
Abstract
Aims:
Liver steatosis is associated with mitochondrial dysfunction and elevated reactive oxygen species (ROS) levels together with enhanced sensitivity to ischemia-reperfusion (IR) injury and limited response to preconditioning protocols. Here, we sought to determine whether the downregulation in the steatotic liver of peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α), a master regulator of mitochondrial metabolism and ROS that is known to play a role in liver metabolic control, could be responsible for the sensitivity of the steatotic liver to ischemic damage.
Results:
PGC-1α was induced in normal liver after exposure to an IR protocol, which was concomitant with an increase in the levels of antioxidant proteins. By contrast, its induction was severely blunted in the steatotic liver, resulting in a modest induction of antioxidant proteins. Livers of PGC-1α−/− mice on a chow diet were normal, but they exhibited an enhanced sensitivity to IR injury and also a lack of response to ischemic preconditioning (IPC), a phenotype that recapitulated the features of the steatotic liver in terms of liver damage, although the inflammatory response differed between both models. Utilizing an in vitro model of IPC, we found that PGC-1α expression was downregulated in hepatic cells cultured at 1% O2; whereas it was induced after reoxygenation (3% O2), and it was responsible for the recovery of antioxidant gene expression after the ischemic period.
Innovation & Conclusion:
PGC-1α plays an important role in the protection against IR injury in the liver, which is likely associated with its capacity to induce antioxidant gene expression. Antioxid. Redox Signal. 27, 1332–1346.
Introduction
L
This challenge has guided the development of strategies to improve IR tolerance of steatotic livers and several preconditioning techniques have been tested, including ischemic preconditioning (IPC), by which prior application of brief IR confer a state of protection against subsequent IR injury. Unfortunately, the limited success of IPC has prevented its widespread use in the clinical setting (30, 44).
The contribution of oxidative stress to the enhanced sensitivity of the steatotic liver to IR injury and the poor response to preconditioning protocols are well established. However, the molecular basis remains a matter of controversy. This study identifies peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) as a key mediator of this effect and elucidates the molecular mechanism involved. This result has clinical significance, as it suggests that evaluation of PGC-1α levels can have predictive value in liver transplantation.
The poor tolerance of the steatotic liver to IR has been attributed, at least in part, to the generation of high levels of reactive oxygen species (ROS) (9, 11, 13). ROS are well-characterized mediators of hepatocyte cell death after reperfusion (18), and it has been hypothesized that because the steatotic liver is under basal oxidative stress, the further increase of ROS during reperfusion drives the massive induction of hepatocyte death in this setting (16, 41).
This hypothesis has led to the evaluation of different antioxidants that are expected to limit IR injury in the steatotic liver. Although results have been partially encouraging, at least for acute liver damage (35, 49), they have not proved to be efficacious in humans, possibly because ROS are long-term preconditioning mediators and their signaling activity is also required for the successful engraftment of the transplant (2, 36). More promising are the results on new experimental approaches that aim at controlling ROS levels, but are not based on antioxidant therapy (5, 31).
Peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) is a transcriptional coactivator that controls the expression of most, if not all, of the metabolic pathways that allow the cellular adaptation to limited nutrient availability or to increased metabolic demand (i.e., cold, exercise) (43). Importantly, PGC-1α boosts the catabolic and oxidative capacity of the cell, and it coordinately induces the expression of a suite of antioxidant enzymes that prevents oxidative damage under these conditions (48). As a result of PGC-1α induction, cells present an enhanced antioxidant capacity and a net reduction in ROS levels.
Obesity and the metabolic syndrome are usually associated with a decrease in PGC-1α activity in all the tissues and cell types where it has been investigated, including the liver (1, 4, 32, 39, 50). Previous results from our group have shown that PGC-1α regulates antioxidant gene expression and ROS levels in hepatocytes (38), pointing to a role for PGC-1α in IR tolerance. This notion is supported by several reports suggesting that PGC-1α may play a protective role against ischemic injury in heart, kidney, and the central nervous system (15, 20, 26).
In the present study, we aimed at elucidating the role of PGC-1α in the poor tolerance of the steatotic liver to IR injury and IPC. We found that IR induces PGC-1α expression in the normal liver and its induction drives the increased expression of antioxidant enzymes; however, this response is absent in the steatotic liver. Furthermore, we demonstrate that the absence of PGC-1α prevents effective IPC. Our findings lead us to conclude that the poor response of the steatotic liver to IR and IPC is, at least in part, attributable to the failure of PGC-1α to induce antioxidant systems in response to IR.
Results
Liver steatosis downregulates PGC-1α expression and leads to a reduction in the levels of antioxidant proteins
To assess the role of PGC-1α in the response of the steatotic liver to IR, we first evaluated the impact of hepatic steatosis for PGC-1α expression and activity. To do this, C57BL/6 wild-type (PGC-1α+/+) mice were fed either a standard chow diet (control) or a high-fat diet (HFD) for 7 weeks. Although HFD had only a marginal effect on the whole body (Fig. 1A) and liver weight (Fig. 1B), after this time, it led to a substantial level of hepatic steatosis as evidenced by the marked paler color of the liver (Fig. 1B). The presence of macrovesicular steatosis in the liver of HFD-fed mice was confirmed by Oil Red O staining of frozen liver sections (Fig. 1C and Supplementary Fig. S1; Supplementary Data are available online at
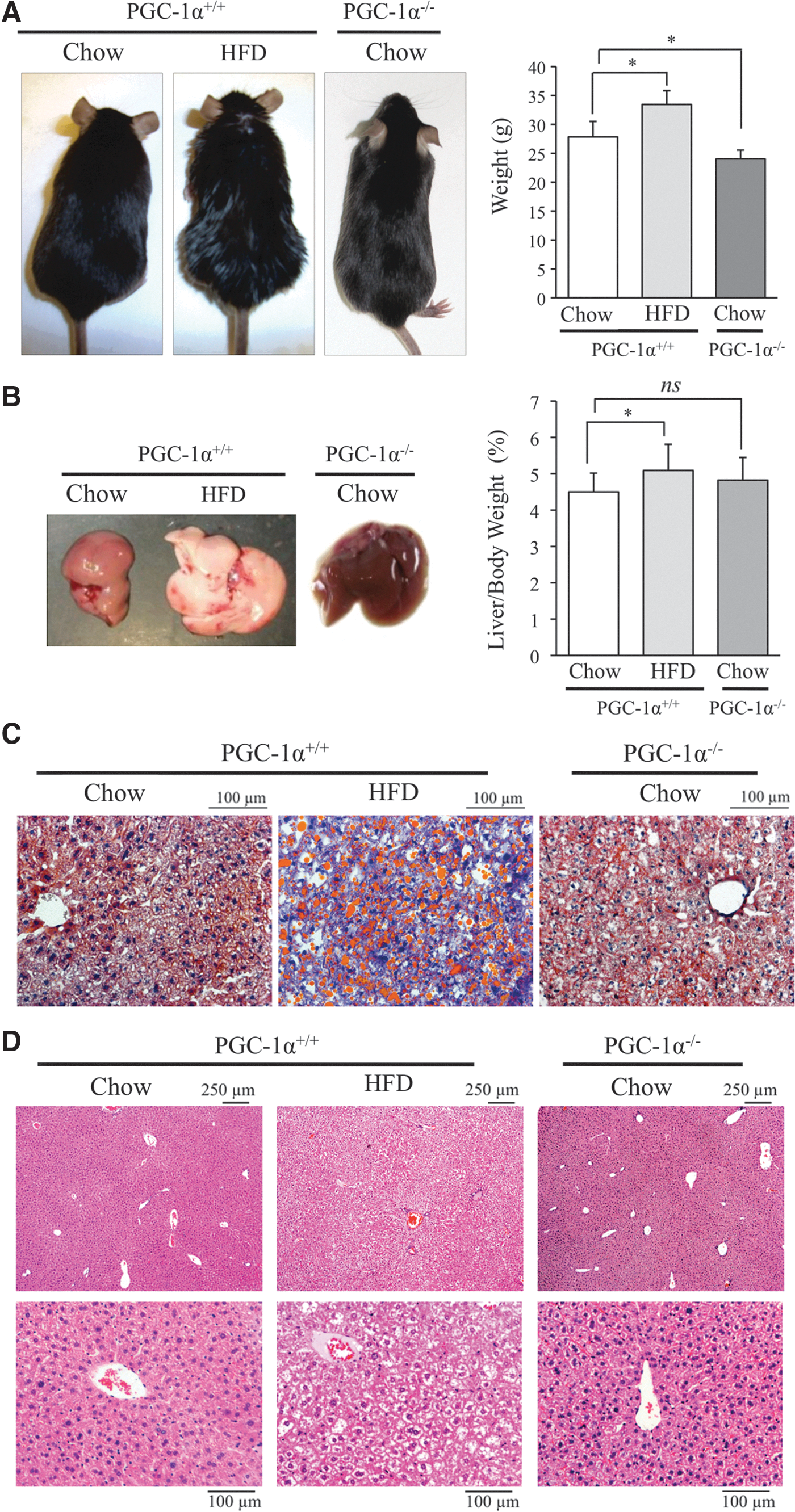
Analysis of liver histology by hematoxylin and eosin (H&E) staining showed a vacuolated pattern that is generally associated with liver steatosis in HFD-fed mice that was absent in the liver of chow-fed mice, and a preservation of the general structure of the parenchyma (Fig. 1D).
We next measured PGC-1α protein expression in the liver of mice on the two diets by Western blotting to assess the impact of HFD on PGC-1α. In agreement with a previous report (1), PGC-1α expression in the liver was significantly lower in HFD-fed mice than in mice on a chow diet (Fig. 2A and Supplementary Fig. S2A), indicating that steatosis downregulates its expression.
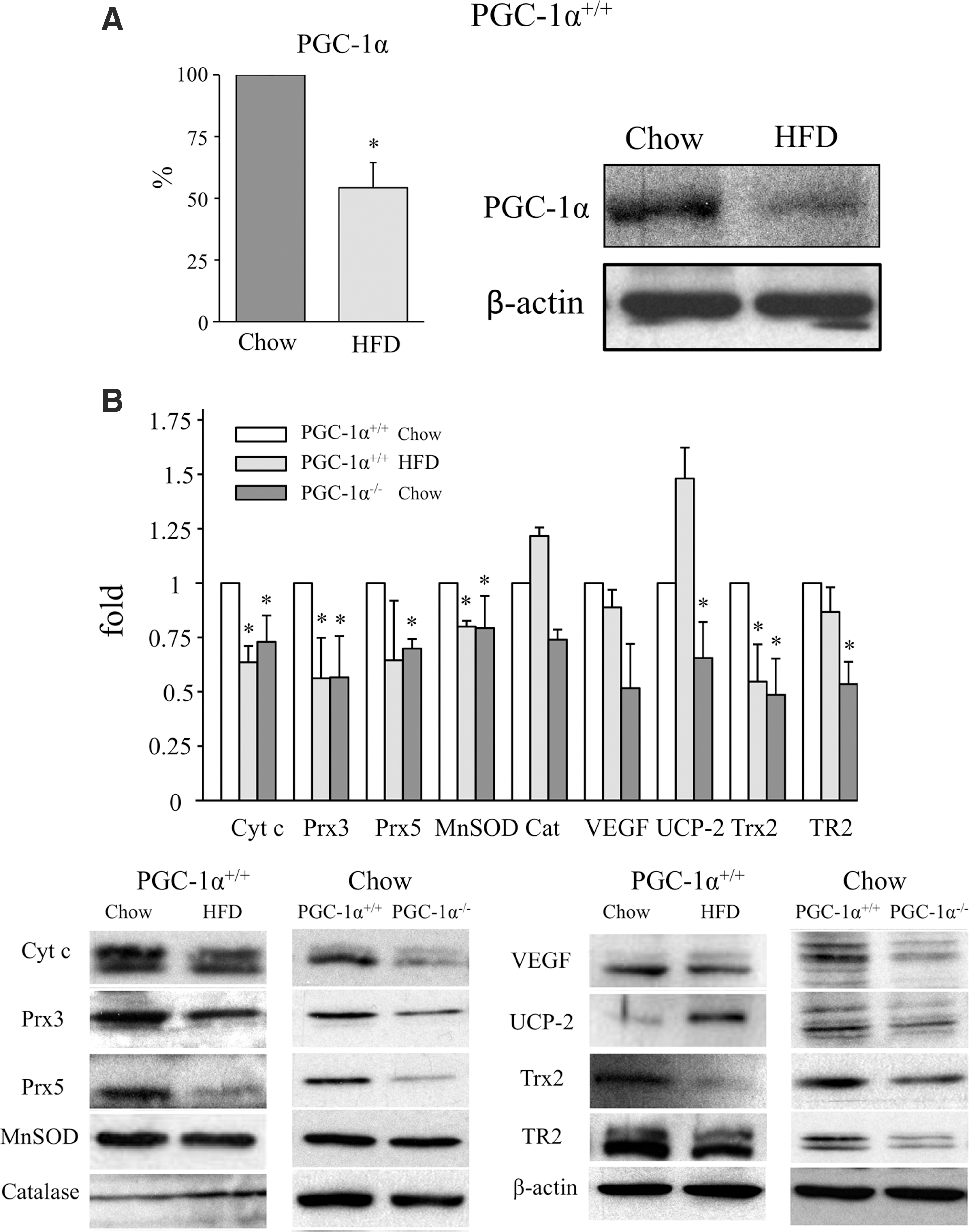
To confirm this finding, we evaluated the impact of liver PGC-1α downregulation on the expression of well-characterized PGC-1α-regulated proteins, including antioxidants. Thus, we compared the impact of liver steatosis in wild-type mice with that of mice with whole-body genetic deletion of PGC-1α. PGC-1α−/− mice are slightly leaner than PGC-1α+/+ mice and do not present liver steatosis under a standard chow diet (Fig. 1A, B). However, quantification of triglyceride (TG) liver content showed that both HFD-fed mice and chow-fed PGC-1α−/− mice had significantly higher levels than PGC-1α+/+ mice on a chow diet (Supplementary Fig. S3).
When wild-type mice on a chow diet were compared with HFD-fed mice and PGC-1α−/− mice, the expression of several antioxidant proteins, including MnSOD, Prx3, Prx5, Trx2, and TR2 together with other PGC-1α-regulated proteins (Cyt c and VEGF-A), was downregulated both in PGC-1α+/+ steatotic liver provoked by HFD and in PGC-1α−/− liver. By contrast, the expression of UCP-2 and catalase was induced in PGC-1α+/+ steatotic liver (Fig. 2B and Supplementary Fig. S3). UCP-2 has been previously reported to be induced in the steatotic liver by ROS and free lipids (14, 22, 46), and catalase is elevated in patients with non-alcoholic fatty liver disease and enhanced oxidative stress (34).
Collectively, these data suggest that the general decrease of antioxidant protein expression in the steatotic liver results from the downregulation of PGC-1α expression and are consistent with a possible role for PGC-1α in maintaining ROS homeostasis in the liver.
PGC-1α is induced in the liver after IR
Elevated generation of ROS after reperfusion has been demonstrated to play an important role in tissue damage by IR (12, 23, 40, 51 –53), and to be a key mediator of the adaptive changes that are associated with IPC (24, 25). It has also been proposed that the elevated levels of ROS produced by the steatotic liver are responsible for its enhanced vulnerability to IR damage (9). Because PGC-1α has been shown to be induced by ROS and hypoxia (3, 29, 42, 48), we next analyzed PGC-1α expression in the liver of wild-type mice on a chow diet and HFD after exposure to IR and IPC protocols.
We found that both PGC-1α mRNA and protein levels were increased by IR and even more so by IPC 6 h after reperfusion in chow-fed mice (Fig. 3A and Supplementary Fig. S4A). By contrast, PGC-1α expression was only marginally increased in the steatotic liver of HFD-fed mice (Fig. 3A and Supplementary Fig. S4A). At 4 h post-IR, PGC-1α levels did not show significant differences among the groups (Supplementary Fig. S5).
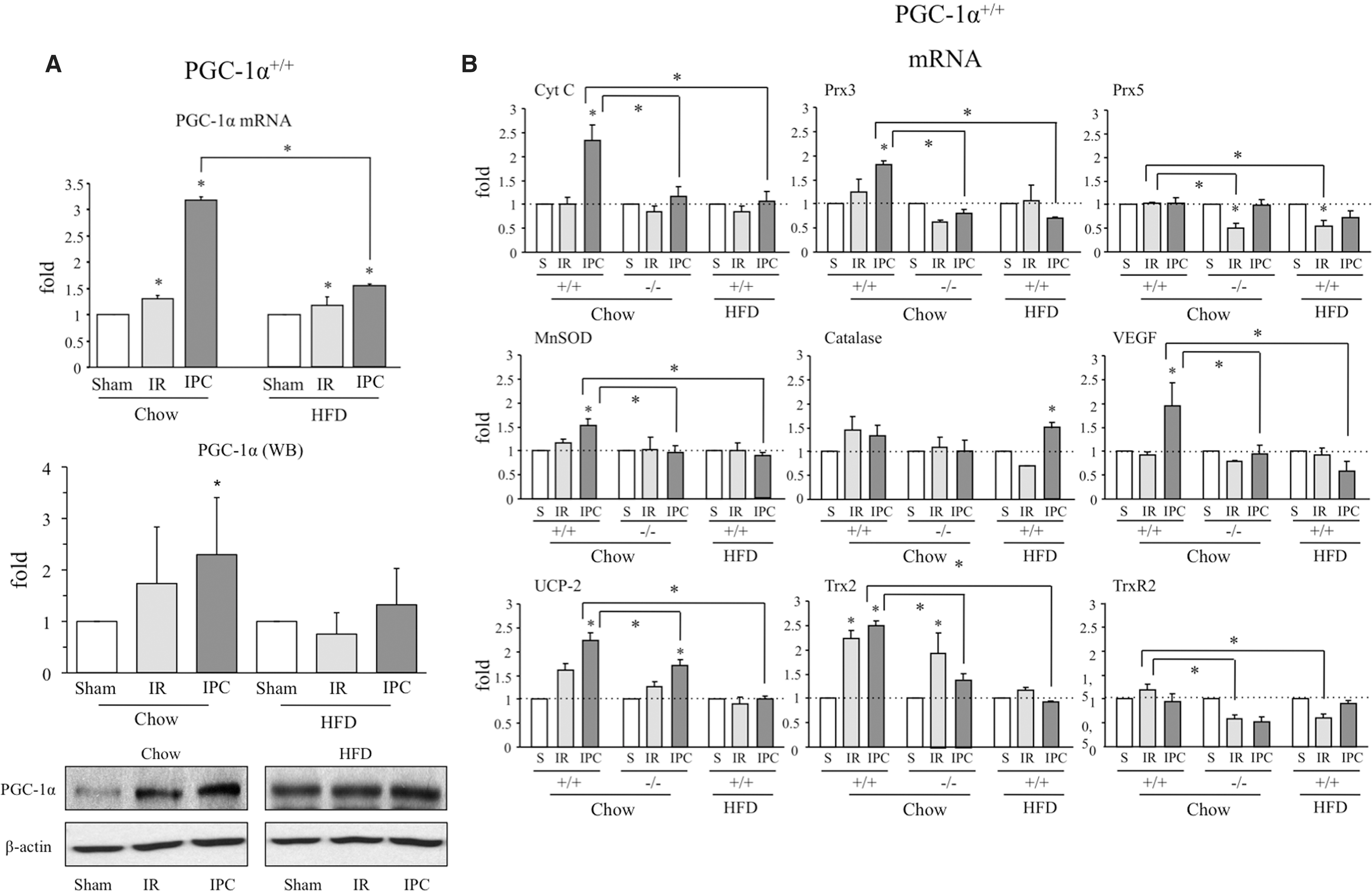
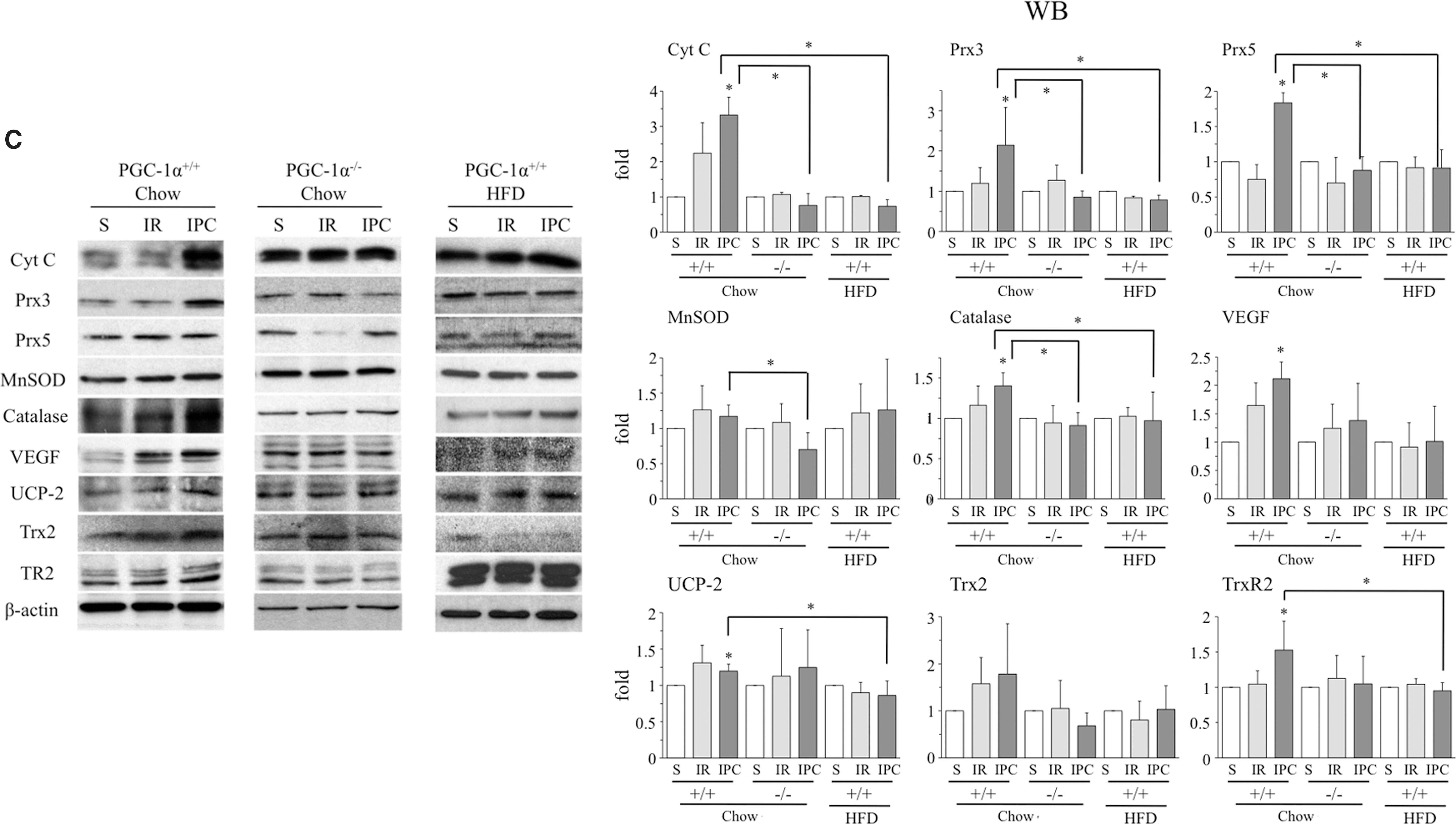
Since PGC-1α regulates antioxidant gene expression in the liver, we next evaluated whether PGC-1α induction was associated with an induction of antioxidant enzymes and other PGC-1α target genes after IR. Among the genes tested in wild-type mice on a chow diet, we detected a significant increase in the mRNA levels of UCP-2 and Trx2 at 24 h after IR, whereas IPC resulted in a significant increase in Cyt c, Prx3, MnSOD, VEGF, UCP-2, Trx2 mRNA, Cyt c, Prx3, Prx5, catalase, VEGF, UCP-2, and TrxR2 protein (Fig. 3B, C and Supplementary Figs. S4B and S6).
The enhanced vulnerability of the steatotic liver to IR damage has been proposed to be the consequence of elevated ROS levels. When we repeated the analysis in HFD-fed mice, the absence of PGC-1α induction in response to IR or IPC was associated with a significantly reduced induction of antioxidant enzymes in the steatotic liver (Fig. 3B, C and Supplementary Fig. S4D), pointing to a role for PGC-1α in this process.
To test this hypothesis, we analyzed antioxidant enzyme induction after IR and IPC in the liver of PGC-1α−/−mice. As anticipated, antioxidant induction was significantly reduced in PGC-1α−/− mice (Fig. 3B, C and Supplementary Fig. S4C). These results strongly suggest that PGC-1α mediates the induction of antioxidant enzymes in response to IR. Importantly, oxidative stress markers such as 4-HNE protein modification and oxidative DNA damage (8-OH-dG) were higher in the liver of PGC-1α−/− mice than in lean PGC-1α+/+ mice (Supplementary Fig. S7).
PGC-1α mediates IPC
Despite significant efforts, IPC has failed to show efficacy in improving the poor tolerance of the fatty liver to IR damage. Given that the enhanced susceptibility of the fatty liver to IR damage has been attributed to its high levels of ROS, we speculated that failure to induce PGC-1α and the consequent reduction in antioxidant defense could be related to the poor tolerance of the steatotic liver to IR and IPC protocols. To test this idea, we compared the liver damage induced by IR with or without prior IPC between PGC-1α−/− and PGC-1α+/+ mice with a normal or steatotic liver. Sham-treated animals were used to determine basal parameters.
Severity of IR injury was first assessed by determining plasma levels of the liver transaminases alanine aminotransferase (ALT/GPT) and aspartate aminotransferase (AST/GOT) 24 h after reperfusion. We found that prior IPC reduced the plasma concentration of the IR liver damage markers ALT/AST in PGC-1α+/+ mice with a normal liver, whereas it increased their concentration both in PGC-1α+/+ mice with a steatotic liver and in PGC-1α−/− mice (Fig. 4A). These results pointed to a requirement of PGC-1α for preconditioning of the liver and suggested that reduced PGC-1α expression in the steatotic liver may be responsible for the failure to induce tolerance to ischemic insult after an IPC protocol.
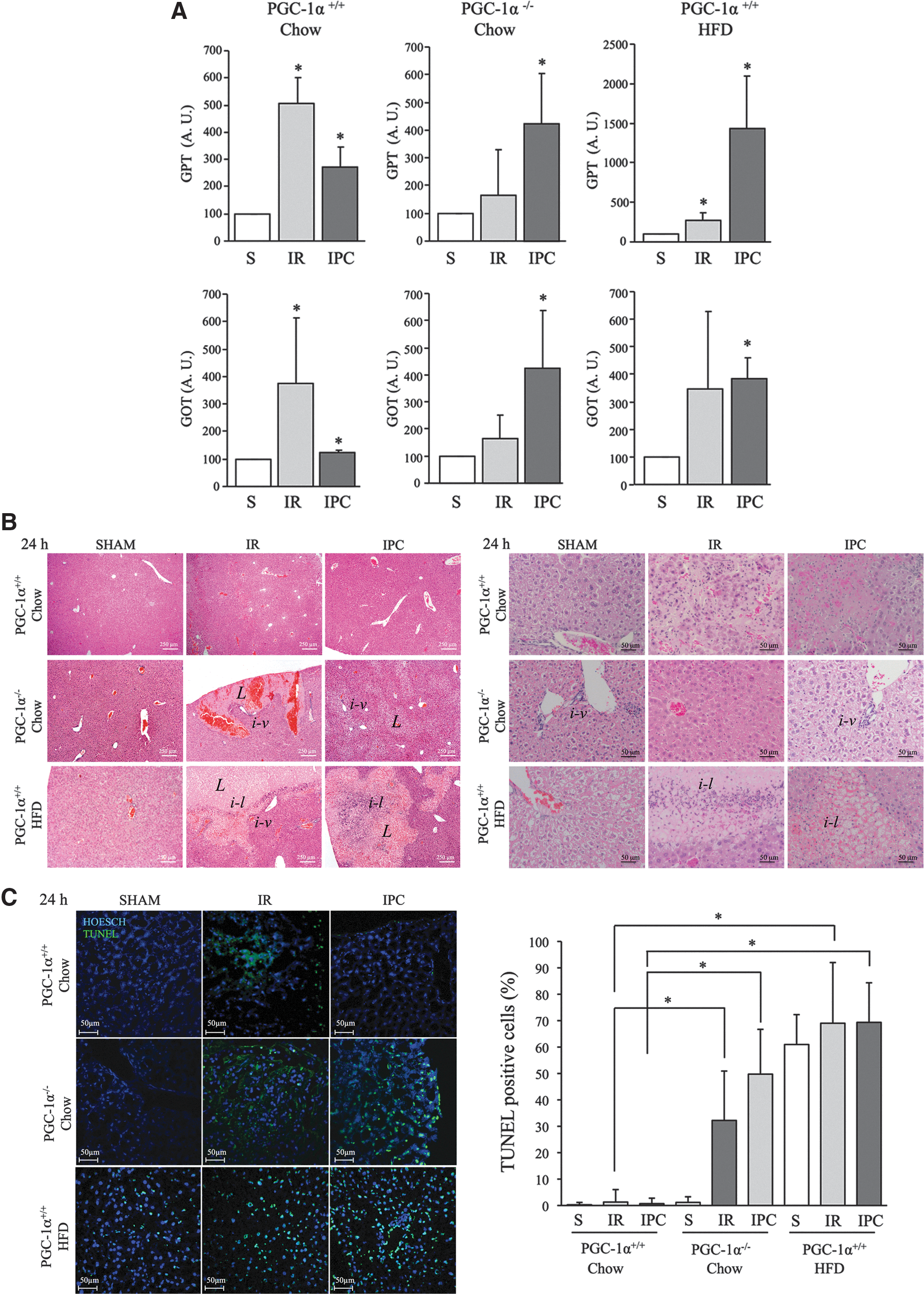
We next evaluated the extent of damage in liver parenchyma in the same groups of mice by histology 24 h and 1 week after IR. At 24 h, we expected to find evidence for hepatocyte cell death, collapsed microvessels, and inflammation, and at 1 week we expected to find evidence for ongoing remodeling, resolution of the inflammatory phase, and regenerative activity, including angiogenesis. H&E-stained sections from 24 h samples of liver from PGC-1α+/+ mice on a chow diet showed that the relatively large lesions (areas of disrupted parenchymal structure) detected after IR were significantly decreased by the IPC protocol. By contrast, in PGC-1α+/+ mice with a steatotic liver and in PGC-1α−/− mice, the IPC protocol increased the lesion size and perivascular inflammatory infiltrates could be identified (Fig. 4B).
However, a marked difference was noted between PGC-1α+/+ steatotic livers and PGC-1α−/− livers regarding the extent of inflammatory infiltration: PGC-1α+/+ steatotic livers had extensive parenchymal infiltrates after IR and IPC protocols, whereas infiltrates were more restricted to perivascular areas in PGC-1α−/− livers (Fig. 4B). Anti-F4/80 IHQ analysis of the number of macrophages present in the liver did not evidence any clear differences among the groups, but showed a statistically significant difference in the response to IR/IPC that could relate to a higher basal level of macrophages in PGC-1α−/− mice and a poorer inflammatory response to IR/IPC (Supplementary Fig. S8A, B).
Quantitative PCR of retro-transcribed cDNA (qRT-PCR) analysis of the main inflammatory mediators showed higher levels of IL-1β and iNOS and lower levels of IL-4 in PGC-1α−/− mice, which could suggest a shift in the M1/M2 ratio in PGC-1α−/− mice (Supplementary Fig. S8C).
Apoptotic cell death at 24 h was evaluated by terminal transferase dUTP nick end-labeling (TUNEL) staining of histological sections. Consistent with the earlier results, the extent of apoptotic cell death (measured as the number of TUNEL-positive cells) after IR in PGC-1α+/+ mice with a normal liver was significantly reduced by the IPC protocol, whereas in both PGC-1α+/+ mice with steatotic livers and in PGC-1α−/− mice, the IPC protocol did not reduce but rather increased the number of TUNEL-positive cells (Fig. 4C).
Moreover, the number of TUNEL-positive cells was higher in PGC-1α+/+ steatotic livers than in PGC-1α−/− livers, a result in accord with the findings of liver damage by plasma transaminase activity (Fig. 4C). Intriguingly, this difference did not seem to correlate with the size of the lesions, as determined by H&E staining (Fig. 4B). Altogether, these findings suggest that PGC-1α induction of antioxidant genes protects the liver from IR damage and is an important mediator of IPC.
Histological analysis of the liver at 1-week post-IR in PGC-1α+/+ mice on a chow diet revealed what appeared to be small fibrotic areas, which were essentially absent in the same group treated with an IPC protocol (Fig. 5A). Consistent with the results from 24 h post-IR, the IPC protocol did not reduce but rather increased the size of the remodeling area both in PGC-1α+/+ mice with a steatotic liver and in PGC-1α−/− mice (Fig. 5A), supporting the presumably protective role of PGC-1α in both acute and long-term IR injury in the liver.
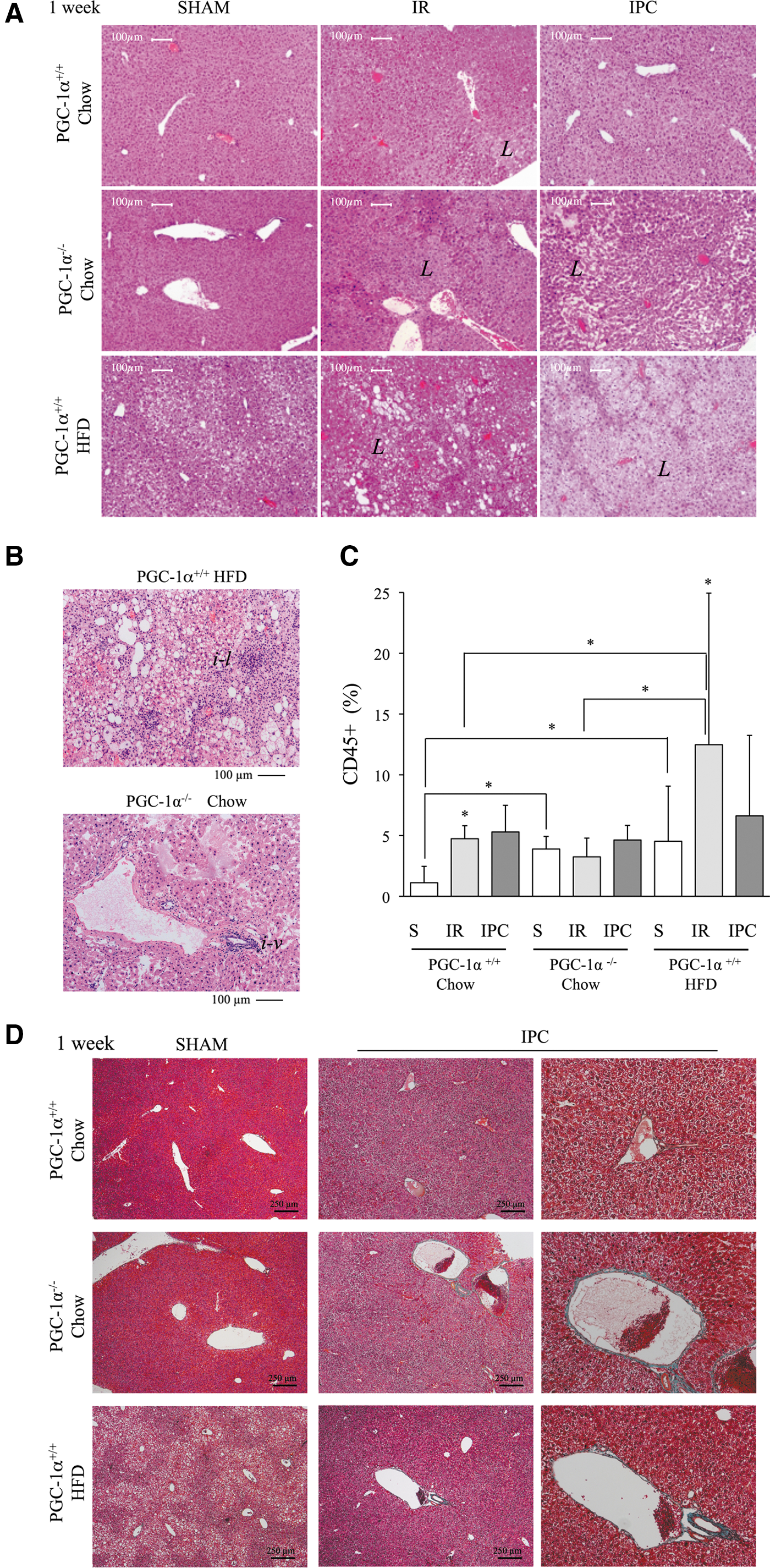
Nevertheless, closer inspection of the liver structure after the IPC protocol revealed a more marked disruption in PGC-1α−/− liver than in PGC-1α+/+ steatotic liver. Two structural differences were evident: Inflammatory cells showed a more dispersed pattern in PGC-1α−/− liver, with the majority of inflammatory foci presenting a perivascular location (Fig. 5B); in addition, the liver of PGC-1α−/− mice had larger blood vessels, which were frequently occluded, whereas vessels were generally smaller in PGC-1α+/+ mice with a steatotic liver and were loaded with erythrocytes (Fig. 5B). These observations likely relate to the low survival rate noted in the group of PGC-1α+/+ mice with a steatotic liver (70% died at day 5 post-IR), whereas all PGC-1α−/− mice survived the entire week despite extensive liver damage.
The extent of leukocyte infiltration 1-week post-IR was quantified in histological sections using the pan leukocyte marker CD45. In sham animals, the number of CD45+ cells was significantly higher in PGC-1α−/− and PGC-1α+/+ mice with a steatotic liver than in normal PGC-1α+/+ mice, suggesting that the low-grade chronic inflammation associated with steatosis is also present in PGC-1α−/− mice (Fig. 5C).
The IR protocol resulted in a significant increase in the number of CD45+ cells in both wild-type and steatotic PGC-1α+/+ mice, but it failed to induce a corresponding increase in PGC-1α−/− mice, suggesting a poor response to normal inflammatory stimuli in these animals that is consistent with the restricted perivascular distribution observed in histology (Fig. 5C). No significant differences were found in the number of CD45+ cells between the IR and IPC protocols in any animal group.
We next evaluated the extent of fibrosis by staining liver sections with Masson's trichrome. Vascular fibrosis was evident in large vessels in IPC-treated steatotic PGC-1α+/+ mice and in PGC-1α−/− mice, but not in the liver from wild-type mice. This was particularly prominent in PGC-1α−/− liver (Fig. 5D).
These results were confirmed by Sirius Red staining, which allowed the quantification of the total fibrotic area and the vascular media thickness and the qRT-PCR quantification of αSMA mRNA levels (Supplementary Fig. S9). These observations could be relevant in terms of IR injury since it has been suggested that IPC protects from IR injury, at least in part through the preservation of microcirculation (17). Taken together, these results confirm that PGC-1α plays a protective role against IR injury in the liver, likely due to its regulation of antioxidant gene expression, and it is also a mediator of IPC.
To test the capacity of PGC-1α to provide protection against IR injury, recombinant adenovirus that drives the expression of PGC-1α or a control adenovirus was administered by iv tail vein injection to steatotic PGC-1α+/+ mice and 72 h later, these animals were subjected to IR/IPC protocols. H&E analysis of histological sections at 1 week showed a reduced disruption of the parenchymal structure in Ad-PGC-1α mice than in control Ad-Shuttle mice. Moreover, the survival rate till 7 days post-IR was significantly higher in the control Ad-Shuttle group than in the PGC-1α overexpressing mice (Supplementary Fig. S10).
Reoxygenation drives PGC-1α-mediated induction of antioxidant genes
The results thus far indicate that PGC-1α is induced in response to IR in the liver. Since PGC-1α is induced by hypoxia and ROS in other systems, we next aimed at assessing whether PGC-1α regulation of antioxidant gene expression was associated with the hypoxic induction of PGC-1α or with the burst of ROS after reoxygenation. To do this, we established an in vitro protocol to analyze the response of primary mouse hepatocytes to hypoxia and reoxygenation. Considering that normal O2 tension in liver parenchyma is ∼3% (10), freshly isolated hepatocytes were maintained in a cell incubator at this tension and cells were serum starved overnight before the initiation of the experimental protocol.
To evaluate the effect of hypoxia only, hepatocytes were transferred to a hypoxic chamber at 1% O2 for 12 h. To evaluate the effect of reoxygenation, cells were first placed at 1% O2 and then incubated at 3% O2 for a further 6 h. After incubation, cells were collected and processed for mRNA and protein analysis. Results showed that the level of PGC-1α mRNA and protein expression was lower in cells incubated at 1% O2 than at 3% O2, but recovered after reoxygenation (Fig. 6A and Supplementary Fig. S11A). These results contrast with previous findings where the reference O2 tension was 21%. This apparent incongruity may relate to the well-established observation that cells at 21% O2 have a reduced respiratory capacity relative to those at lower oxygen tensions, presumably because the electron flux in the mitochondrial electron transport chain produces massive amounts of ROS (45).
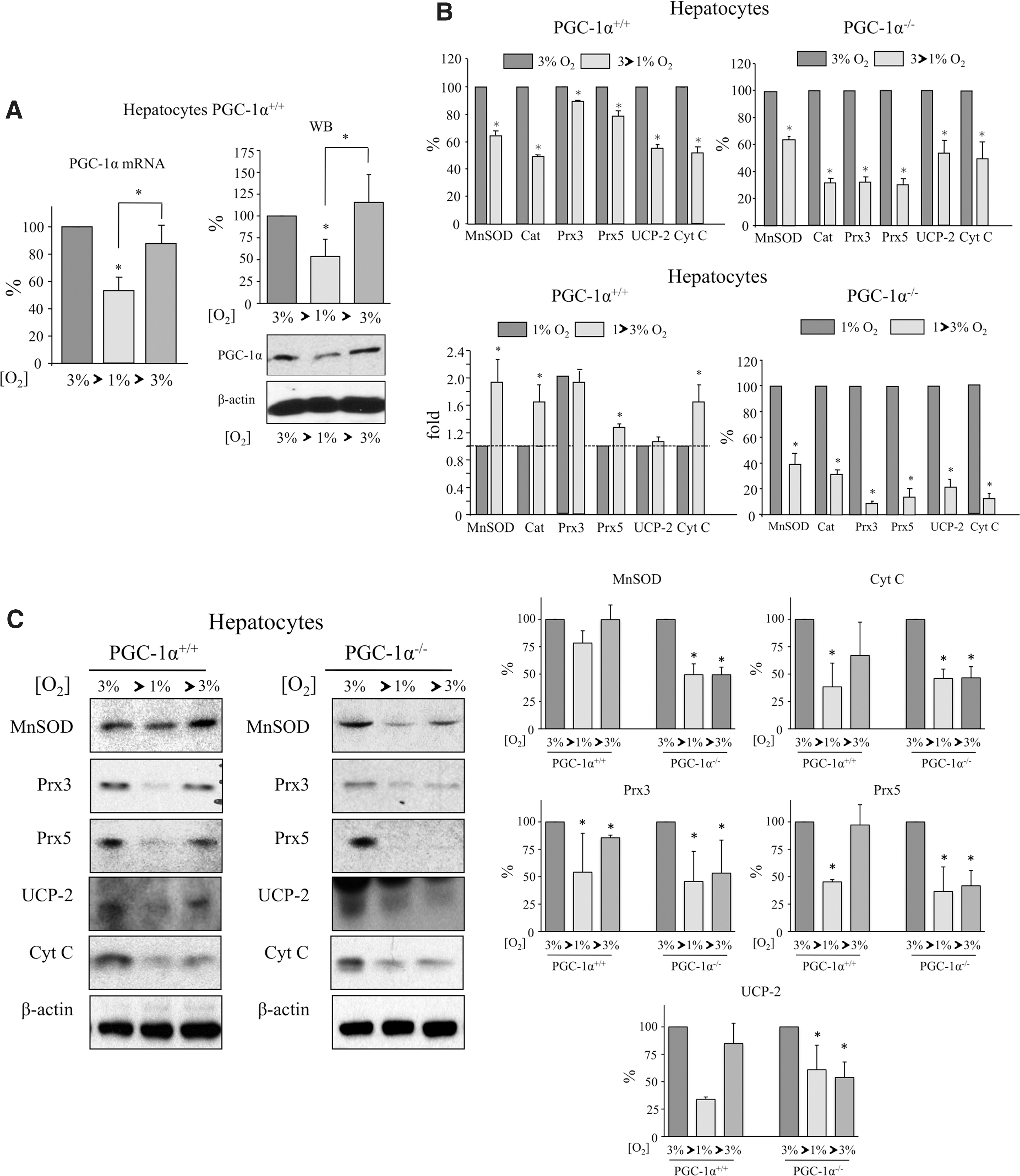
We then analyzed the response to hypoxia and the reoxygenation of PGC-1α target genes. Hypoxia downregulated mRNA (Fig. 6B) and protein levels (Fig. 6C and Supplementary Fig. S11) of antioxidant proteins and Cyt c in both PGC-1α+/+ and PGC-1α−/− hepatocytes, suggesting that antioxidant gene expression was modulated by hypoxia in a PGC-1α-independent manner (Fig. 6B). However, reoxygenation led to an induction in the mRNA and protein levels of antioxidants and Cyt c in PGC-1α+/+ hepatocytes, but this recovery was blunted in PGC-1α−/− hepatocytes (Fig. 6B, C and Supplementary Fig. S11). This result strongly suggests that PGC-1α is necessary for the recovery of antioxidant gene expression after reoxygenation.
Discussion
The mechanisms that mediate IR injury are complex and have been investigated for decades. It is well established that ROS are required for IPC and are also important mediators of IR-induced cell death in various organs, including the liver. The elevated level of oxidative stress in the steatotic liver is considered a key element in its enhanced sensitivity to IR damage. However, the inability of the steatotic liver to respond to IPC has so far puzzled researchers. In this study, we show that PGC-1α is an important mediator of IPC. In the context of liver steatosis, PGC-1α expression is downregulated and the resulting failure to restore antioxidant gene expression after reperfusion may underlie both the hypersensitivity of the steatotic liver to IR injury and its inability to respond adequately to IPC.
We found that IR induces PGC-1α expression in the normal liver, which is enhanced by IPC. This observation is consistent with a previous study showing that IR and IPC protocols induce PGC-1α levels in the heart (20). However, in the steatotic liver, PGC-1α basal levels are low and fail to be induced by IR and/or by preconditioning. Accordingly, IR induced PGC-1α-regulated antioxidant enzyme expression, and this induction was generally boosted by IPC in the normal liver but not in the steatotic liver. We used PGC-1α−/− mice to evaluate how its absence and the resultant reduction in antioxidant defense could impact liver IR injury. We found that in the extent of liver damage by IR, PGC-1α−/− mice behaved similarly to PGC-1α+/+ steatotic mice, with an enhanced sensitivity to IR damage and a loss of response to IPC.
These results demonstrate the protective role of PGC-1α activity in IR injury and suggest that this activity may be related to the induction of antioxidant systems. Furthermore, our findings suggest that the lack of induction of PGC-1α target genes in the steatotic liver could explain its poor response to IPC and its enhanced sensitivity to IR damage.
Finally, we aimed at elucidating the mechanism involved in the induction of PGC-1α and downstream antioxidant genes in response to changing oxygen tensions. Using a hypoxia-reoxygenation protocol in primary hepatocytes, we found that PGC-1α was downregulated by hypoxia and upregulated by subsequent reoxygenation, possibly suggesting that PGC-1α could be activated in vivo during the reperfusion phase.
Importantly, when we examined antioxidant gene expression in PGC-1α+/+ and PGC-1α−/− hepatocytes, we noted that although hypoxia resulted in a general reduction in the expression of antioxidant genes in both groups, their induction by reoxygenation was generally blunted in PGC-1α−/− hepatocytes, indicating that PGC-1α does not mediate the hypoxic downregulation of those genes but is rather involved in their reactivation during the reoxygenation phase. Thus, PGC-1α activity could be particularly relevant in vivo to sustain antioxidant gene expression after organ reperfusion.
Interestingly, metformin administration has been shown to have protective effects against IR injury in the heart (19), an effect that is dependent on the activation of AMPK and correlates with the induction of PGC-1α. This observation is particularly relevant while taking into account that regulation of PGC-1α by AMPK and metformin administration has been amply demonstrated to be functionally relevant in the liver (8).
In conclusion, we show that PGC-1α is a mediator of IPC that is downregulated in the steatotic liver, blunting the induction of protective antioxidant enzymes. In light of the general failure of classical antioxidant treatments to translate into clinical settings, targeting PGC-1α activity and/or expression might be a therapeutic option to improve the acute failure of steatotic liver transplants.
Materials and Methods
Animals
Male C57BL/6 and C57BL/6 PGC-1α−/− mice were used in this study. Mice at 8–10 weeks of age were placed on a standard chow diet or an HFD (Harlan TD88137) for 7 weeks. Animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the CNIC and the CSIC. All procedures conformed to the Declaration of Helsinki. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratoriy Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (No. 86-23 revised 1985).
Cell culture
Primary hepatocytes from 12- to 16 week-old female C57BL/6 wild-type and PGC-1α−/− mice were isolated and cultured as described (38). Dispersed cells were seeded onto collagen-coated plates (0.2% gelatin, 1% collagen) and cultured in Williams E Medium containing 10% fetal bovine serum, 2 mM L-glutamine, 100 nM dexamethasone, 100 nM insulin, and antibiotics. Fresh medium was added 4 h after plating. In some experiments, hepatocytes were cultured in a humidified hypoxia incubator at 37°C with 3% O2 and 5% CO2 for 24 h and serum starved overnight. Hepatocytes were then transferred to a hypoxia chamber set at 3% or 1% O2 for 12 h. To test the effect of reoxygenation, hepatocyte cultures were re-exposed to 3% O2 for 6 h.
Liver IR and IPC
An established protocol of total warm hepatic IR and IPC was used (33), with minor modifications. For the IR protocol, an atraumatic clip was applied to interrupt the blood flow to the liver lobes. The clip was removed after 30 min to restore blood flow. The IPC protocol involved 5 min of ischemia followed by 10 min of reperfusion before 30 min of ischemia. Mice were anesthetized with 2% isofluorane inhalation. Buprenorphine (0.1 mg/kg) was administered subcutaneously 6 h before surgery and every 24 h for 3 days after surgery for analgesia. Experimental groups: (1) Sham operated, PGC-1α+/+ Chow, n = 13; PGC-1α+/+ HFD, n = 8; PGC-1α−/− Chow, n = 11. (2) IR, PGC-1α+/+ Chow, n = 13; PGC-1α+/+ HFD, n = 8; PGC-1α−/− Chow, n = 11. (3) IPC, PGC-1α+/+ Chow, n = 13; PGC-1α+/+ HFD, n = 8; PGC-1α−/− Chow, n = 11.
Adenoviral infection
Ad-Shuttle and Ad-PGC-1α have been previously described (48); 1010 pfu in PBS was injected through the tail vein. Seventy-two hours after the infection, the mice were subjected to laparotomy and total warm hepatic IR and IPC procedures or were sham operated.
Histological analysis
Liver sections were fixed in 10% buffered formalin and embedded in paraffin or in OCT™ (optimal cutting temperature). For histology, 4-μm sections were stained with H&E. To assess the presence of liver infiltrating macrophages and neutrophils, 6-μm sections were immunostained with an anti-CD45 antibody (30-F11, No. 553081; BD Biosciences), followed by incubation with a fluorescent tagged secondary antibody. Positive cells were counted from a random selection of × 10 images that were acquired with a Nikon fluorescence microscope by using ImageJ software (NIH). Nuclei were stained with TO-PRO-3 or Hoechst 33258.
Fibrosis analysis
Four-micrometer sections were stained with Masson's trichrome and Sirius Red. For Sirius Red staining, 4 μm paraffin-included slides were de-waxed in xylol, hydrated with serial washes of ethanol (100%, 96%, and 70%), and stained with Sirius Red Solution for 1 h. Then, they were washed in acidified water and dehydrated with ethanol. Photographs were taken with a Nikon 90i microscope (Nikon) and analyzed with ImageJ (NIH).
Hepatic TG content was determined as described (47). In brief, hepatic lipids were extracted as described (6). After purification, lipids were re-suspended in isopropyl alcohol and TG were analyzed with a colorimetric kit (Biosystems). Hepatic TG content was also evaluated by Oil Red O staining of liver cryosections.
TUNEL assay
The In Situ Cell Death Detection Kit Fluorescein (Roche) was used to label apoptotic cells in frozen histological sections by following the manufacturer's instructions. Images were acquired with a fluorescence microscope. The presence of nicks in the DNA was identified by the addition of fluorescein-labeled dUTPs by the enzyme terminal deoxynucleotidyl transferase (TdT). Apoptotic nuclei were counted in a random selection of × 10 images that were acquired with a Nikon fluorescence microscope by using ImageJ software.
Immunohistochemistry
Four-micrometer paraffin-included slides were de-waxed in xylol and hydrated with serial washes of ethanol (100%, 96%, and 70%). Antigen retrieval was made with citrate buffer in a microwave for 5 min, and endogenous peroxidase activity inhibition was done with 0.3% H2O2 in methanol for 30 min. The slides were blocked by following the manufacturer's instructions of the ABC Kit (Vector) and incubated with the primary antibody O/N: 8-OH-deoxyguanosine (1:100, NB600-1508; Novus Biologicals) or F4/80 (1:150, MCA497; AbD Serotec). For secondary antibody and DAB incubation, ABC Kit and DAB Kit (Vector) were used, respectively. Photographs were taken with a Nikon 90i microscope (Nikon) and analyzed with ImageJ (NIH).
Determination of plasma transaminase
Blood samples were collected by puncture of the caudal cava vein, and enzymatic determinations of alanine aminotransferase (GPT) and aspartate aminotransferase (GOT) in plasma were performed by using diagnostic kits (GN 41125, GN 40125 RAL) following the manufacturer's instructions.
RNA isolation and qRT-PCR
Total mRNA was isolated from frozen samples of liver tissue that were homogenized in 1 ml of TRIZOL (Thermo Fisher Scientific). The protocol for qRT-PCR and several primers used have been previously described (7, 29, 38, 48).
Non-previously described qPCR oligonucleotides:
αSMA: Forward 5′-GCTGAAGTATCCGATAGAAC ACG-3′
Reverse 5′-GGTCTCAAACATAATCTGGGTCA-3′
IL-4: Forward 5′-CGAAGAACACCACAGAGAGTGA GCT-3′
Reverse 5′-GACTCATTCATGGTGCAGCTTATCG-3′
iNOS: Forward 5′-GAGCTGGGCTGTACAAACCTT-3′
Reverse 5′-CATTGGAAGTGAAGCGTTTCG-3′
TNFα: Forward 5′-ATGAGAAGTTCCAAATGGCC-3′
Reverse 5′-TGGTTTGCTACGACGTGGG-3′
IL-1β: Forward 5′-GCTGAAAGCTCTCCACCTCA-3′
Reverse 5′-AGGCCACAGGTATTTTGTCG-3
Arg-1: Forward 5′-CTCCAAGCCAAAGTCCTTAGAG-3′
Reverse 5′-AGGAGCTGTCATTAGGGACATC-3′
Protein extraction and Western blotting
Whole liver extracts were prepared from frozen samples of liver tissue that were homogenized in 1 ml of lysis solution and analyzed by Western blotting as previously described (28).
Antibodies used for Western blot
HNE (1:1000, HNE11-S; Alpha Diagnostic). All other antibodies used have been previously described (7, 29, 38, 48).
Image analysis
ImageJ software was used to analyze Western blots and to determine the number of nuclei in histological section fields.
Statistics
Data are expressed as means ± SD. Statistical significance was evaluated by one-way analysis of variance or by the two-tailed unpaired t-test. ANOVA was used to evaluate whether the effect of IR/IPC was affected by the PGC-1α genotype and/or diet. t-test was used for simple direct comparisons (e.g., PGC-1α+/+ vs. PGC-1α−/−). Values were considered statistically significant at p < 0.05; n ≥ 3 for all experimental conditions.
Footnotes
Acknowledgments
The authors thank Dr. Santiago Lamas (CBMSO, Madrid, Spain) for a careful reading of the article and Dr. Mercedes Ricote (CNIC, Madrid, Spain) for qPCR primers to quantitate Arg-1 and IL-1β mRNA levels. Editorial support was provided by Dr. Kenneth McCreath. This work was supported by grants from the Spanish “Ministerio de Economía y Competitividad” (MINECO) and FEDER funds (Grant Nos. SAF2012-37693, SAF2015-63904-R [MINECO/FEDER], CSD 2007-00020 & SAF2015-71521-REDC, SAF2015-65267-R), the “Comunidad Autónoma de Madrid” (grant No. S2010/BMD-2361), and CIBER (ISCIII).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.