
Abstract
Aims:
Iron overload (IO) is a life-threatening complication of chronic hemolytic disorders such as β-thalassemia. IO results in severe cellular oxidative damage, leading to organ failure. Peroxiredoxin-2 (Prx2), a typical 2-cysteine-(Cys)-peroxiredoxin, is an important component of the cytoprotective system, but its response to IO is still to be fully defined.
Results:
We studied the effects of IO on Prx2-knockout mice (Prx2−/−). The absence of Prx2 enhanced toxicity due to IO on erythropoiesis. We found that IO failed to induce the typical hepcidin (Hamp) upregulation in Prx2−/− mice due to its failure to activate the signal transducer and activator of transcription-3 (STAT3) with intact Jak2 signaling. In Prx2−/− mice, the loss of Hamp response was also observed after administration of a single dose of oral iron. When lipopolysaccharide (LPS) was used to explore IL6-STAT3 activation in Prx2−/− mice, STAT3 activation and Hamp upregulation were once again defective. Treatment with PEP-fusion-recombinant-Prx2 (PEP Prx2) significantly increased STAT3 activation with upregulation of Hamp expression in both IO- and LPS-exposed Prx2−/− mice. We also confirmed the beneficial effects of PEP Prx2 on Hamp expression through STAT3 activation in β-thalassemic mice.
Innovation:
We propose that Prx2 plays a key role in responding to cytotoxicity of IO, directly targeting STAT3-transcriptional factor in a Jak2-independent fashion and regulating Hamp in response to canonical stimuli.
Conclusion:
Collectively, our data highlight a novel role of Prx2 in iron homeostasis. Prx2 is a key cytoprotector against IO that is induced either by iron supplementation or due to chronic hemolysis as in β-thalassemia. Prx2 is required to support STAT3 transcriptional activity and regulation of Hamp expression. Antioxid. Redox Signal. 28, 1–14.
The absence of peroxiredoxin-2 (Prx2) enhances toxicity due to iron overload as a consequence of lack of hepcidin expression. Prx2 is a key cytoprotector of the active form of signal transducer and activator of transcription 3 (STAT3) in response to canonical stimuli for upregulation of hepcidin expression. Recombinant fusion protein PEP Prx2 controls oxidative stress, modulating targets that are sensitive to redox and iron homeostasis through the hepcidin-erythroferrone axis.
Introduction
I
Since liver is the main site for iron accumulation, redox-sensitive transcription factor such as signal transducer and activator of transcription 3 (STAT3) might be activated in response to iron accumulation and oxidative stress (19, 39 –41, 47). Recent studies have noted that physiological levels of ROS promote STAT3 activation and upregulate Hamp expression, independently of the well-documented role of IL-6 signaling in regulating Hamp expression (39). Importantly, high reactive oxygen species (ROS) levels downregulate Hamp expression (40). It is of note that STAT3 transcription activity has been recently linked to peroxiredoxin-2 (Prx2) in the presence of low levels of H2O2 (47).
Prx2, a typical 2-cysteine (Cys) peroxiredoxin, is part of the cytoprotective systems involved in the tight control of the levels of ROS generated during a cell's life span or related to cellular stress such as in IO (9, 21, 29, 35). Since ROS might also function as a second messenger through transient oxidation of cysteine residues in signaling targets, it has been proposed that the redox state of cysteine residues of Prxs might control the redox state of partner proteins, possibly through the on-off switching mechanism of proteins that are sensitive to redox conditions (3, 47).
Recently, we reported that Prx2 is a key anti-oxidant system in both stress and pathologic erythropoiesis characterized by oxidative damage, such as β-thalassemia (8, 9, 15, 16, 34). The ablation of Prx2 in β-thalassemic mouse genotype (Prx2−/−Hbb3th/+) worsened the ineffective erythropoiesis with increased ROS accumulation, amplified cell apoptosis, and increased extra-medullary erythropoiesis compared with β-thalassemic (Hbb3th/+) mice (34). In addition, Prx2−/−Hbb3th/+ mice display higher iron accumulation in the liver compared with either Prx2−/− or Hbb3th/+ mice (34). Administration of Prx2 fused to cell-penetrating carrier PEP (cell-penetrating peptide; PEP Prx2) significantly ameliorated anemia in Hbb3th/+ mice, whereas it decreased the extent of liver and spleen IO (34).
To further explore the mechanistic basis for the role of Prx2 in iron homeostasis, in this study, we explored the effects of IO in mice genetically lacking Prx2. The absence of Prx2 results in severe anemia and is associated with the lack of Hamp induction in response to either IO or after the administration of a single dose of oral iron. The lack of Hamp induction in response to IO is through the loss of STAT3 activation. Treatment with fused recombinant PEP Prx2 restored both STAT3 activation and Hamp expression in response to IO. These findings were also confirmed in Hbb3th/+ mice, used as a mouse model of β-thalassemia. We, thus, identified Prx2 as a novel regulator of iron homeostasis, acting through activation of STAT3 and upregulation of liver Hamp expression in response to either IO or acute iron (AI) administration. These findings suggest a novel therapeutic strategy to control clinical consequences of IO through modulation of Prx2 activity.
Results
Prx2 is an important cytoprotective factor in modulating erythropoiesis under condition of excess iron
Ablation of Prx2 (Prx2−/− mice) is associated with early development of a more severe anemia compared with wild-type mice after IO (Fig. 1A). As shown in Figure 1A, Prx2−/− mice showed a rapid drop in hemoglobin (Hb) levels with an associated increase in mean cell volume (MCV), red cell distribution width (RDW; Supplementary Fig. S1A; Supplementary Data are available online at
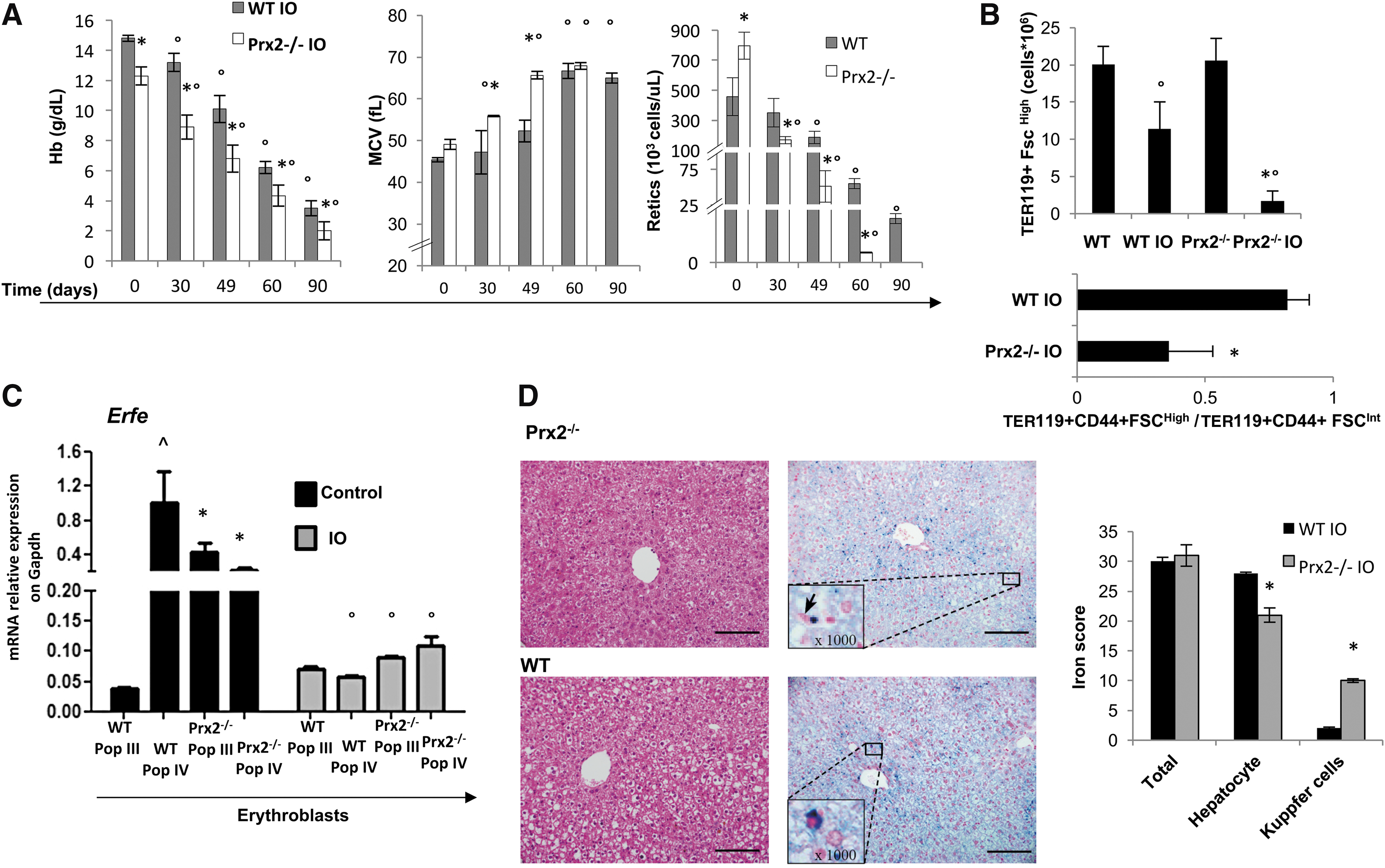
Since erythroferrone (Erfe; Fam132b) has been linked to iron homeostasis and erythropoiesis (23, 24), we evaluated Erfe expression in sorted erythroblasts from both mouse strains. We used the CD44-TER119 strategy to isolate and purify erythroid cells (2, 28, 34). In wild-type mice on a standard diet, we found upregulation of Erfe gene expression in orthochromatic erythroblasts (Pop IV) compared with polychromatic erythroblasts (Pop III) (Fig. 1C). Conversely, we observed upregulation of Erfe expression in Prx2−/− polychromatic erythroblasts (Pop III) and downregulation of Erfe in Prx2−/− orthochromatic erythroblasts (Pop IV) compared with wild type (Fig. 1C). This altered expression of Erfe might be related to ineffective erythropoiesis in Prx2−/−, which is similar to that described in β-thalassemic mice (Hbb3th/+) (34). Our data are in agreement with upregulation of Erfe previously reported in polychromatic erythroblasts from Hbb3th/+ mice (23). IO promoted downregulation of Erfe to a similar extent in both mouse strains (Fig. 1C), and this is associated with almost undetectable soluble levels of Erfe in both IO mouse strains (data not shown). Collectively, these data suggest that the absence of Prx2 is associated with amplified iron cytotoxicity.
Iron-overloaded Prx2−/− mice show reduced STAT3 activation with downregulation of hepcidin expression
We then evaluated liver iron concentration (LIC), which was similarly increased in both mouse strains exposed to IO (data not shown). However, Pearl's staining analysis revealed different patterns of iron distribution in cellular components of the liver. In Prx2−/− mice, iron accumulated in hepatocytes and Kupffer cells, whereas iron deposits were present only in hepatocytes of wild-type liver (Fig. 1D). This may be related to hyper-activation response of macrophages lacking Prxs, as previously shown in other models (18, 43). Iron distribution pattern in the spleen of Prx2−/− mice was similar to that of the liver (Supplementary Fig. S2A). Oxyblot analysis and quantification of malondialdehyde (MDA) levels were used to evaluate the degree of protein oxidation and lipid peroxidation, respectively, in the liver. Both oxidative processes were significantly increased in IO Prx2−/− compared with wild-type mice (Fig. 2A). The levels of ferritin H chain, the expression of which increases in response to IO and oxidative stress (7, 45), were higher in Prx2−/− mice compared with wild-type mice (Supplementary Fig. S2B).
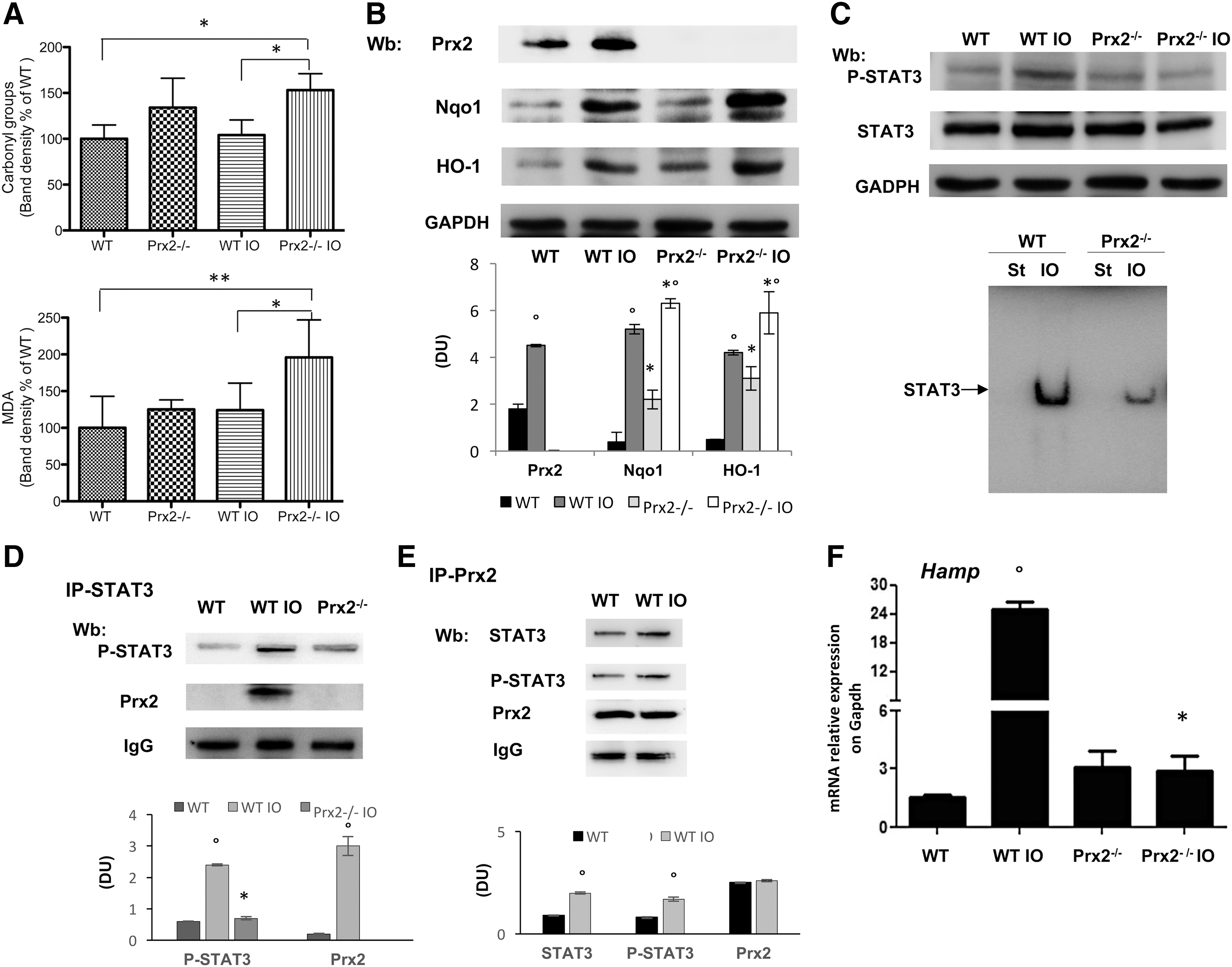
Expression of Prx2 in liver was significantly increased in IO wild-type mice compared with animals under a standard diet, indicating that Prx2 is part of the adaptive cellular response of the liver to IO (Fig. 2B and Supplementary Fig. S2C). Indeed, we found an increase in other known cytoprotective systems, including Nqo1 and HO-1 (Fig. 2B and Supplementary Fig. S2B). It is interesting to note that upregulation of Nqo1 and HO-1 in response to IO was higher in Prx2−/− mice than in wild type, whereas basal levels were similar in the two strains (Fig. 2B and Supplementary Fig. S2C). These findings suggest that in the absence of Prx2 there is a severe oxidative stress in the liver of Prx2−/− compared with IO wild-type mice due to IO.
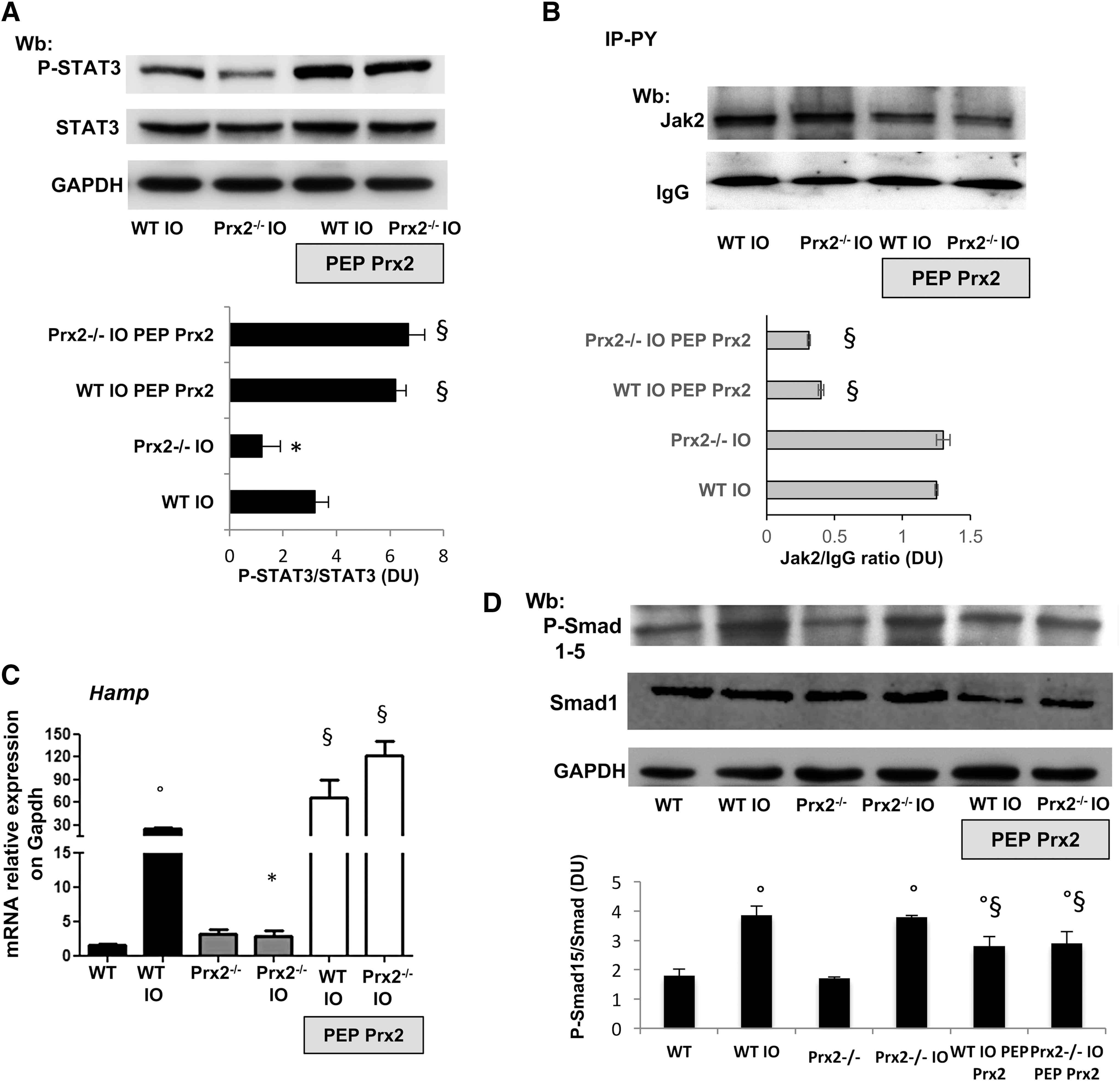
Since Prx2 has been recently linked to STAT3 function and oxidative stress, we evaluated the activity of STAT3, which contributes to Hamp expression (39, 40). In IO wild-type mice, we found increased activation of STAT3 compared with mice on a standard diet. However, this activation was absent in IO Prx2−/− mice (Fig. 2C, upper panel; Supplementary Fig. S2C, D), as confirmed by different methodologic approaches (DNA-binding activity of STAT3, Fig. 2C, lower panel; immune-precipitation of STAT3, then revealed with P-STAT3, Supplementary Fig. S2F). No major change in liver IL-6 expression was detectable in either of the IO mouse strains (data not shown, see also Supplementary Fig. S8A). Jak2 activation in response to IO was similar in liver from both mouse strains (data not shown, see also Fig. 5B). This finding indicates the integrity of the Jak2-mediated signaling pathway in IO Prx2−/− mice, but it decreased STAT3 phosphorylation in the absence of Prx2. Indeed, we found that Prx2 co-immunoprecipitated (IP) with STAT3 and phospho-STAT3 (P-STAT3) in livers from WT exposed to IO compared with vehicle-treated animals, whereas, as expected, such a complex was undetectable in IP from liver of Prx2−/− IO mice (Fig. 2D and Supplementary Fig. S3A). Previous in vitro studies documented a functional linkage between Prx2 and STAT3 and between STAT3 activation and H2O2 levels (39). In a hepatic cell line (Huh7), escalating doses of H2O2 promoted Prx2 dimerization to cope with H2O2-mediated oxidative stress (Supplementary Fig. S3C). Under the same experimental condition, we confirmed STAT3 activation in response to H2O2 and upregulation of Hamp in agreement with previous studies (39, 47). To verify the association between STAT3 and Prx2 in our IO model, we evaluated whether active STAT3 co-immunoprecipitates with an equal amount of Prx2 from liver of WT exposed to vehicle or IO. As shown in Figure 2E, increased amounts of active STAT3 are associated with Prx2 in IO animals compared with vehicle treatment (see also Supplementary Fig. S3B). As expected, we noted upregulation of Hamp in IO wild-type mice, whereas no change in Hamp expression was observed in IO Prx2−/− compared with standard diet Prx2−/− mice (Fig. 2F). To better understand the role of Prx2 in the signaling pathway involved in Hamp regulation, we evaluated Smad 1/5 activation and Id1 expression in IO mice (25). Smad activation and upregulation of Id1 in response to IO was similar in both mouse strains (Supplementary Fig. S3C and Fig. 3A). We found reduced staining for Fpn1 in C-duodenal tissue from IO WT mice compared with standard diet, whereas no changes were observed in Prx2−/− mice with and without IO (Fig. 3B).
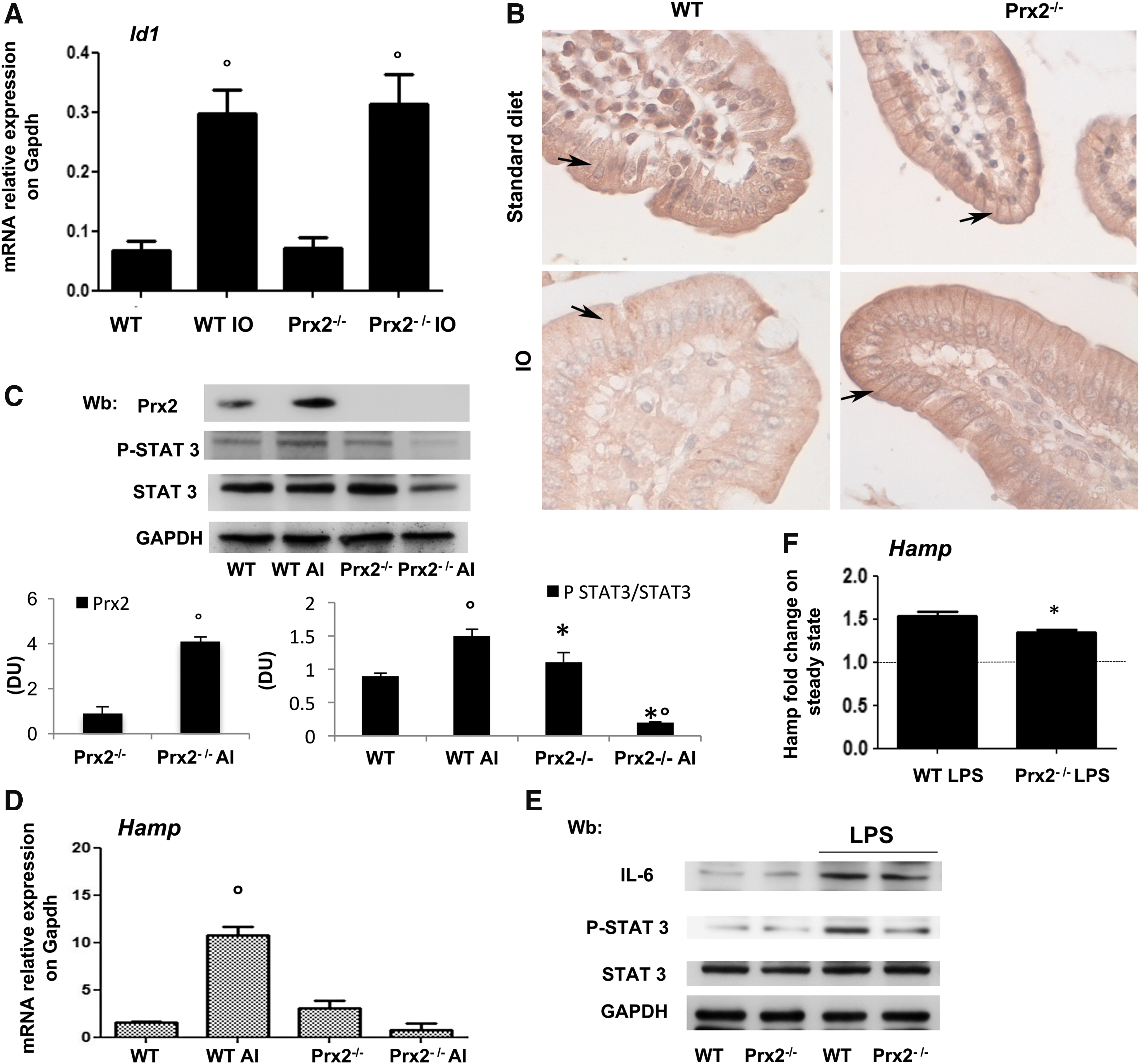
Prx2 is required to ensure adequate hepcidin expression in response to canonical stimuli requiring STAT3 activation
To better understand the interplay between Prx2 and Hamp in response to canonical stimuli, we studied the effects of a single dose of oral iron (AI) or lipopolysaccharide (LPS). Prx2 liver expression significantly increased in WT mice at 24 h after AI (Fig. 3C and Supplementary Fig. S4A). STAT3 was activated in WT but not in Prx2−/− mice treated with AI (Fig. 3C and Supplementary Fig. S4A). No change in IL-6 levels was observed in either of the mouse strains exposed to AI (data not shown). As expected, we found significantly increased Hamp expression in liver from WT mice but not in Prx2−/− mice (Fig. 3D and Supplementary Fig. S4A). No changes were noted in plasma levels of Erfe in response to AI administration in either of the mouse strains (data not shown). Id1 expression was similar in both mouse strains after AI (data not shown).
When Prx2−/− mice were treated with LPS, a known inflammatory trigger of IL-6–STAT3 system (20, 32, 39), as shown in Figure 3E, LPS induced IL-6 expression and STAT3 activation in WT mice but not in Prx2−/− animals (see also Supplementary Fig. S4B). Hamp expression in response to LPS was lower in Prx2−/− mice compared with WT animals (Fig. 3F).
We then evaluated the effects of low-iron (LI) diet on hematologic phenotype and Hamp expression in both mouse strains. Mice were studied at 3 and 7 weeks of LI. Hamp expression was significantly reduced in both mouse strains at 3 and 7 weeks of LI (Supplementary Fig. S5A for Pearl's staining; Supplementary Fig. S5B). Soluble Erfe levels were significantly increased after 7 weeks of LI diet in both mouse strains (data not shown). EPO expression was significantly increased at 7 weeks of LI in both mouse strains (Supplementary Fig. S5C).
Collectively, these data indicate that in Prx2−/− mice Hamp response to canonical stimuli such as IO, AI, or LPS is absent and is associated with loss of STAT3 transcription activity, validating an important role for Prx2 in regulating STAT3 function.
Recombinant fused PEP Prx2 ameliorates the IO-induced anemia and is associated with increased Erfe expression
Recently, we showed that PEP Prx2 is able to reduce ineffective erythropoiesis, ameliorate anemia, and decrease liver IO in a mouse model for β-thalassemia (34). To determine whether PEP Prx2 also protects erythroid cells against the toxicity of IO, we administrated PEP Prx2 to WT and Prx2−/− mice under an IO diet. PEP Prx2 administration was started at the same time as the IO diet. In both IO wild-type and IO Prx2−/− mice, PEP Prx2, to a large extent, rescued the IO-induced hematologic phenotype with (1) amelioration of red cell morphology and reduction in hemichromes (Fig. 4A, Supplementary Fig. S6A); (2) increased Hb levels and reduced RDW (Fig. 4B upper panel, Supplementary Fig. S6B); and (3) increased reticulocyte count (Fig. 4B, lower panel). In agreement, we found a significant increase in total erythroblasts (CD44+TER119+FSChigh) in both mouse strains exposed to IO and treated with PEP Prx2 (Fig. 4C, upper panel). We also noted a significant reduction in the amount of IO-induced apoptotic orthochromatic erythroblasts in both mouse strains treated with PEP Prx2 (Fig. 4C, lower panel). The decrease in IO-induced ineffective erythropoiesis by PEP Prx2 was associated with upregulation of Erfe in both polychromatic and orthochromatic erythroblasts from wild-type and polychromatic erythroblasts from Prx2−/− mice compared with erythroid precursors from IO-treated mice (Supplementary Fig. S6C).
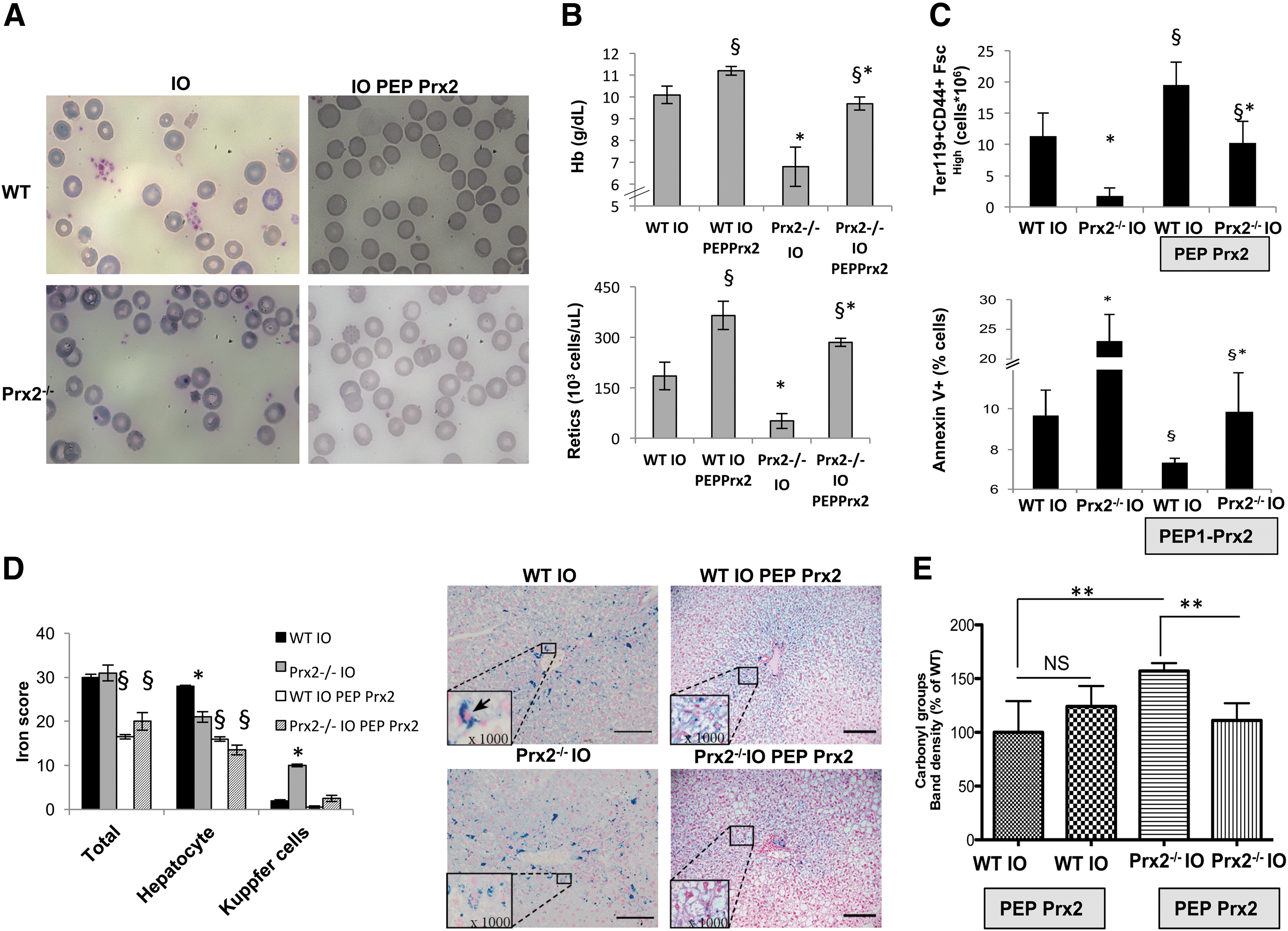
PEP Prx2 treatment supports STAT3 activation with upregulation of Hamp expression in response to IO
PEP Prx2-treated IO mice showed a marked reduction of IO in liver (Fig. 4D) and reduced protein oxidant damage (Fig. 4E). In both PEP Prx2-treated IO mouse strains, we found a marked activation of STAT3 compared with vehicle-treated mice (Fig. 5A and Supplementary Fig. S7A). Jak2 activation was decreased in IO-exposed mice treated with PEP Prx2, in agreement with previous reports linking oxidative stress and modulation of Jak2 signaling (27, 37) (Fig. 5B and Supplementary Fig. S7B). Hamp expression was significantly increased in both PEP Prx2-treated IO mouse strains (Fig. 5C). Smad activation was reduced by PEP Prx2 treatment in both IO mouse strains compared with vehicle-treated IO mice (Fig. 5D and Supplementary Fig. S7C). No changes in IL-6 expression were observed in liver from PEP Prx2-treated IO mice (Supplementary Fig. S8A). These findings imply that PEP Prx2 blunts IO-related Smad activation, but it promotes STAT3 activation with upregulation of Hamp expression in both mouse strains in a Jak-2-independent fashion.
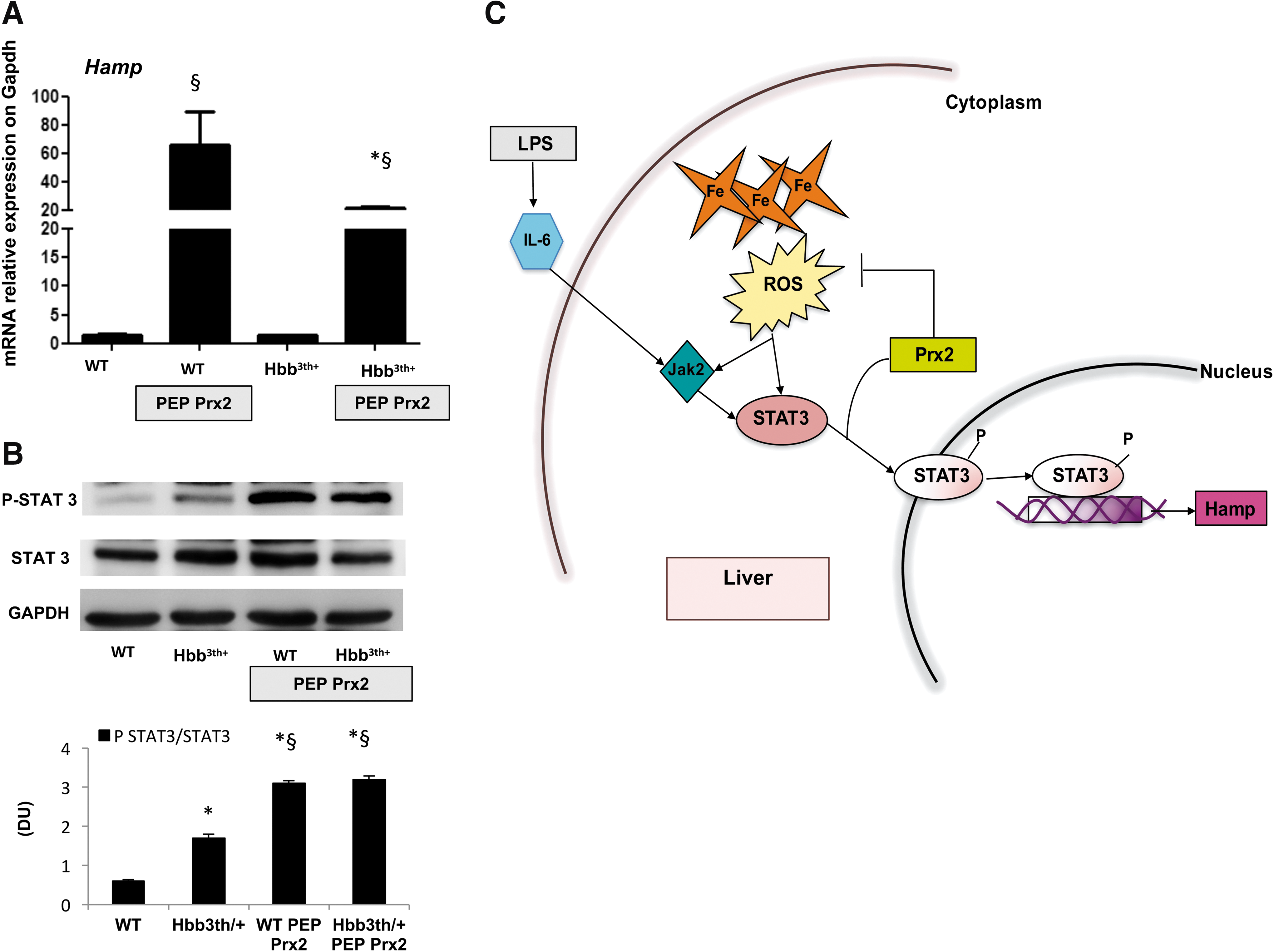
In β-thalassemic mice, PEP Prx2 treatment upregulates Hamp expression in liver and is associated with STAT3 activation
We previously reported that PEP Prx2 administration significantly ameliorates β-thalassemic anemia in conjunction with decreased ineffective erythropoiesis and reduced liver IO (34). In both PEP Prx2-treated Hbb3th/+ and wild-type mice, we found increased Hamp mRNA levels compared with vehicle-treated mice (Fig. 6A). STAT3 was significantly activated in both PEP Prx2-treated mouse strains compared with vehicle-treated groups (Fig. 6B and Supplementary Fig. S8B). These data collectively suggest that Prx2 modulates STAT3-regulated hepatic Hamp expression in both Hbb3th/+ and wild-type mice.
Discussion
Our study documents a critical role for Prx2 as a cytoprotector against toxic effects of IO. Absence of Prx2 and associated loss of the expected iron-induced increase in Hamp expression in Prx2−/− mouse liver, in part, account for deleterious effects of iron accumulation. IO Prx2−/− mice displayed reduced STAT3 activation compared with WT, in the presence of the preserved Jak2 signaling pathway. Previous studies have shown that ROS levels significantly influence the activation status of STAT3, affecting Hamp expression in response to IO or to the IL-6 mediated inflammatory system (39, 40). Thus, the failure in STAT3 activation in IO Prx2−/− mice represents the key event responsible for the lack of Hamp upregulation in response to IO. This conclusion is bolstered by our finding that in contrast to STAT3, Smad activation was similar in both IO mouse strains. In addition, the reduced expression of Erfe in both mouse strains exposed to IO indicates that Erfe is not directly involved in the loss of Hamp expression observed in IO Prx2−/− mice. By exploring the effects of AI administration and LPS treatment on Hamp expression in Prx2−/− mice, we documented that the absence of Prx2 affects Hamp expression and is associated with reduced STAT3 activation. The effects of PEP Prx2 treatment on STAT3 activation in response to IO as well as documentation that Prx2 directly associates with active STAT3 support the important cytoprotective role of Prx2 and imply a robust interplay between iron and the Prx2 Hamp pathway.
The loss of the functional connection between Erfe and Hamp in the absence of Prx2 is another novel element in the scenario of iron homeostasis and ineffective erythropoiesis in the presence of adequate EPO levels. Indeed, the administration of PEP Prx2, which we previously reported to ameliorate ineffective erythropoiesis in both Prx2−/− and Hbb3th/+ mice (34), prevented the IO-induced cytotoxic effect on erythropoiesis and was associated with upregulation of Erfe in both mouse strains. This effect was more pronounced in WT than in Prx2−/− mice, most likely related to the fact that the exogenous PEP Prx2 does not fully compensate for the complete absence of Prx2 in Prx2−/− mice.
IO is a life-threatening complication of chronic hemolytic red cell disorders such as β-thalassemia. Our data on PEP Prx2 treatment in Hbb3th/+ mice further support a functional connection between Prx2 and STAT3 toward Hamp expression, supporting a key role of Prx2 in iron homeostasis in red cell disorders that are characterized by IO. It is of note that a previous report on proteomic analysis of erythroid precursors has identified Prx2 in complexes with iron storage protein ferritin (1). Thus, the beneficial effects of PEP Prx2 treatment in both models of exogenous IO and spontaneous IO in β-thalassemic mice might be also related to the potentiation of the cellular function of Prx2 as a cytoprotective system, an H2O2 sensor, and a chaperone-like molecule. In addition, the results of rescue experiments with PEP Prx2 suggest PEP Prx2 as a new interesting therapeutic modulator of iron metabolism with a possible synergistic effect with other novel therapeutic tools under development for clinical practice such as mini-hepcidins (5, 17, 44).
In summary, we propose that Prx2 controls oxidative stress and modulates targets that are sensitive to redox conditions, including STAT3 (Fig. 6C). Thus, Prx2 levels might act as an on-off switch of STAT3 transcription activity, profoundly affecting Hamp expression in response to IO in a Jak-2-independent fashion (Fig. 6C). The effects of PEP Prx2 on STAT3 activation and Hamp expression in either IO mice or β-thalassemic mice further support this working model. The amelioration of IO-induced ineffective erythropoiesis in PEP Prx2-treated mice supports the role of Prx2 as “big brother” in controlling IO-induced cytotoxicity and optimizing the functional pathway involved in iron homeostasis. These findings suggest a novel therapeutic strategy to control clinical consequences of IO through modulation of Prx2 activity.
Materials and Methods
Drugs and chemicals
NaCl, Na3VO4, bicine, benzamidine, β-mercaptoethanol, bromophenol blue, sodium dodecyl sulfate (SDS), NaF, EDTA, May-Grunwald stain, Giemsa stain, phenylhydrazine hydrochloride, albumin from bovine serum (BSA), and glycerol were obtained from Sigma/Aldrich (St Louis, MO); protease inhibitor cocktail tablets were from Roche (Basel, Switzerland); dithiotreithol (DTT) was from Fluka (Buchs, Switzerland); Triton X-100 was from GE Healthcare (Little Chalfont, United Kingdom); 40% Acrylamide/Bis Solution, 37.5:1 was from BIO-RAD (Hercules, CA); Dulbecco's phosphate-buffered saline (DPBS) was from Lonza (Verviers, Belgium); and Luminata Forte and Luminata Classico Hrp solutions were from Merck Millipore (Armstadt, Germany).
Mouse strains and design of the study
The Institutional Animal Experimental Committee of University of Verona (CIRSAL) and the Italian Ministry of Health approved the experimental protocols. Two-month-old female wild-type (WT) and Prx2−/− mice were fed with either standard diet or a diet containing 2.5% carbonyl-iron (IO) for 3 months. A subset of mice was fed with LI diet for either 3 or 7 weeks. Whenever indicated, WT and Prx2−/− mice were treated with a single dose of AI (4 mg/kg by gavage). Mice were studied 24 h after AI administration. LPS was administered at the dosage of 1 mg/kg/mouse, and LPS-treated mice were sacrificed at 6 h after LPS administration as previously reported (20). Hbbth3/+ mouse strain was used as a mouse model of β-thalassemia intermedia. Where indicated WT, Prx2−/− and Hbbth3/+ mice were treated with PEP Prx2 (in PBS) at a dose of 3 mg/kg/day ip or vehicle (PBS) (34). We evaluated hematologic parameters, red cell indices, and reticulocyte count in mouse strains at baseline and at 30, 49, 60, and 90 days during IO. Hematological parameters, red cell indices, and reticulocyte count were determined as previously reported (13, 14, 34). Blood was collected with retro-orbital venipuncture in anesthetized mice by using heparinized microcapillary tubes (11). Hematological parameters were evaluated on a Bayer Technicon Analyser ADVIA. Hematocrit and hemoglobin were manually determined (2, 10). Determination of hemichromes in mouse red cells was carried out by FACS analysis as previously reported (34, 36).
Flow cytometric analysis of mouse erythroid precursors and molecular analysis of sorted erythroid cells
Flow cytometric analysis of erythroid precursors from bone marrow and spleen of mice from the four strains was carried out as previously described by using the CD44−TER-119 strategy (2, 28, 34). Analysis of apoptotic orthochromatic erythroblasts was carried out on the CD44−TER-119 gated cells by using the Annexin-V PE Apoptosis detection kit (eBioscience, San Diego, CA) according to the manufacturer's instructions (2, 16, 34). Sorted cells were used for (1) immuno-blot analysis with specific antibodies against Prx2 (Clone 1E8; AbCam, Cambridge, United Kingdom) and actin (Sigma-Aldrich) used as a loading control; and (2) real-time polymerase chain reaction (RT-PCR) analysis (34). Details of RT-PCR and immune-blot protocols used for the analysis of sorted erythroblasts were previously described (30, 33).
Generation of recombinant-PEP Prx2 fusion protein (PEP Prx2)
The fusion protein PEP Prx2 was generated as previously reported (6, 34).
Histological analysis of spleen and liver
Immediately after dissection, spleen and liver were formalin fixed and paraffin embedded. From each paraffin block, 3 μm-thick section were cut and stained with hematoxylin eosin, Masson's trichrome, and May-Grunwald-Giemsa. Tissue iron was stained by using Pearl's Prussian blue stain after treatment with diluted hydrochloric acid to prevent ferric ions from binding proteins. The analysis of iron staining was performed on four different fields at a magnification × 200 by two pathologists (A.J., C.L.) blinded to the experimental groups. Results were expressed as the mean number of cells loaded with iron based on Pearl's staining (12).
Molecular analysis of liver
Protocols used for RNA isolation, cDNA preparation, and quantitative RT-PCR have been previously described (12, 34). Detailed primer sequences are available on request and shown in Supplementary Table S1.
Liver Immuno-blot analysis
Frozen livers from each studied group were homogenized and lysed with iced lysis buffer (LB containing: 150 mM NaCl, 25 mM bicine, 0.1% SDS, 2% Triton X-100, 1 mM EDTA, protease inhibitor cocktail tablets [Roche], 1 mM Na3VO4 final concentration) followed by centrifugation for 30 min at 4°C at 12,000 g. Proteins were quantified and analyzed by mono-dimensional SDS polyacrylamide gel electrophoresis. Gels were transferred to nitrocellulose membranes for immuno-blot analysis with specific antibodies. When indicated, we carried out an immunoprecipitation assay as previously described (31). The following antibodies were used: Anti-Heme-Oxygenase-1 (HO-1), anti-NAD(P)H: Quinone Oxidoreductase-1 (NQO-1), anti-STAT3 (STAT3), and Phospho-Tyrosine (clone PY99) were from SCBT (Santa Cruz, CA); anti-peroxiredoxin-2 (Prx-2, clone 1E8) was from AbCam. Anti-GAPDH and anti-actin from Sigma-Aldrich were used as loading controls. Secondary donkey anti-rabbit IgG and anti-mouse IgG HRP conjugated were from GE Healthcare Life Sciences; secondary donkey anti-goat IgG HRP conjugated was from SCBT. Blots were developed by using the Luminata Forte Chemiluminescent HRP Substrate from Merck KGaA, and images were acquired by using Image Quant Las Mini 4000 Digital Imaging System (GE Healthcare Life Sciences). Densitometric analyses were performed by using the ImageQuant TL software (GE Healthcare Life Sciences) (22, 46).
EMSA
Liver nuclear extract was used for nonradioactive electrophoretic mobility shift assay. Electrophoretic mobility shift assay (EMSA) was performed on liver nuclear protein extracts by using the NonRadioactive EMSA-STAT3 Kit (Viagene Biotech, Inc., Tampa, FL), following the manufacturer's instructions. Briefly, livers were homogenized in ice cold Buffer A (10 mM Hepes pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, protease inhibitor cocktail [Roche], 1 mM Na3VO4) to which 10% Nonidet P-40 (Sigma) was added by using the Tissue Master 50 homogenizer (Thermo Fisher Scientific). Nuclear proteins were extracted in Buffer B (20 mM Hepes pH 7.9, 25% Glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, protease inhibitor cocktail [Roche]) as previously described (38) and quantified by using the DC protein assay (BioRad). Fifteen micrograms of nuclear extracts was used for the Non-Radioactive EMSA-STAT3 Kit (Viagene Biotech, Inc.) (38).
Ferroportin-1 quantification
Ferroportin (Fpn1) quantitation was carried out as previously reported (2). Five-micrometer-thick duodenum sections were de-paraffinized, treated in citrate buffer (1 mM, pH 6.0) for 15 min, washed, and incubated with 3% H2O2 for 10 min. Slides were blocked in 1% BSA for 1 h at room temperature, followed by overnight incubation with rabbit anti-ferroportin antibody (alpha diagnostics MTP11-A), then with biotinylated goat anti-rabbit antibody (Vector Laboratories) for 30 min at room temperature. The stain was visualized with 3,3′-Diaminobenzidine (DAB) (Sigma-Aldrich) (2).
Ferritin quantification
Ferritin content was determined by ELISA as previously described (45).
Detection of oxidized proteins and peroxidation of lipids
Oxidized proteins were monitored by using the Oxyblot Protein Oxidation Detection Kit (EMD Millipore) as previously reported (7, 45). The extent of lipid peroxidation was determined by measuring MDA levels.
Statistical analysis
Prism software (GraphPad software, Inc., La Jolla, CA) was used for statistical analysis. Comparisons between two groups were performed with two-sided Welch t-test and among >2 groups with one- and two-way ANOVA followed by the Bonferroni post-test. A value of p < 0.05 was considered significant.
Footnotes
Acknowledgments
The authors are grateful to E. Nementh for her support of their work and many fruitful discussions. This work was supported by PRIN (L.D.F. and A.I.: 201228PNX83) and FUR_UNIVR (L.D.F.).
Authors' Contributions
A.M., L.D.F., N.M., and I.A. designed the experiments, analyzed data, and wrote the article; T.G. designed the experiments, wrote the article, and carried out Erfe measurements; F.C. and S.L. analyzed the data and wrote the article; S.M. and I.S. carried out in vitro exposure and wrote the article; A.J. and C.L. carried out the histologic analysis; A.M., A.S., E.F., and L.D.F. carried out the experiments; L.D.F. and M.B. performed the molecular experiments and analyzed the data; G.F. carried out the analysis on LIC; CSY and KDW generated the PEPPrx2; S.L. and A.C. carried out the analysis of liver oxidative stress and the determination of high ferritin levels; and D.M. and C.C. carried out EMSA experiments and analyzed data.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.