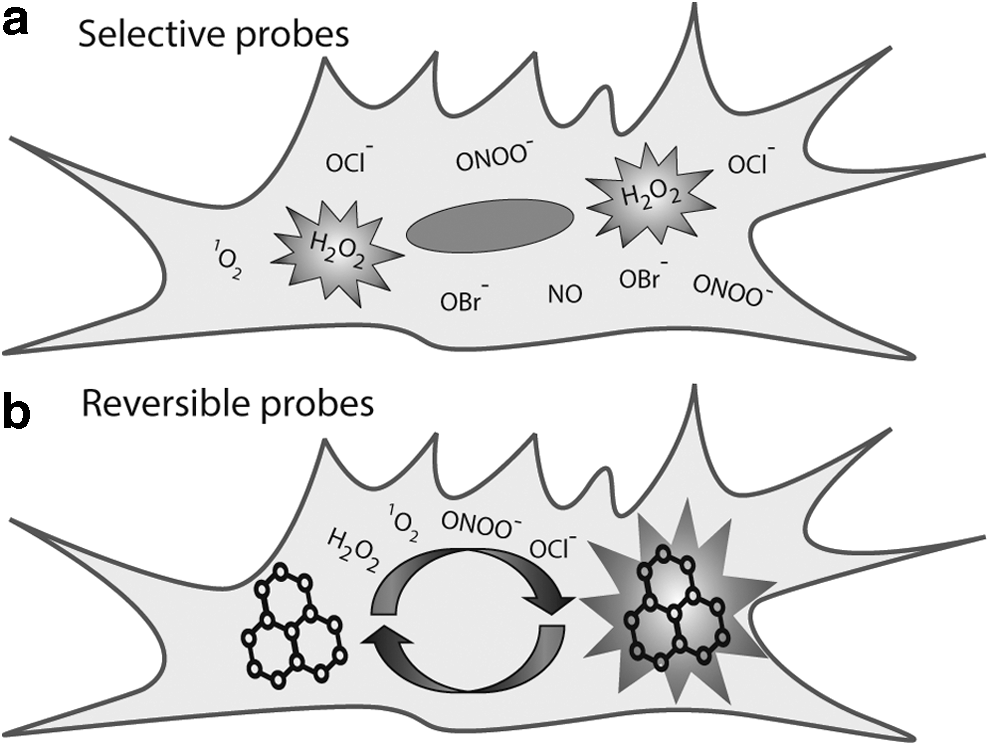
Selective Probes for Redox Signaling
The specificity of biological targets of ROS/RNS is primarily dictated by structural features: access to an enzyme active site, for example, depends heavily on the shape and charge distribution of the channel. This strategy can be readily employed in the design of fluorescent protein-based ROS/RNS probes, but is not feasible for small-molecule sensors, where size limitations preclude such designs. Instead, alternative strategies must be explored to develop small molecules that exhibit selectivity for an individual ROS/RNS.
Most selective probes rely on the selective reaction of a chemical functionality with a specific ROS/RNS. Selective probes usually contain a profluorescent molecule bearing a chemical scaffold, which reacts chemoselectively with the ROS/RNS of interest, resulting in molecular transformations that generate the fluorescent molecule, hence allowing the visualization of the reactivity of specific ROS/RNS. Since the sensing mechanism of such probes is based on a selective chemical reaction, these probes are also referred to as reaction-based probes. Selective probes can provide vital information about the identity and localization of the ROS/RNS involved in a particular signaling cascade. An important consideration in preparing reaction-based probes is that the kinetics of reaction must be fast enough to ensure that sufficient reaction occurs within the lifetime of the ROS/RNS (37).
The particular challenge in the design of selective sensors for ROS/RNS is therefore the identification of reactions that are specifically induced by a single species. This is no straightforward task as ROS and RNS bear many structural and electronic similarities. There has been a vast array of selective small-molecule probes for ROS/RNS reported in the literature, which have been summarized on a number of occasions (15, 16, 19, 22, 37, 51, 94, 140, 147, 149).
One key drawback in this field is that despite the prolific preparation of new probes, there is a lack of comprehensive structure–activity studies to assess the true selectivity or generalizability of certain design strategies. This is a significant impediment to the rational design of highly selective tools. In the following discussion, we focus on the key synthetic strategies for achieving selectivity and response and will highlight only key examples of each strategy. Strategies have been organized by the nature of the chemical transformation induced by the ROS/RNS on the probe molecule, and we critically analyze each strategy to provide insight into the origins of selectivity for individual ROS/RNS and to inspire further research in this direction.
Radical Reactions
Many of the roles of ROS/RNS in both health and disease arise from their free radical nature. One of the primary ways in which ROS/RNS can transform their reaction partners is through radical-mediated reactions. The radical oxidation of probes by ROS/RNS, which primarily leads to radical-mediated restoration of a π-conjugated fluorescent system, is therefore a key mechanism for selective sensing.
Probably the most widely explored strategy for the development of redox fluorescent probes is a reconstitution of a π-conjugated chromophore upon a radical oxidation. This has been the case for 2′,7′-dichlorodihydrofluorescein (DCFH) (59), which is one of the most extensively utilized fluorescent probes to measure cellular oxidative capacity (Fig. 2a). This initially nonfluorescent molecule undergoes a two-electron oxidation to the fluorescent 2′,7′-dichlorofluorescein (142). Although originally reported as an H2O2 probe, it can also be oxidized by other ROS/RNS as well as by enzymatic and metal-catalyzed reactions. A key drawback of DCFH is that radical oxidation leads to formation of a free radical intermediate, which can undergo various side reactions, leading to nonfluorescent by-products, and can generate further ROS/RNS (67, 112, 140, 150). While other shortcomings, such as reduced cellular retention of the oxidized form and photosensitivity, stem more from the nature of fluorophore, the former are intrinsic to the mechanism of interaction between the ROS/RNS and the probe. Therefore, other probes operating by a similar mechanism of action can potentially suffer from same limitations. For example, dihydrorhodamine operates by a similar mechanism to DCFH and, despite being commonly applied to detect ONOO−, is known to also respond to hypochlorous acid (HOCl) as well as other reactive species (51, 141).
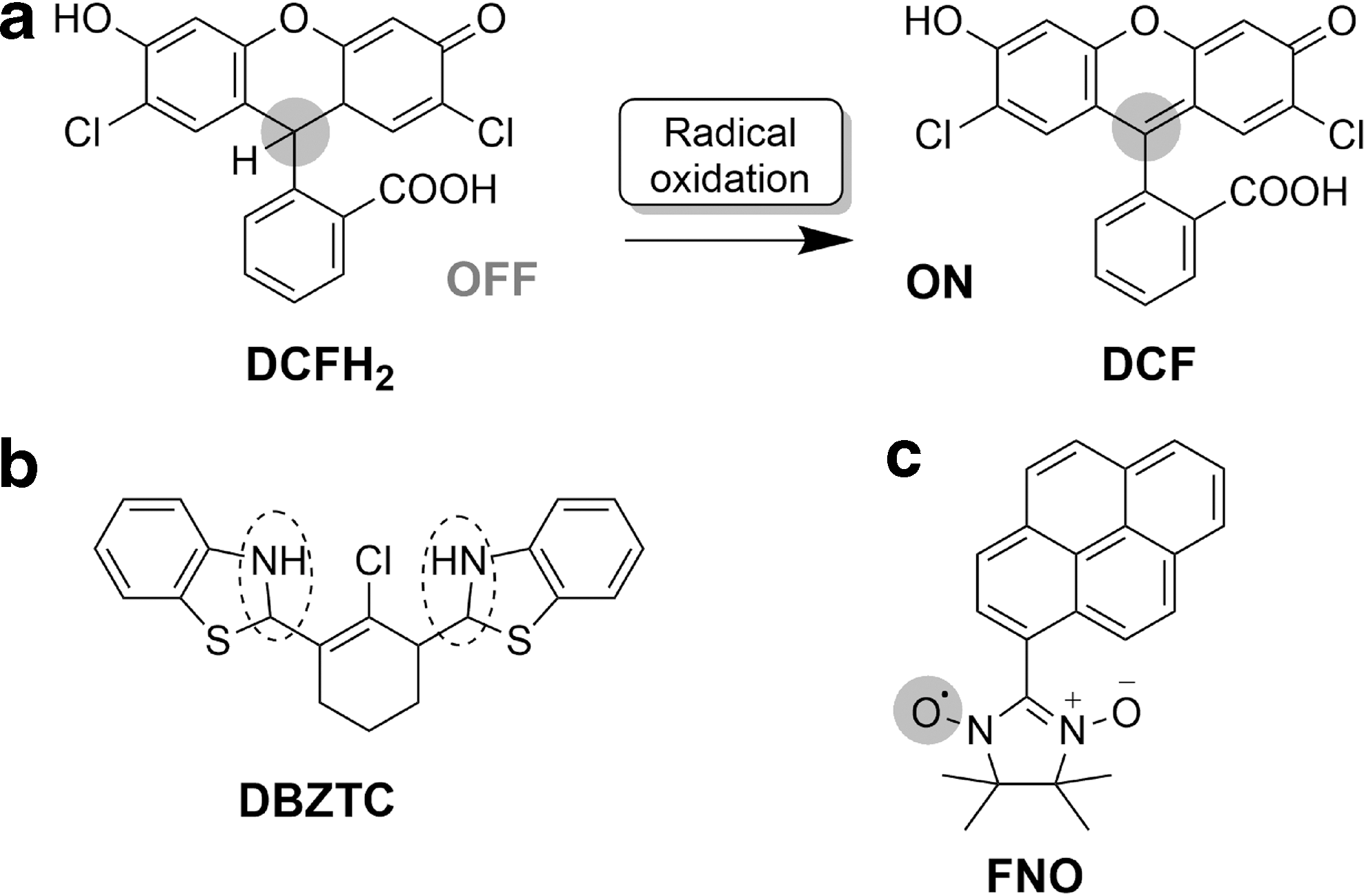
As this mechanism involves radical oxidation, it is perhaps best suited to the development of selective probes for radical ROS/RNS such as superoxide. The superoxide radical, a precursor for the generation of other ROS/RNS, is a product of one-electron reduction of molecular oxygen. While not much has been reported about its role in signaling pathways, high concentrations of superoxide are known to damage cellular membranes and tissues.
The detection of intracellular levels of O2 •− has mainly involved the use of dihydroethidine (DHE) and its mitochondrially targeted analog, MitoSOX, capable of detecting O2 •− quantities as low as 1.5 pmol (102, 111, 170). Initially, it was believed that the reaction between dihydroethidine and O2 •− results in the conversion of the aniline moiety to the iminium cation, leading to the restoration of a conjugated π-system in ethidium, which shows enhanced fluorescence properties upon binding to DNA. However, more recently reported studies demonstrate that the product is in fact 2-hydroxyethidium and not ethidium (170).
The use of dihydroethidine as an ROS marker has been extensively assessed and reviewed, with the key finding that it can also be oxidized by other ROS/RNS, including •OH and ONOO− (171). The products of oxidation with different ROS/RNS cannot be readily distinguished by fluorescent detection, and instead require high-performance liquid chromatography analysis, precluding utility for in cellulo studies (34, 171). Despite the challenges of selectivity, DHE can be considered a suitable probe to detect general changes in oxidants as it does not self-oxidize or generate ROS/RNS (51). However, oxidation of DHE by cytochrome C (11), H2O2 via peroxidases (105), and its own catalysis of the dismutation of superoxide (11) present further limitations.
Hydrocyanine-based probes such as HydroCy7, Hydro-IR783, and Hydro-ICG exploit a similar mechanism of restoration of an extended π-conjugated system (69). While these probes have near-infrared (NIR) fluorescence properties for applications in animal imaging, they respond to O2
•− as well as other radical oxidants (•OH and •OtBu). Improved selectivity toward O2
•− over •OH, H2O2, and other biologically relevant ROS/RNS has been achieved in 2-(2-pyridyl)benzothiazoline
DCFH and DHE are among the most studied and scrutinized of all ROS/RNS probes. The in-depth understanding of these radical oxidation probes enables identification of the key design features that must be achieved for future offerings and also provides a comprehensive array of properties that should be assessed of each new probe. The advantage of this class of probes lies in the potential for extremely high sensitivity: even considering the very low in cellulo concentrations of the ROS/RNS to be detected, the nanomolar detection limits of the latest probes should ensure sufficient sensitivity. Furthermore, sites for radical oxidation can be built into a wide range of fluorescent scaffolds, ensuring versatility of photophysical and biological behavior.
The main challenge in designing probes based on radical oxidation is the achievement of selectivity, with cross-reactivity observed in particular with O2 •− over •OH, H2O2, and ONOO−. The Tang group has reported recent progress toward achievement of selectivity (130), but more extensive structure–activity studies might shed valuable light on key design features that enable specific reactions.
Key among the potential drawbacks of probes of this type are the possibility of side reactions of the free radical intermediate, oxidation of the probe by other species (whether enzymes or metals), and the further generation of free radicals following reaction. It is essential that each of these scenarios be investigated in assessing potential probes.
One radical-based strategy for the development of reversible probes, which will be discussed in a later section of this review, is the use of the nitroxyl radical. One factor that affects the true reversibility of such probes is the possibility for the recombination of two radicals, thus fatiguing the system. However, this property has also been utilized in the design of irreversible reaction-based probes. By this strategy, the Likhtenshten group reported the development of nitronyl nitroxide-based probes such as the pyrene nitronyl nitroxide (FNO; Fig. 2c), which is nonfluorescent due to the intramolecular quenching effect of the nitroxide (95). Reduction of FNO by O2 •− generates a fluorescent molecule. While this probe has been shown to detect submicromolar levels of O2 •−, it suffers from poor selectivity as it responds to the presence of •OH as well as some cellular antioxidants.
The Abramson group developed 4-chloro-7-nitrobenzofurazan (NBD-Cl) as a superoxide sensor by this same mechanism, but solubility issues and its reactivity with thiols and amines limit its biological applications (103). For probes of this type, it is likely that the hydroxyl radical, and cellular antioxidants, would be the main interferents.
Oxidative Nucleophilic Reactions of ROS/RNS
In addition to being oxidizing species, many ROS/RNS also have nucleophilic character, a property that can be exploited in the design of selective ROS/RNS probes. For example, hydrogen peroxide has a pKa of 11.7 and will therefore be largely deprotonated at biological pH. The deprotonated hydroperoxide form has a lower basicity than hydroxide, but is ∼200 times more nucleophilic in aqueous solutions due to the α-effect of two adjacent electronegative atoms, which lowers the solvation energy of hydroperoxides in comparison with the more charge-dense hydroxide (6). The challenge in designing probes based on nucleophilic reactions is to achieve selectivity through judicious design of the electrophile on the probe (89).
The most common realization of this strategy involves masking of a phenolic group on the fluorophore with a nucleophile-responsive hydrolysable moiety. The nucleophilic reaction will lead to hydrolysis and unmasking of the fluorophore and hence restoration of the push–pull mechanism of fluorescence.
One of the most commonly used cleavable masking groups for phenols are aryl boronates due to the electrophilic nature of electron-poor boron (68). One or more boronic esters are incorporated onto a fluorophore to give a profluorescent probe. Upon hydrolysis, originally envisaged to be selective to H2O2, the fluorescent molecule is released (Fig. 3a), as helpfully reviewed elsewhere (76, 77). Initial attempts in this direction resulted in the preparation of aminocoumarin (AMC) boronate ester (82) and peroxyfluor-1 (PF1, Fig. 3b) (17), both of which possess a pinacol boronic ester that masks the phenolic oxygen, resulting in low fluorescence. H2O2 chemoselectively oxidizes the boronic ester to a hydroxyl group, restoring fluorescence.
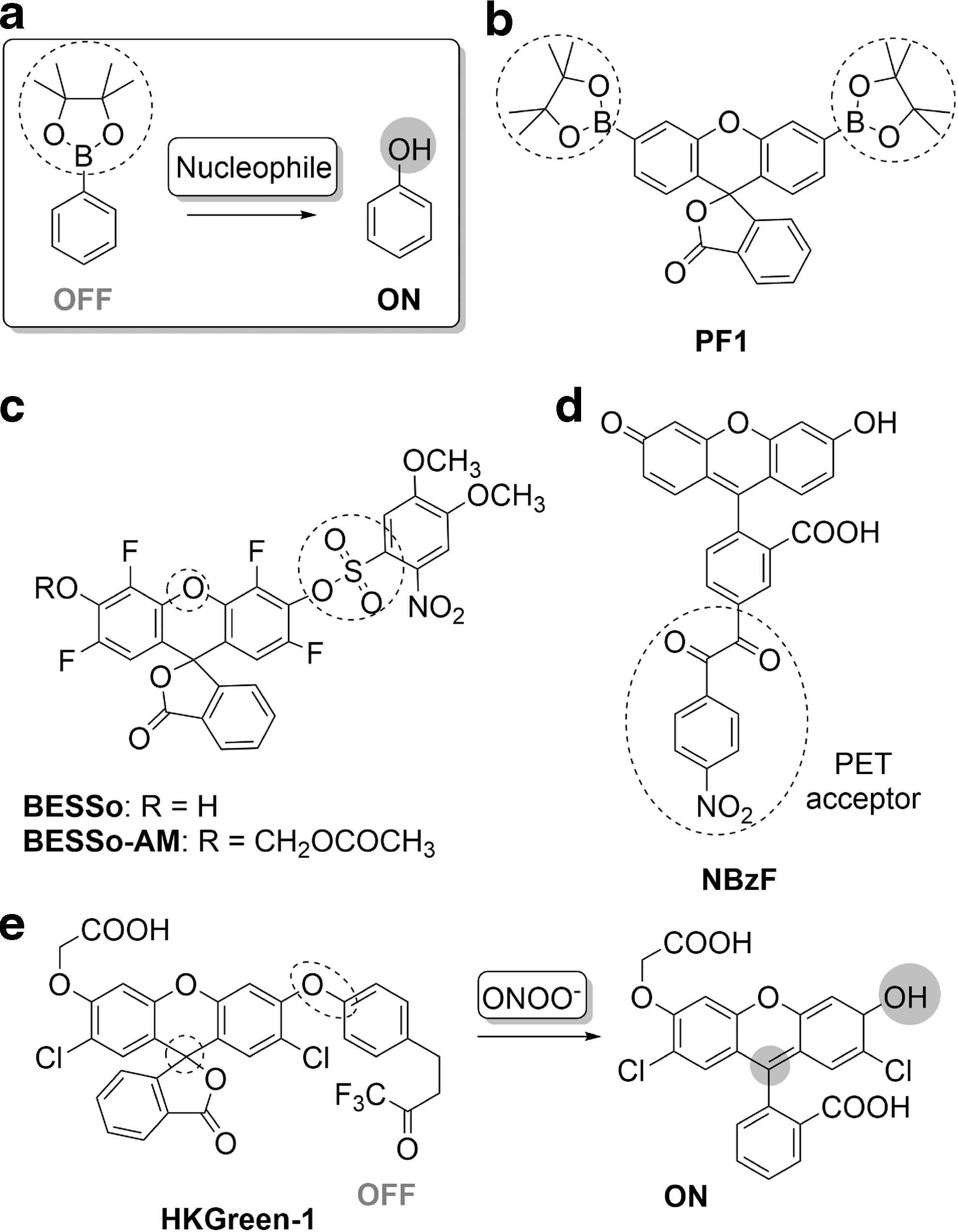
Building off this strategy, a myriad of fluorescent probes has been developed, with excitation–emission profiles that span the entire visible spectrum (4, 23, 25, 41, 42, 97, 98, 113, 123). An important addition to this category was ratio-peroxyfluor-1 (RPF1), a Förster resonance energy transfer (FRET)-based ratiometric fluorescent probe for H2O2, comprising a coumarin donor and a fluorescein acceptor that bears a boronic ester. Reaction with H2O2 gives an eightfold increase in the fluorescence intensity ratio (λ517/λ464) (5). RPF1 shows selectivity for H2O2 over some oxidizing species and experiments with viable yeast mitochondria suggested that it can determine endogenous H2O2 production.
Boronate-based probes have also been successfully targeted to subcellular organelles such as mitochondrially localizing MitoPY1 (21), nuclear NucPE1 (24), and lysosomal probe, LNB (61). However, it is now understood that in a direct reaction, peroxynitrite hydrolyses boronates nearly a million times faster than hydrogen peroxide, with ClO− (almost 104 times faster than H2O2) and potentially other hydroperoxides being other interferents (119). In this context, boronates emerge as promising probes for detection of ONOO− in biological samples with rapid kinetics of the reaction promising real-time detection (172). Only 85% of peroxynitrite acts as a nucleophile toward boronates, with the remaining 15% decomposing to reactive species further generating phenyl and phenoxyl radical reactive intermediates (172). This will further confound quantification of ROS/RNS levels.
Sulfonates have also been extensively explored as nucleophilically cleavable masking groups. Fluorescein-pentafluorobenzenesulfonyl ester (FPBS) (88) initially adopts the nonfluorescent lactone form. The strongly electron-withdrawing pentafluorophenyl moiety activates the sulfonyl group for hydrolysis by H2O2, resulting in ring opening and enhanced fluorescence intensity. FPBS showed selectivity toward H2O2, with detection limits of 4.6 pM, although some interference (one-third of the H2O2 signal) from NO was observed. Further developments in this strategy involved the incorporation of sulfonyl groups on fluorescein (FS-1 and FS-2) (151) and naphtho-fluorescein (NFDS-1) (154) scaffolds.
Interestingly, the use of the benzenesulfonyl group instead of pentafluorophenyl in masking the phenol on fluorescein in probes, such as DNS-F (90) and BESSo-AM (Fig. 3c) (89), made it susceptible to preferential hydrolysis by O2 •−, generating a highly fluorescent fluorescein molecule. Although probes, DNS-F and BESSo-AM, are based on the same strategy, BESSo-AM was reported to have a 10-fold higher detection limit and better selectivity toward O2 •− over other ROS/RNS. While, through detailed studies of the effects of varying the nature of the aryl group (electrophile), the authors successfully limited cross-reactivity of BESSo-AM not only against ROS/RNS but also glutathione (GSH), the concentration of the latter (50 μM) was significantly lower than the intrinsic GSH concentration found in cells (89).
The replacement of the electron-withdrawing group on the sulfonyl moiety by trifluoromethane in the HKSOX series of probes for O2 •− has been shown to have a detection limit of 23 nM, enabling a detection of superoxide in live animals with excellent selectivity, including against GSH (45).
Diphenylphosphinate can also be used to protect phenols, such as in the PF1 probe, wherein the reaction with O2 •− results in cleavage of the diphenylphosphinate to regenerate fluorescein. PF1 exhibited over 10-fold selectivity over other ROS/RNS and GSH and high sensitivity with a detection limit of 4.6 pM (153). A naphthofluorescein analog, PNF-1, has also been developed for NIR imaging (152).
Another effective fluorescence masking group is the benzil moiety, which acts as a photoinduced energy transfer (PET) donor quencher. This group can be hydrolyzed by ROS to the hydrolytically labile anhydride, as explored in the design of the NBzF probe (Fig. 3d) (2). In a Baeyer–Villiger-type reaction with H2O2, the nitro-benzil group yields a benzoic anhydride, which on further hydrolysis releases the fluorescent 5-carboxy fluorescein. The probe was shown to be selective toward H2O2 over other ROS, but a small fluorescence increase with tBuOOH or ONOO− was observed. NBzF was employed to monitor H2O2 production in phorbol 12-myristate-13-acetate (PMA)-stimulated RAW 264.7 murine macrophages and in A431 epidermoid carcinoma cells stimulated with an EGF. NBzF-BG, a SNAP-tag derivative of NBzF, was also developed and applied for the visualization of phagosomal H2O2 (1).
Peroxynitrite, formed in vivo by the reaction of NO with the superoxide radical, is a short-lived and highly reactive oxidant, which has been found to mimic potassium peroxymonosulfate in a unique oxidation of an ethyl trifluoromethyl ketone group to a dioxirane intermediate, in a process initiated by nucleophilic attack by the oxidant.
HKGreen-1, which contains a fluorescein scaffold linked to an ethyl trifluoromethyl ketone by an aryl–ether bond, exploits this strategy (Fig. 3e) (156). The ketone is oxidized to a dioxirane intermediate that brings about the ether bond cleavage and release of the fluorophore within an hour. HKGreen-2 and HKGreen-3 probes also respond to ONOO− via formation of a dioxirane intermediate, leading to phenol–quinoid transformation (HKGreen-2) (106, 128) or cleavage of aryl–amine bond (HKGreen-3) (106, 128), unmasking probe's fluorescence. Interestingly, the main interferent in the detection of ONOO− by this strategy is ClO− and the •OH radical, suggesting potentially an alternative, not purely nucleophilic, mechanism of action. The biggest challenge in further development of these probes lies in improving the efficiency of the transformation from ketone to fluorescent species, which is currently only 10–50%.
Utilization of the nucleophilic nature of selected ROS/RNS is therefore a valuable strategy in the development of responsive fluorescent probes. However, it is clear that true selectivity is difficult to achieve, with the main interferents being other potentially nucleophilic species (most notably H2O2 or other hydroperoxides, ONOO−, and sometimes superoxide). However, other non-ROS/RNS nucleophiles could also potentially interfere with the probe response, and it is therefore essential to test the selectivity of these probes against biologically relevant concentrations of other cellular nucleophiles containing thiol and amine groups (e.g., lysine). More extensive structure–activity relationship studies in the future might serve to bring further improvement in selectivity.
The boronates, despite the lack of initially envisaged true selectivity for hydrogen peroxide, emerge now as promising nearly quantitative tools for rapid detection of peroxynitrite. This class of probes also illustrates the great diversity that can be gained for a single sensing mechanism through variation of the fluorophore scaffold. Such an approach would be valuable for other probes, ensuring the breadth of emission colors and subcellular localizations necessary for a detailed biological study.
Electrophilic Nonradical Oxidations
The principal property of ROS/RNS is an oxidative character. As oxidants, they are themselves reduced and therefore have a propensity to accept electrons. ROS/RNS can therefore be considered to have electrophilic character.
A key example of an electrophilic ROS is HOCl. It is produced by the myeloperoxidase-H2O2-Cl− system in phagocytes, giving these cells the ability to kill a wide range of pathogens. Although HOCl is essential for proper immune response, uncontrolled production within the phagocytes is detrimental to the host tissue (109). Its damaging effect stems from the fact that it reacts very rapidly with endogenous nucleophiles, in particular with thiols and sulfides (at rates of∼108 M −1s−1 at pH 7.4 at 22°C) (126), as well as with amines, although less rapidly (rates of 105–106 M −1s−1) (30). HOCl also reacts with ascorbate in a nonradical manner, although∼10 times slower than with GSH. These reactions clearly suggest the potential designs for the probes that harness the reactivity of HOCl (30).
When alone, HOCl does not act via a radical mechanism, although it can generate free radical species upon interaction with hydrogen peroxide (in alkaline media), superoxide, or upon redox-active metal catalysis. However, the latter reaction is much slower than with nucleophiles or ascorbate (30).
Hypochlorous acid has a pKa of 7.53 and so it exists as an equilibrium between protonated and deprotonated forms in physiological conditions. While being a strong oxidant, its electrophilic character is pronounced in its protonated form, which is a source of electrophilic Cl+, with OH− as a leaving group. Upon deprotonation, no suitable leaving group is available in OCl−, therefore limiting the rate of Cl+ transfer. However, a decrease in pH can also lead to the protonation of the nucleophile, preventing the reaction. Therefore, a pH close to the pKa of the HOCl seems to be optimal for this type of transformation.
Many of the probes for the detection of HOCl are therefore based on its electrophilic oxidative capacity. This strategy is key in the development of reversible sensors, through oxidation of chalcogenides to chacogenoxides, which will be discussed in a later section. A number of irreversible sensors also use the electrophilic oxidative capacity of HOCl, with the most widely used strategy being induction of a ring opening of fluorescein and rhodamine derivatives by destabilization of spirolactam analogs upon initial Cl+ transfer to nucleophilic S or N and subsequent oxidation or hydrolysis to the carboxylic acid form (Fig. 4a).
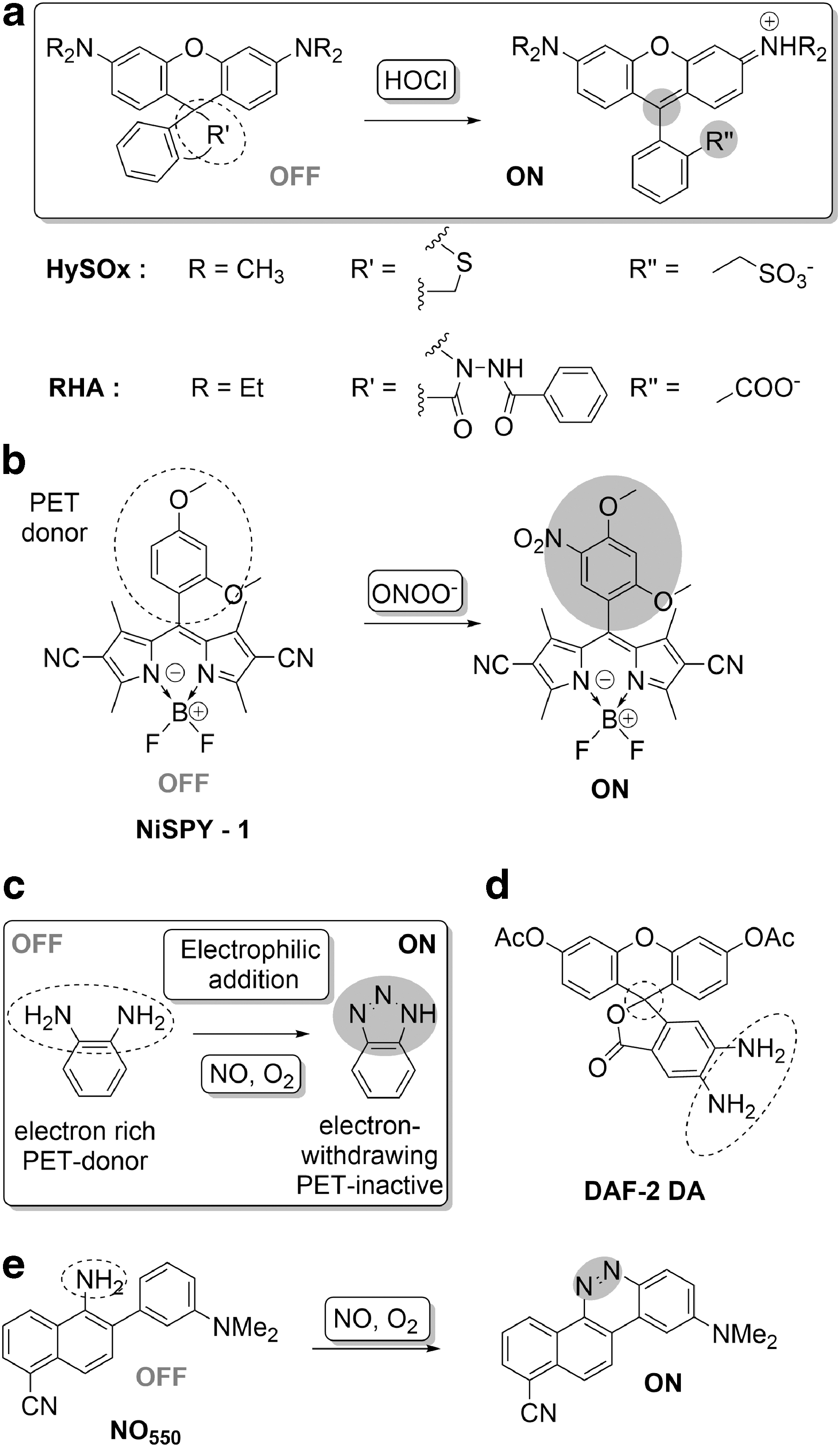
This was first demonstrated in HySOx, a fluorescent probe selective for HOCl detection (Fig. 4a) (58). Reaction of HOCl with the thioether moiety in the rhodamine scaffold resulted in ring opening to give the corresponding sulfonate and a simultaneous increase in fluorescence. HySOx was reported to have good selectivity toward HOCl and was also employed for the detection of HOCl in porcine neutrophils during phagocytosis. Subsequent work yielded thiolactone (R19S and R101S) and selenolactone (R19Se) containing rhodamine probes for HOCl (18). These probes exploited the HOCl-selective oxidation of the thio/selenol lactones, facilitating ring opening and an enhanced fluorescence response. While R19S and R101S showed excellent selectivity for HOCl over other ROS/RNS, the selenollactone in R19Se was observed to be more susceptible to oxidation by other oxidants.
Other approaches in this direction include spirolactam ring-opening mechanisms, as for rhodamine hydroxamic acid (RHOA) (159) and rhodamine hydrazide (RHA)-based probes (Fig. 4a) (19).
The oxidative reactivity of HOCl toward N and O-bearing functional motifs has also been harnessed in an HOCl-induced deoximation, which acts as an oxidative deprotection. This strategy gave rise to a ratiometric fluorescent probe (PAI) based on a phenanthroimidazole moiety bearing oxime functionality (127). In this case, oxidative hydrolysis of oximes mediated by HOCl leads to formation of the electron-withdrawing aldehyde group and a consequent intramolecular charge transfer-derived red-shift in fluorescence. This ratiometric probe was reported to be highly selective for HOCl, with a 10-fold increase in the ratio (I509/I439). A similar probe (TPA) based on a deoximation reaction was reported, utilizing a triphenylamine scaffold.
In addition to HOCl, other ROS/RNS can mediate electrophilic oxidations. For example, peroxynitrite can cause nitration reactions. Although nitration of fluorophores generally leads to fluorescence quenching, it can lead to a turn-on of fluorescence if occurring on a PET-quenching moiety. This is utilized in the NiSPYs (Fig. 4b), BODIPY-based probes with a quenching phenyl moiety (134). Rapid nitration by peroxynitrite alleviates the PET quenching, leading to fluorescence turn-on.
Another electrophilic oxidation is the N-nitrosation of aromatic amines, which has proved to be an effective strategy for sensing NO, which, under aerobic conditions, undergoes rapid oxidation to a highly reactive nitrosating species such as N2O3, which in turn can transform a PET-quenching O-phenylenediamine to a triazole, with restoration of fluorescence (Fig. 4c). Using this principle, diaminofluorescein-2-diacetate (DAF-2 DA, Fig. 4d) was developed as a selective probe for NO. This probe bears diacetyl groups for better cell permeability and exhibits a more than 100-fold increase in fluorescence upon reaction with NO (64). Although DAF-2 DA demonstrated the utility of O-phenylenediamine as an NO-responsive scaffold, subsequent probe development sought to overcome issues related to pH and retention and has given rise to probes with a broad palette of emission colors (32, 47, 63, 65, 107, 114).
A sequence of transformations initiated by the N-nitrosation of an aromatic amine in the presence of oxygen enabled detection of NO by the probe, NO550. An initial nitrosamine intermediate transforms to hydroxyhydrazine via electrophilic aromatic substitution (158). Dehydration of the hydroxyhydrazine results in the formation of a green fluorescent product with 1500-fold higher intensity. NO550 was also shown to be selective for NO over a range of oxidants. NO550 was also used for the visualization of nitric oxide in stimulated astrocytes.
Electrophilic reaction on a nucleophile is a central process in organic chemistry, yet it affords remarkable selectivity for individual ROS/RNS. While HOCl selectively oxidizes S-centers, NO has selectivity for N-centered oxidation and peroxynitrite to aromatic rings. While good selectivity has been demonstrated for these oxidants over other ROS/RNS, it is nevertheless crucial that selectivity be assessed against a broader array of electrophiles. In addition, involvement of radical-mediated oxidation of these probes by the radical species formed upon generation of electrophiles from ROS/RNS (e.g., formation of NO2 upon NO oxidation) and selectivity against other highly reactive radical species should further be examined.
In practice, HOCl reacts very rapidly with thiols, amines, and ascorbate such that the response can be generally observed within seconds. Similarly, nitrosation and nitration reactions are also relatively fast and the fluorescent products, upon the reaction with the probes, can be observed within few minutes or even seconds (114, 134). The rapid response to the presence of ROS/RNS perfectly positions these reactions for the real-time monitoring of ROS/RNS that can directly act as electrophiles or can rapidly form electrophilic species. It is essential that the in cellulo stability of probes of this type be assessed—the dianiline probes, for example, could potentially react with any carbonyl group, therefore confounding results.
Metal-Centered Reactions
The use of metal-centered reactions to sense ROS/RNS has had some success, as summarized by McQuade and Lippard (94). Paramagnetic metal complexes can be used in the design of turn-on fluorescent probes. In such systems, the metal ion will quench the fluorescence of a coordinated (or proximal) fluorophore, which can be restored when the fluorophore is released from this coordination sphere.
An example of a probe employing this strategy is [Co(DATI-4)] (Fig. 5a), containing a cobalt(II)aminotroponiminate complex tethered to a dansyl moiety (31). One of the coordinating arms bearing a fluorophore can be replaced by NO ligands due to the steric strain imposed by a coordination of bulky substituents to form a cobalt-dinitrosyl product, enhancing fluorescence. Other probes involving ruthenium (72) and rhodium (43) complexes based on this strategy have been reported, but the in vivo application of these probes is limited by slow reaction with NO and the interference of water in the coordination sphere.
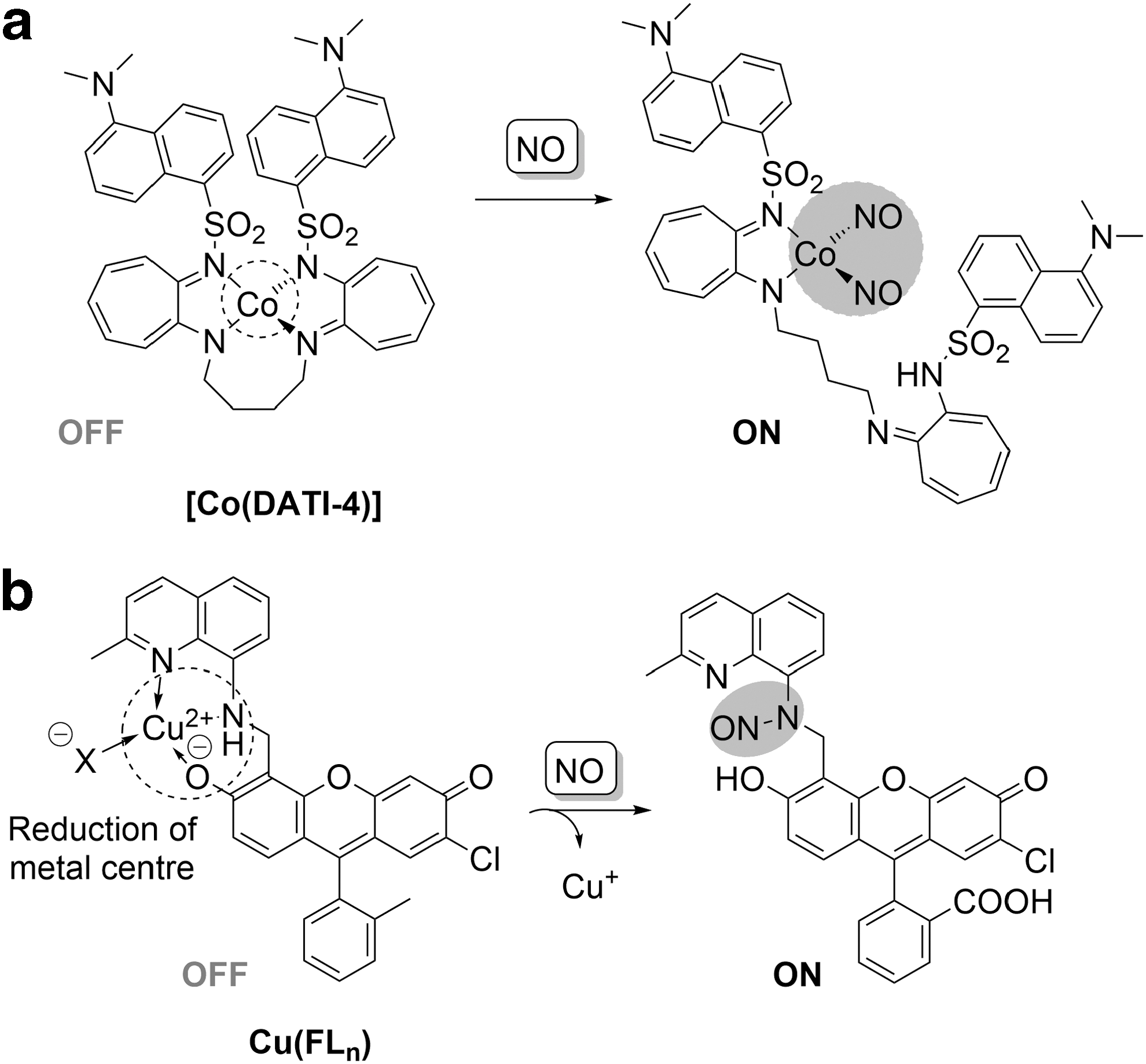
An interesting approach to explore N-nitrosation of arylamines for the detection of NO has been demonstrated in the development of a biocompatible Cu(II)-containing probe, Cu(FL n ) (Fig. 5b), with ligands bearing quinoline–fluorescein conjugates (73 –75). NO-mediated N-nitrosation of a coordinating motif fluorescein with simultaneous chemoselective reduction of copper(II) to copper(I) results in the release of the metal ion from the chelate, removal of the metal quenching effect, and a subsequent 16-fold increase in fluorescence (73 –75). With a detection limit of 5 nM, Cu(FL n ) was utilized for visualizing nitric oxide production in lipopolysaccharides and IFN-γ-stimulated RAW 264.7 macrophages. This strategy was expanded by developing ratiometric (121) and π-conjugated polymer (120)-based probes for NO.
This strategy, of metal-centered reactions, has not been widely utilized, but is nonetheless very interesting. There are considerable advantages to using metal coordination for sensing purposes, best demonstrated to date in the anion sensing literature (7), based around the fact that systems are sensitive not only to coordination but also to geometry, charge, and even redox potential. The main challenge in designing ROS/RNS-sensing metal complexes is to design a complex that is stable enough in biological media, but at the same time can undergo ligand exchange with the analyte. This ligand exchange process will destabilize the complex toward other competitive ligands. In assessing potential sensing systems, it is essential to test selectivity not only toward other ROS/RNS but also toward other potential ligands that are likely to be present in biological systems.
Reversible Probes for Redox Signaling
The second crucial aspect of redox probes for studying redox processes is the reversibility of the response to redox stimuli to enable temporally resolved monitoring of both oxidation and reduction processes in real time. While such time-resolved studies are possible with some redox-responsive fluorescent proteins (26, 99, 143), it still remains a challenge for small-molecule probes, spurring recent efforts to bridge this gap. The prerequisite for a reversible probe is that it can cycle back and forth between fluorescently different oxidized and reduced forms without loss of signal.
Reversible redox probes reported to date can be classified into three groups according to the mechanism by which the target-induced redox transformation is translated into an optical readout, namely a redox reaction of a target with a probe can lead to a change in fluorescence/luminescence due to the following: (i) an energy/electron transfer mechanism—the target induces transformation of a redox-responsive motif, leading to a change in its electron/energy transfer capabilities, resulting in quenching or enhancement of the fluorescence of a tethered fluorophore; (ii) a conformational mechanism—the target interacts with a redox-responsive motif, causing a change in the spatial proximity of fluorophore/luminophore and a quencher/enhancer; and (iii) a chromophore-centered mechanism—the redox reaction with the target is centered on the chromophore, leading to its formation/destruction.
The following section will cover reversible tools for studying redox processes in live systems arranged according to the above-mentioned classification.
Electron/Energy Transfer
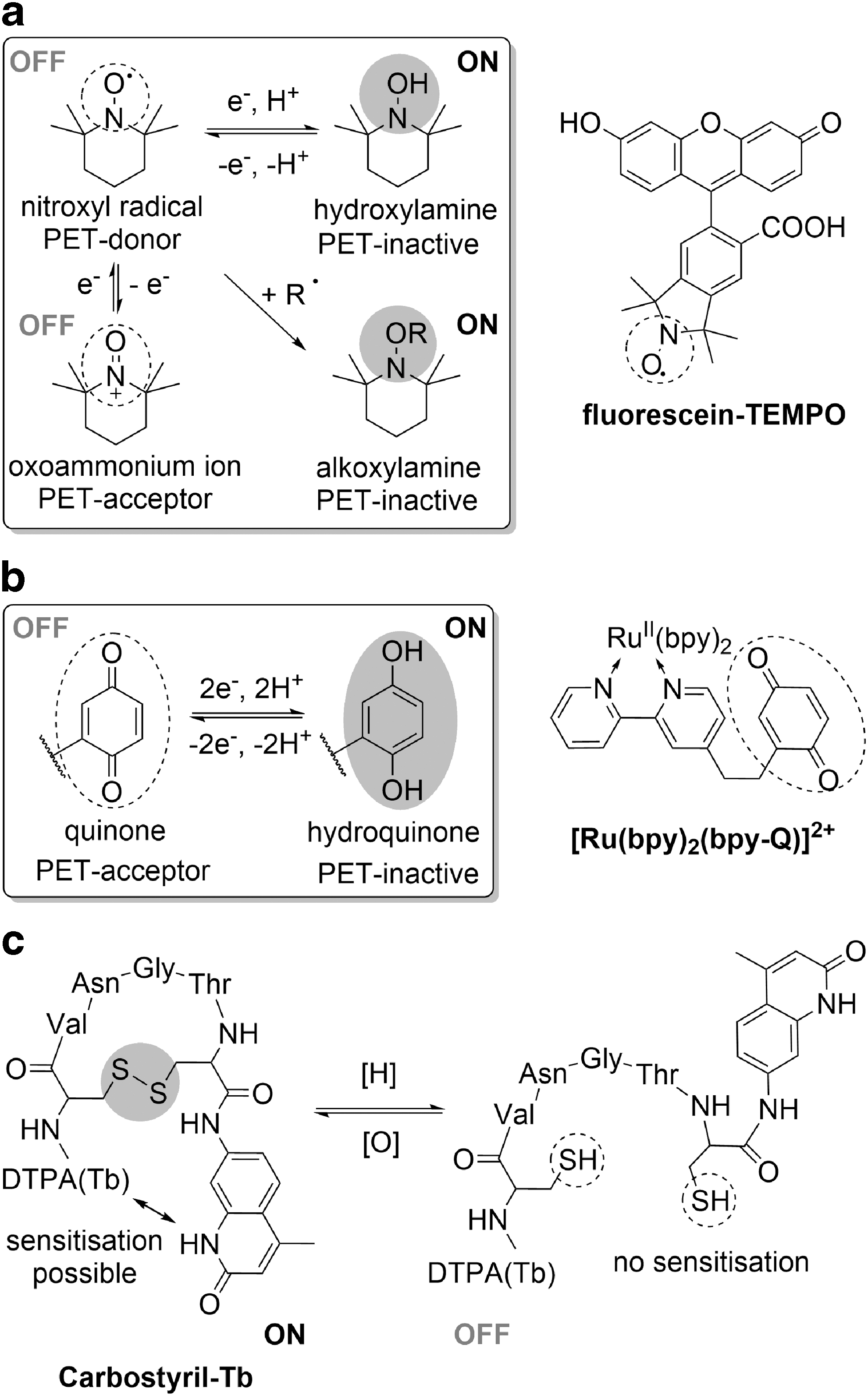
One of the most common methods utilized in the development of responsive fluorescent sensors is an analyte-induced change in electronics. Among redox-responsive probes of this class are nitroxide-based probes, which take advantage of redox-mediated reversible switching between the fluorescence quenching, stable paramagnetic nitroxyl radical (oxidized form) and the fluorescence-benign diamagnetic hydroxylamine (Fig. 6a).
Since the proof of concept was reported in 1988 (13), various fluorophores have been linked to a nitroxyl radical moiety, yielding a wide range of redox-responsive profluorescent nitroxide probes (PFNs) with a spectrum of optical properties (12, 14, 35, 86). Of these, only a few structures exhibit the sufficiently long excitation and emission wavelengths and high quantum yields required for biological applications. Among these probes is the BODIPY-based green fluorescent probe, TMB, used to report on oxidative stress in H2O2-stimulated HepG2-cells (80) while being hydrolytically resistant to trypsin activity in vitro over 1 h (81).
While red-emitting (700 nm) Si-phthalocyanine R2c turns on in response to ascorbate in HeLa cells, both TMB and R2c probes suffered from small (∼20 nm) Stokes shifts. A fluorescent redox-induced response with red excitation, NIR emission, and significant (90 nm) Stokes shift was achieved by incorporation of cyanine dye in spatial proximity of nitroxyl radicals in the polymeric dual magnetic resonance imaging/fluorescence ORCAFluor probes, enabling in vivo detection of ascorbate levels in mice (122). NIR excitation was also achieved by developing two-photon intensity-based probes, TEtNO-Anthracene and TEtNO-Fluorescein, used successfully to report on H2O2-induced oxidative stress in Chinese hamster ovarian cells (3).
Nevertheless, despite the potential reversibility of response of PFNs, this property has only been experimentally demonstrated for Fluorescein-TEMPO (Fig. 6a) (100) and its rhodamine analog, ME-TRN (110). The reduction-induced increase of Fluorescein-TEMPO fluorescence intensity, followed by fluorescence quenching upon reoxidation on air, could be observed for up to three cycles in vitro. However, an increase in the basal level of fluorescence in the oxidized form suggests that with each cycle, a fraction of the probe becomes irreversibly activated by permanent destruction of a radical, likely by reaction between two radical species.
While the probe responded selectively to superoxide over H2O2 and hydroxyl radical in vitro, the authors interpreted the localized fluorescence of the probe in the membrane and lysosomes of hTERT immortalized fibroblasts as an indicator of response to metabolism-related overall oxidative capacity. In turn, the rhodamine analog, ME-TRN, with improved pH-stability of fluorescence was successfully used to study time-resolved changes in stress-induced oxidative capacity of rat retina, boding well for further in vivo application of this type of probes (110).
Quinones are another class of redox-reversible motifs, which differ by the quenching capacity of reduced and oxidized form and therefore have also been considered in the design of redox-responsive probes that involve electronic modulation of a redox-active group. In the oxidized form, the electron-deficient quinone motif can act as a PET acceptor, quenching the dye's fluorescence, which can be restored upon reversible reduction to the more electron-rich hydroquinone. Sequential cycling between the two forms was achieved in vitro for multiple examples (10, 38, 166), notably for the ruthenium-based luminescent probes [e.g., [Ru(bpy)2(bpy-Q)]2+, Fig. 6b (38)], but only a few dye–quinone constructs have been tested in live systems.
While oxidized rhodamine-quinone probe, RhQ, was reversibly reduced by cysteine in vitro, it could not be reoxidized in cells, probably due to the strong reducing capacity of the intracellular environment. The authors suggest that future synthetic modifications to increase the redox potential of the quinone motif should yield a cellularly responsive reversible fluorescent redox probe (60). The redox-mediated fluorescent response in cellulo was achieved with the DA-Cy probe, which showed selectivity toward H2O2 and hydroxyl radical, but its reversibility was limited to only one cycle (163).
A robust redox cycling of the nicotinamide moiety in nicotinamide adenine dinucleotide (NAD) in biological environments is crucial for its role in mediating natural redox processes as an enzyme cofactor and mobile electron carrier, making it highly attractive for the design of reversible redox probes. The only probe based on the nicotinamide motif, perylene-NAD, undergoes a 10-fold increase in fluorescence upon oxidation attributed to the alleviation of PET quenching, but this probe has only been evaluated in vitro.
Conformational Mechanism
The reversible reduction of disulfide and diselenide bridges is a key biological transformation in vivo, involved in controlling the structure and/or function of proteins (such as in the cysteine–cystine equilibrium or RSeH/RSeSeR transformations in the glutathione peroxidase [GPx] active site) or buffering of an intracellular redox environment. Therefore, this chalcogenide–dichalcogenide equilibrium is well suited for sensing purposes, as exemplified by the widespread use of endogenous RSH/RSSR ratios as an indicator of oxidative capacity in biology. Nevertheless, despite the advantage of reversibility in a biological environment, only a limited number of fluorescent probes based on this transformation have been reported.
In the carbostyril-Tb ratiometric probe, oxidation is accompanied by an increase in the luminescence of the terbium complex due to formation of a disulfide bond that brings the terbium into close proximity of its sensitizer, carbostyril (Fig. 6c) (71). While the probe has not been tested in living systems, its reduction potential lies within the biologically relevant range for RSH/RSSR pairs (−243 mV) and can be further tuned by modifying the linker, but the probe uptake and proteolytic resistance of the linker require further investigation. The FSeSeF probe operates according to a similar mechanism; in this probe, two fluorescein molecules are tethered with a diselenide linker that quenches their fluorescence. The fluorescence can be restored upon GSH-mediated cleavage, yielding two separate dye molecules (85) that can diffuse apart, making reoxidation unlikely, and hence limiting the practical reversibility of the response.
Chromophore-Centered Mechanism
While for probes operating according to the latter two mechanisms the redox-responsive motif and the dye were distinct moieties, there are a number of chromophores that are intrinsically susceptible to the activity of redox species. Again, only a handful of such probes operate in a reversible manner and in a biological environment. Such groups, which simultaneously play the role of redox-responsive element and signal emitter, usually lose their fluorescence upon reduction due to destruction of the chromophore. Therefore, unlike the two previously discussed probe classes, where the oxidation event primarily influences emission efficiency, the redox response for probes of this type arises from a target-induced change in absorption. Consequently, this strategy promises greater differences in the fluorescence intensity of oxidized and reduced forms, but at the same time, the design of new probes of this type remains more challenging as it does not allow for a combinatorial approach in which a fluorophore can be simply linked to a quenching motif.
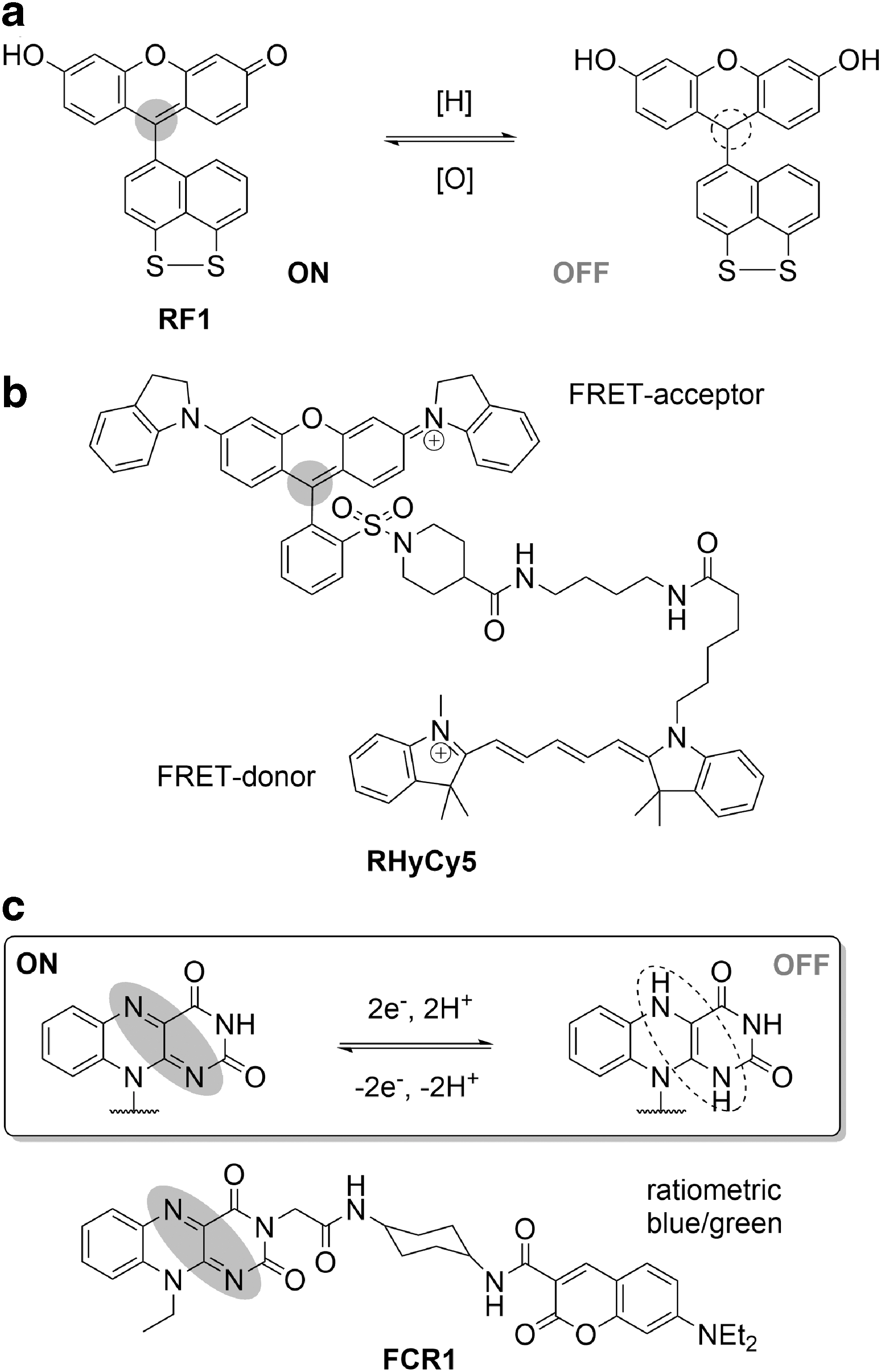
One of the most common examples of such redox-active chromophores is xanthene, the core of fluorescein and rhodamine-type dyes. While fluorescein alone cannot be back-reduced in biological environments, this was achieved in RF-1, in which a disulfide motif was integrated into the structure (Fig. 7a). RF-1 provided the first example of imaging of reversible redox cycles in cells by a small-molecule probe. The reversible disappearance of the characteristic absorption of fluorescein chromophore upon reduction suggests a chromophore-centered redox process, breaking the electronic communication between push and pull poles of the fluorophore. In another design, the NIR-absorbing RHyCy5 bears a rhodamine QSY-21 chromophore that functions as an FRET acceptor for covalently linked NIR-emitting cyanine 5, quenching its fluorescence (Fig. 7b). In hypoxic conditions, one-electron enzymatic reduction of QSY-21 eliminates its NIR absorbance, enabling detection of NIR emission from the Cy5 FRET donor. Consequently, RHyCy5 was successfully used to image multiple cycles of normoxia/hypoxia in A549 human breast cancer cells (129).
The recently reported Rs-HFB-Rs is the first example of a pro-probe, which upon irreversible reaction with biothiols releases a resorufin moiety in situ, capable of reversible switching between fluorescent, oxidized, and nonfluorescent reduced forms (87). This reversible response was successfully demonstrated in macrophages, where the sulfide-activated resorufin was reduced upon glucose/GOx treatment and reoxidized by placing cells in basic pH in air and, as such, was claimed to be a suitable indicator of intracellular redox homeostasis.
Further inspiration from nature has led to the design of redox-active fluorescent probes based on vitamin B2 (riboflavin) structural motifs. As reversible redox-responsive chromophores, flavins are unique and highly important cofactors crucial for controlling intracellular redox homeostasis. The redox switching in biologically relevant conditions and inherent lack of toxicity prompted the development of flavin-based redox probes, which have been successfully used in various biological settings. As the reversible reduction of flavins results in loss of fluorescence (Fig. 7c), such probes promise a large difference in fluorescence between oxidized and reduced forms. The first redox-responsive fluorescent probe of this type, CMFL-BODIPY, consists of carboxymethylflavin tethered to a BODIPY dye, and exhibits a reduction potential of −240 mV, similar to endogenous flavins. This probe underwent a ninefold decrease in fluorescence upon reduction in vitro and was successfully used in HeLa cells.
Our group has recently reported a naphthalimide-flavin probe, NpFR1 (160), and its mitochondrially localized analog, NpFR2 (54), which were shown to respond reversibly over a number of redox cycles to a range of biologically relevant oxidizing agents, with over a 100-fold change in fluorescence intensity. These probes have been proposed for the measurement of general oxidative stress in biological models. In particular, NpFR1 was successfully used to observe a relationship between glucose levels and oxidative stress in 3T3-L1 adipocytes (160). NpFR2, in turn, was validated as a useful flow cytometry marker of mitochondrial oxidative status to distinguish cell populations within mouse spleen, bone marrow, and thymus.
An important development in devising flavin-based reversible redox probes was a ratiometric FRET probe, FCR1, in which a redox-sensitive, flavin (green fluorescence) FRET acceptor was linked to a redox-insensitive coumarin (blue fluorescence) FRET donor (Fig. 7c) (55). The blue-to-green fluorescence ratio of FCR1 is a direct indicator of overall oxidative capacity of the environment, that is, it increases with reduction of a flavin chromophore. Our probe is compatible not only with flow cytometry assays and classic confocal microscopy but it can also sense changes in intracellular redox status via a change in the fluorescence lifetimes, expanding its versatility.
Reversible and Selective Probes for Redox Signaling
As mentioned in the first part of this review, the majority of selective sensors of ROS/RNS involve irreversible reactions that lead to the irreversible transformation of the starting material. Such strategies necessarily preclude their use as reversible sensors as they are insensitive to immediate concentration of the analyte, relying instead on a build-up of reacted probe over time. On the other hand, the reversible probes discussed above, based on several redox-induced reversible transformations operable in a biological environment, enable imaging of oxidation–reduction cycles in cells, but without providing more specific information on the nature of redox-active species. In addition to these two distinct classes of probes, there are several examples of probes that demonstrate both selectivity and reversibility of response to a specific ROS/RNS. These probes are distinct from the reversible probes above because they are oxidized by a specific ROS/RNS, and then reduced back to the original structure by one or more cellular reducing agents, generally thiols. In the following discussion, we highlight the redox-responsive groups used and the degree of selectivity to the selected target over other tested species.
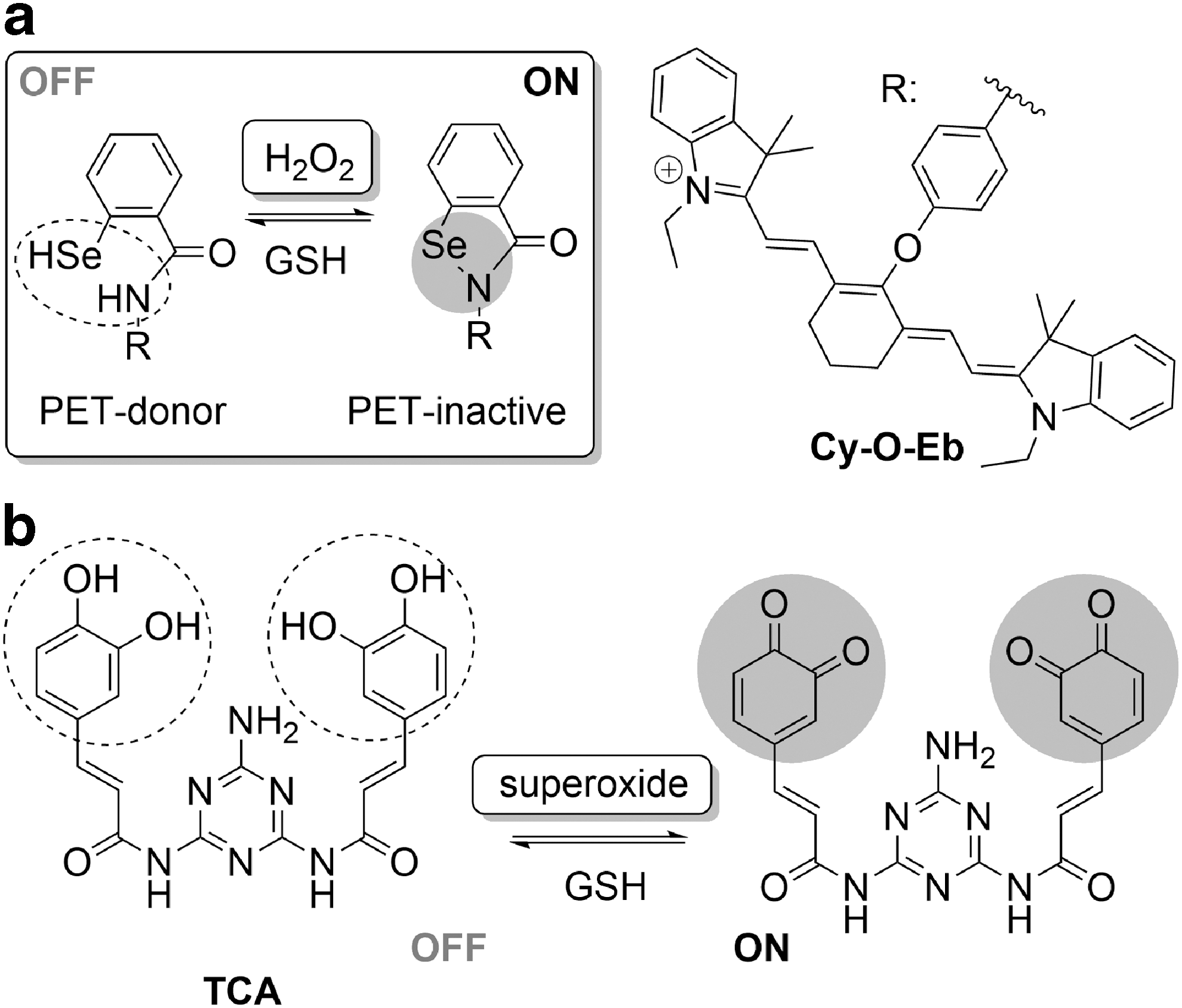
Hydrogen peroxide is arguably the most widely studied ROS due to its profound implications in a wide range of processes in live systems. Despite its strongly oxidizing character, H2O2 reacts slowly with most redox-responsive moieties due to a high activation barrier of a two-electron process (146), but can react rapidly with selenium-containing compounds. This fact was used in the development of Cy-O-Eb, prepared by integration of ebselen, a small-molecule mimic of the GPx enzyme with a redox-active Se-N bond, into a cyanine dye (Fig. 8a) (155).
This probe experiences a reversible decrease in NIR fluorescence intensity upon reaction with thiols due to suggested PET quenching of cyanine fluorescence. The probe was demonstrated to have in vitro selectivity toward H2O2 over ClO− and tBuOOH, but the previously suggested reactivity of ebselen toward other ROS (115) has not been investigated for Cy-O-Eb. The robust reversibility of fluorescence response to GSH/H2O2 redox cycles, as well as its high photostability and excellent cellular retention, was demonstrated in Hep2G cells, together with mitochondrial localization of Cy-O-Eb, and the probe was successfully used to investigate changes in oxidative stress in wounded zebrafish (155).
Reversible probes for superoxide were recently prepared by utilizing a quinone-type transformation of pyrocatechol motifs integrated into the chromophore in the one/two-photon excitable probes, TCA (Fig. 8b) (167) and PY-CAO (169). These probes could be used for the highly selective and sensitive (down to 3.2 nM) detection of superoxide in cultured cells, zebrafish and mice, and could be back-reduced with GSH. Only minor interference from high excesses of other ROS/RNS was observed. In addition to the greater than 1000-fold increase in fluorescence intensity and the excellent cellular retention, these probes were shown to be highly photostable in cellular environments.
Another probe, Bis(BODIPY)diselenide, combining two BODIPY molecules via a diselenide linker, undergoes a selenide–selenoxide transformation that is selectively induced by superoxide over other ROS, leading to a biothiol-reversible increase in green fluorescence intensity of probe, with demonstrated efficacy in cultured cells (91). Exciting results in the detection of hypoxic conditions in cellular and animal studies have been achieved with metal-based NIR-luminescent constructs exploiting a luminescence quenching effect of molecular oxygen (66, 101). This mechanistically unique response to a target molecule is based on a direct interaction of O2 with an excited-state luminescent complex, reducing its emission efficiency by providing a nonradiative mechanism of relaxation. While the ruthenium-phenanthroline chelate (Ru-Py) experienced a decrease of orange luminescence upon increase in oxygen levels (66), the PS-NPs nanocapsules, containing a Pd-based oxygen-sensitive complex and an organic reference dye, enabled a ratiometric readout (101).
A large proportion of the reversible probes with demonstrated ROS/RNS selectivity were inspired by the discovery of the mechanism of action at the GPx active site. These probes utilize the reversible switching between the fluorescence quenching PET donor chalcogenide (sulfide or selenide) and PET-inactive chalcogenoxide (sulfoxide or selenoxide) upon ROS/RNS oxidation, with reduction mediated by biothiols (Fig. 9a). This design has been successfully used to prepare a range of probes, with selectivity to peroxynitrite, hypochlorous, or hypobromous acids, as helpfully reviewed by Lou et al. (83).
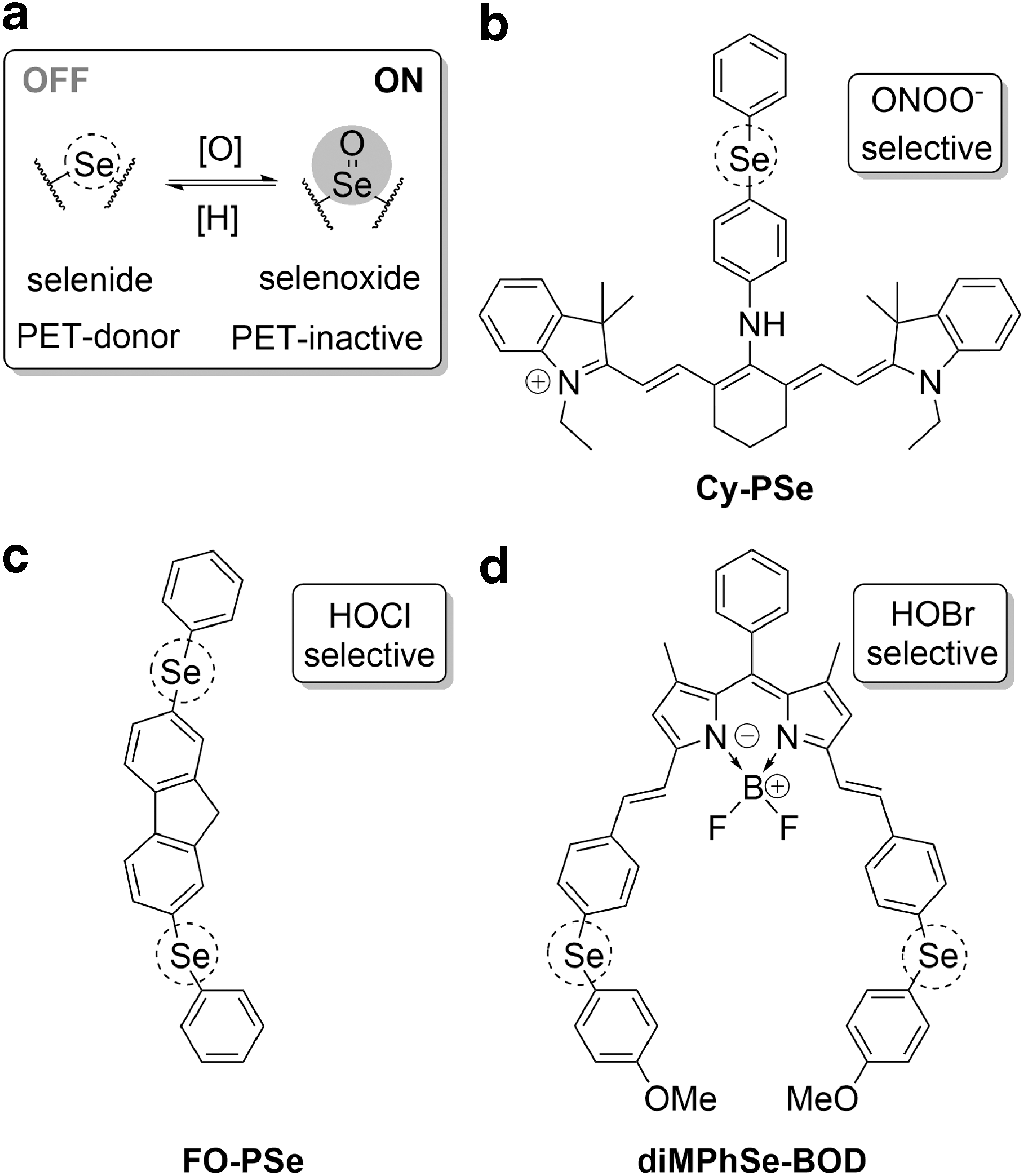
The first probe of this kind, the diphenylselenide-cyanine conjugate, Cy-PSe (Fig. 9b), showed a 23-fold increase in cyanine NIR fluorescence intensity in vitro upon reaction with ONOO− over a range of other ROS with limited interference from H2O2, NO, and •OH (162). The robust reversibility of its response to the peroxynitrite/biothiols redox pair (GSH, cysteine, metallothioneins) was demonstrated in activated macrophages, where GSH was believed to be the major reductant (162). Slightly lower selectivity toward ONOO− over peroxides and ClO− was observed upon replacement of the selenide with a telluride in Cy-NTe (164) probe, which was successfully used to study oxidative stress in mice. However, the 2Me TeR probe, with tellurium replacing an oxygen atom of a xanthene moiety of rhodamine dye, could not discriminate between ONOO− and ClO− (62). Nevertheless, the in vitro reversibility, with only a moderate 20% decline of a maximal signal over three cycles due to bleaching, enabled the authors to qualitatively monitor fluxes of H2O2-induced myeloperoxidase-dependent oxidative stress in HL-60 cells.
Following these reports, several probes exploring similar chemical transformation of selenides and sulfides to their monoxides with a range of optical properties have been reported and successfully used in live systems (78, 79, 84, 138, 169). In particular, large Stokes shifts of NI-Se (84), the S-to-S = O-type luminescent sensor [Ru(bpy)3 2+]-PTZ (78), and the first two-photon excitable probe, FO-PSe (168), (Fig. 9c) enabled their use in vivo. While all of these probes showed good selectivity to hypochlorite/hypochlorous acid, diMPhSe-BOD (137) (Fig. 9d) exhibited a reversible and ratiometric response to hypobromous acid over a few cycles both in vitro and in cellulo. However, it is important to note that despite high selectivity of diMPhSe-BOD to HOBr over other ROS, it exhibits poor selectivity for HOBr over HOCl, which can induce approximately a fifth of the response measured for HOBr.
This limited selectivity over H2O2 and HOCl was also observed with cyanine-based sensors exploiting PET-inactive hydroxylamines that could be reversibly oxidized to the PET-accepting oxoammonium form. Cy-TemOH, with an intensity-based response, and the ratiometric mCyTem-OH, with an oxidation-quenched red fluorescence and redox-insensitive, but pH-dependent, yellow emission, showed selectivity to HOBr over other ROS/RNS. The ascorbate-mediated reversibility of their redox response, with limited interference from thiols, enabled detection of fluxes of hypobromous acid and ascorbate in macrophages (165).
Conclusions
This survey of currently available probes reveals the vast potential of the field. There is a crucial role for chemists in the provision of molecular imaging tools to those studying redox signaling. Although it is well established that ROS/RNS play a vital role in physiological and pathological processes within the cell, unraveling the mechanistic details with respect to their production, build-up, localization, trafficking, and response still remains a challenge. A better understanding of such processes will require the elucidation of specificity (or otherwise) of redox signaling processes (possible with selective probes) and the identification of the tipping point between reversible signaling and irreversible oxidation-induced toxicity (achievable with reversible probes).
While the scientific community has turned greatest attention to the first of these properties, the latter is also important, and more recent work is providing exciting tools in this field (57, 83, 92). In both distinct fields, it is essential to build up clear protocols for assessing and utilizing probes. Recent work highlighting the preferred selectivity toward peroxynitrite in the sensing of peroxide (119) exemplifies that it is essential for all putatively selective sensors to be tested against the full range of ROS/RNS and against other possible interferents. For reversible probes, it is essential that measurements are made over multiple cycles of oxidation and reduction, with reversibility also demonstrated in cellular studies. Furthermore, for both classes of probes, analysis of the kinetics of probe response is essential in correlating observed changes with the timescale of cell signaling events.
Ideally speaking, a probe that could achieve both aims (of selectivity and reversibility) would be the most powerful tool to monitor cellular redox processes with high spatiotemporal resolution, begging the question of whether it is possible to achieve true selectivity and reversibility. We have identified a number of probes for which oxidation can be achieved preferentially by one ROS/RNS over others and which can subsequently be reduced by cellular thiols. While this class of probes is exciting for its potential for simultaneous and reversible sensing, the relationship between probe structure and selectivity is not well understood, and probe development tends to follow a library-based approach rather than rational design (83). Furthermore, in many cases, the selectivity for one ROS/RNS over another is only very small, and it is not clear whether any such probes can truly be classed as selective.
It would certainly be valuable to investigate other avenues that could possibly lead to reversible and selective redox sensors. Metal complexes may hold the key to such sensor design as they can reversibly bind to small molecules (139, 161). It is important to consider, however, the extremely low concentrations and short lifetimes likely to be involved in redox signaling processes, particularly for free radicals, which persist for 10−9 to 10−6 s (140). While reaction-based selective sensors can circumvent this challenge by enabling the accumulation of activated probe over time, a reversible selective sensor would need to have an extremely low limit of detection and very rapid kinetics of response to be able to find such use: it is unclear, at present, whether this is possible.
Instead, the way forward may be to study both parameters by simultaneously using a selective probe with a reversible probe. Due to the vast array of probes on offer, particularly those of the former type, it should be straightforward to select two probes with nonoverlapping fluorescence emissions. Furthermore, covalent tethering of the two probes would ensure colocalization of the two sensing groups. An exciting field of future research would therefore involve studies of the viability of such a strategy, that is, testing for the compatibility of appropriate probes.
This review has focused only on small-molecule fluorescent sensors and their potential for reversible and/or selective study of redox signaling. It is worth acknowledging that the field of fluorescent protein-based redox sensors has, at this point, better met the challenges of simultaneously sensing selective ROS/RNS in a reversible manner (96). In addition to their advantages such as biocompatibility and photostability, it is possible to design fluorescent proteins such as HyPer, which are selective for H2O2 and respond in a reversible manner (9). However, there remain many applications for which fluorescent proteins are not suitable, and there is therefore a need for small-molecule fluorescent probes to reach these standards set by fluorescent proteins.
While small-molecule probes, unlike protein-based sensors, do not require an invasive genetic modification of the system, all exogenous probes may potentially modify the studied environment to some extent. Unlike probes for metal ions, for example, which reversibly bind the target and therefore do not change its nature upon interaction, most redox sensors reported to date react with an oxidizing agent of interest, diminishing its effective levels and possibly disrupting or altering the system. On the other hand, the probe, or its photoexcitation during detection, can lead to the further generation of ROS/RNS. Therefore, measuring and minimizing the effects of the sensor on cellular redox homeostasis, largely neglected to date, remain the biggest challenges in designing and evaluating new tools to study redox processes in live systems.
While much has been learnt about the role of redox signaling in health and disease, much remains to be uncovered, and chemists stand to play a key role in the provision of more sophisticated tools for such studies. In this review, we have highlighted the elegant strategies that have been designed to date to achieve selectivity, reversibility, or both, and we have presented the key directions for future research in this exciting field.
Footnotes
Acknowledgments
The authors gratefully acknowledge the support of the Fondation ARC pour la Recherche sur le Cancer for Postdoctoral Outgoing Fellowship SAE (J.L.K.), University of Sydney for a World Scholars Scholarship (A.K.), the Australian Research Council for a Discovery Early Career Researcher Award (E.J.N.) and the Ramaciotti Foundation for an Establishment Grant.