
Abstract
Significance:
Glutathione is the most abundant antioxidant molecule in living organisms and has multiple functions. Intracellular glutathione homeostasis, through its synthesis, consumption, and degradation, is an intricately balanced process. Glutathione levels are often high in tumor cells before treatment, and there is a strong correlation between elevated levels of intracellular glutathione/sustained glutathione-mediated redox activity and resistance to pro-oxidant anticancer therapy.
Recent Advances:
Ample evidence demonstrates that glutathione and glutathione-based systems are particularly relevant in cancer initiation, progression, and the development of anticancer drug resistance.
Critical Issues:
This review highlights the multifaceted roles of glutathione and glutathione-based systems in carcinogenesis, anticancer drug resistance, and clinical applications.
Future Directions:
The evidence summarized here underscores the important role played by glutathione and the glutathione-based systems in carcinogenesis and anticancer drug resistance. Future studies should address mechanistic questions regarding the distinct roles of glutathione in different stages of cancer development and cancer cell death. It will be important to study how metabolic alterations in cancer cells can influence glutathione homeostasis. Sensitive approaches to monitor glutathione dynamics in subcellular compartments will be an indispensible step. Therapeutic perspectives should focus on mechanism-based rational drug combinations that are directed against multiple redox targets using effective, specific, and clinically safe inhibitors. This new strategy is expected to produce a synergistic effect, prevent drug resistance, and diminish doses of single drugs. Antioxid. Redox Signal. 27, 1217–1234.
Introduction
G
The control of intracellular glutathione homeostasis through its synthesis, consumption, and degradation is an intricately balanced process (Fig. 1). Cellular GSH is supplied by de novo synthesis from precursor amino acids (glutamate, cysteine, and glycine), reduction of GSSG by glutathione reductase (GR), and cellular uptake of extracellular glutathione. This important tripeptide is synthesized in the cytoplasm via two consecutive ATP-dependent reactions (88). In the first step, glutamate-cysteine ligase (GCL, also known as gamma;-glutamylcysteine synthetase) catalyzes the unconventional attachment of cysteine to the γ-phosphate of glutamate to produce γ-glutamylcysteine. In the second step, GSH synthetase (GS) catalyzes the attachment of γ-glutamylcysteine to glycine to produce GSH. Under physiological conditions, GSH is much more abundant (>98%) than its oxidized form, GSSG (37). Indeed, GSSG can be reduced to GSH by GR, which uses reduced nicotinamide adenine dinucleotide phosphate (NADPH) as an electron donor. Alternatively, it can be exported out of the cell by multidrug resistance-associated proteins, such as multidrug resistance protein 1 (MRP1) (11, 22). Oxidation of glucose in the pentose phosphate pathway (PPP) provides NADPH, the major reducing equivalent for thiol-dependent antioxidant defenses. GSH is consumed by oxidation into GSSG, conjugation to diverse endogenous and exogenous compounds, such as certain xenobiotics, and hydrolysis (Fig. 1). GSH can be directly oxidized by ROS and RNS or indirectly oxidized by assisting GSH-dependent peroxidases. Conjugation with endogenous and exogenous electrophiles consumes a substantial portion of cellular glutathione. In addition, cells may lose glutathione due to export of its reduced, oxidized, or conjugated forms into extracellular spaces (134). Effluxed GSH and GSSG, or GSH conjugates, can be hydrolyzed by γ-glutamyl transpeptidase (GGT), an enzyme that is capable of hydrolyzing the γ-glutamyl bond, which eventually leads to the release of glutamate, cysteine, cystine, and glycine (57). The hydrolysis products are then taken up by cells, either as individual amino acids or as dipeptides, to replenish GSH synthesis. In addition to this extracellular GGT-initiated glutathione salvage pathway, a newly discovered cytosolic pathway has been identified for the degradation of glutathione. The tightly regulated ChaC1 and constitutively expressed ChaC2, members of the γ-glutamylcyclotransferase family, share the same specificity for GSH in the cytosol (25, 71). These new players of glutathione degradation are not only an integrated component of glutathione homeostasis, but they are also likely to play a role in redox signaling (see review by Bachhawat and Kaur in this Forum).
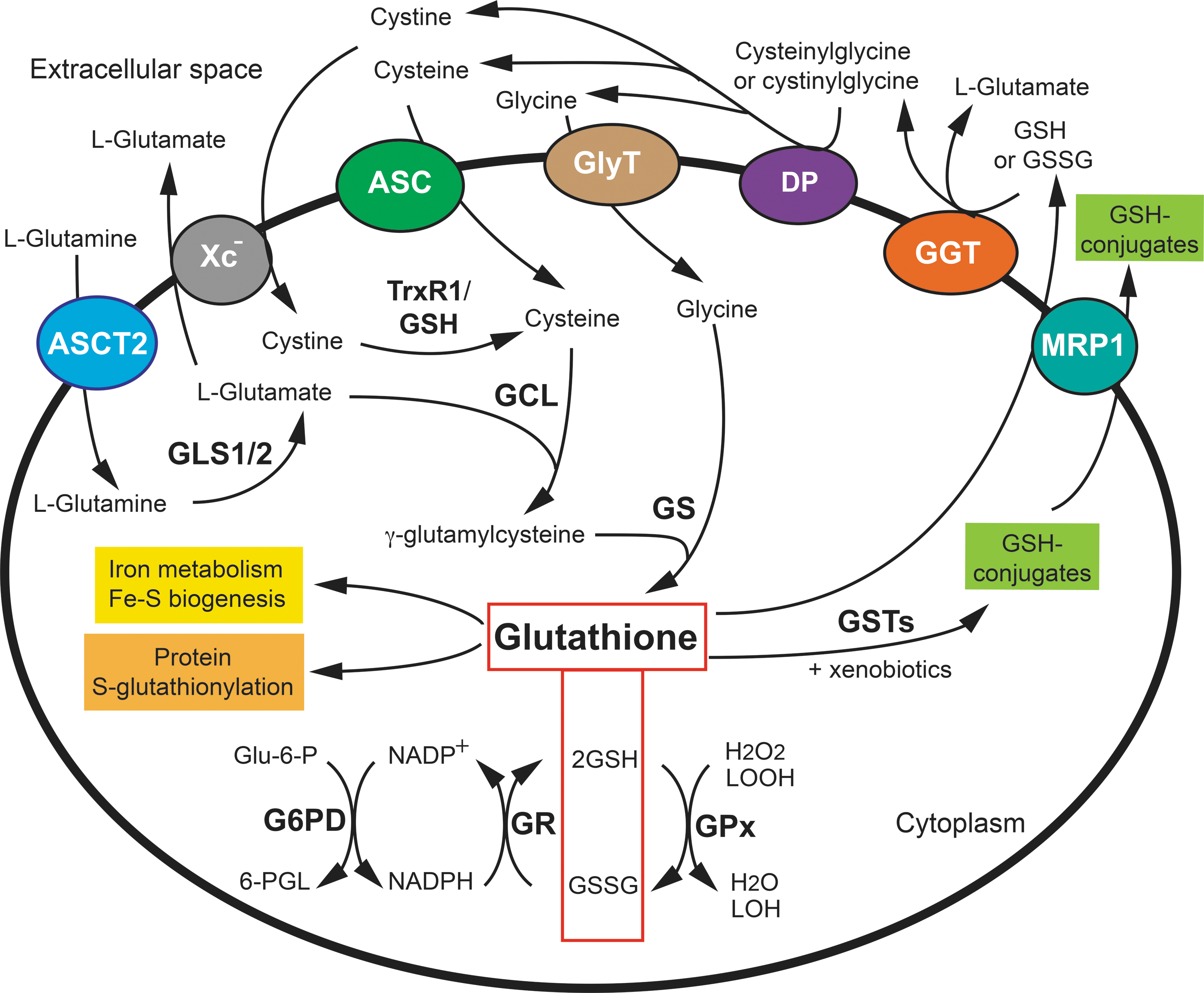
Glutathione is at the heart of the glutathione-based systems, encompassing glutathione-dependent enzymes that are involved in the maintenance of the cellular redox state and numerous cellular processes (Fig. 1). Among them, glutathione peroxidases (GPx) use GSH for the reduction of hydrogen peroxide (H2O2) or organic H2O2 to water or the corresponding alcohol (153). GPx1 is one of the most abundant isoforms of GPx, and it is ubiquitously expressed in mammalian tissues. It is also one of the main H2O2-degrading enzymes in mitochondria and has been reported to be even more efficient than catalase at degrading intracellular peroxides under physiological conditions (6, 16, 93). The family of glutaredoxins (GRXs) catalyze thiol/disulfide exchange reactions to reduce disulfide bonds in substrate proteins (58). One oxidized GRX is reduced by two GSH molecules. In addition, the condensation of GSH to cysteine residues, which are oxidized in the sulfenic acid form, leads to the formation of Cys-glutathione adducts in a process called S-glutathionylation (8), thus protecting cysteine residues from irreversible oxidation. S-glutathionylated adducts are generally reduced by GRXs. S-glutathionylation is also an important post-translational modification that regulates cell signaling (98). Further, GSH acts together with GSH S-transferases (GSTs) to detoxify various xenobiotics. GSTs catalyze the conjugation of GSH with a wide variety of compounds, resulting in the formation of the corresponding GSH conjugates, subsequently facilitating their clearance from the cell (134). This GSH-driven detoxification mechanism underlies the resistance against several chemotherapeutic agents. Finally, in addition to the various roles of GSH in the glutathione antioxidant system, other functions of GSH should not be overlooked, notably in redox-independent iron metabolism and iron-sulfur biogenesis (24, 76). The essential role of GSH for yeast viability appears to be linked to this function.
It has long been known that glutathione metabolism and the glutathione-based systems are altered in cancer cells. Glutathione levels are often high in tumor cells before treatment, and many studies have shown a strong correlation between elevated levels of intracellular glutathione and resistance to pro-oxidant chemotherapy (10, 36). This review highlights the multifaceted roles of glutathione and the glutathione systems in carcinogenesis and anticancer drug resistance. We also summarize mechanism-based pharmacological approaches that target glutathione and glutathione systems as potential anticancer strategies.
Glutathione in Cancer Initiation, Progression, and Metastasis
ROS are widely recognized as one of the major triggers for cancer initiation and progression through their role in promoting both genome instability and activation of signaling pathways (67, 142). Thus, the use of antioxidants or the stimulation of cellular antioxidant activity has been considered a reasonable strategy for cancer prevention. However, experimental data and clinical trials have provided inconsistent results. Further, a number of studies suggest that increased glutathione levels are associated with the proliferation of both normal and malignant cells and show a direct correlation between glutathione levels and cellular proliferation and metastasis. For example, an early study showed that an intrasplenic injection of B16 melanoma cells (B16 M) with high GSH content, obtained by GGT overexpression, showed higher metastatic activity in the liver than cells with low GSH content (102). More recently, the role of glutathione in cancer initiation and progression was investigated by using various mouse models (59). Disruption of GCLM (the gene encoding the GCL modifier subunit) in mice (Gclm−/−) that develop spontaneous mammary tumors resulted in a significant delay in tumor onset, relative to controls. Gclm−/− mice have only 10–25% of the glutathione levels of wild-type mice, but they are completely viable. This finding was validated in two independent Gclm−/− mouse models that developed sarcomas, lymphomas, and thymomas, and mouse models in which GSH synthesis was chemically inhibited by oral delivery of L-buthionine-(S,R)-sulfoxime (BSO), a potent inhibitor of GSH synthesis, immediately after weaning. Paradoxically, the inhibition of GSH synthesis on tumor onset by BSO failed to reduce the tumor burden. This intriguing result was explained by the increased activity of the thioredoxin (Trx) antioxidant pathway that compensated for the lack of glutathione in established tumors (59). An independent study reported that dietary supplementation with the antioxidants N-acetyl-cysteine (NAC) and vitamin E markedly increased tumor progression and reduced survival in serine/threonine protein kinase B-Raf (BRAF)- and Kirsten rat sarcoma viral oncogene (KRAS)-induced lung cancer mouse models by reducing ROS levels, DNA damage, and p53 expression in the cancer cells (127).
The roles of ROS and glutathione in the process of metastasis have also been evaluated. Metastasis is a multistep process involving separation from the primary tumor, invasion through tissues around the initial lesion, entry into blood or lymphatic vessels and survival in circulation, reaching distant organs, and proliferation (162). A recent study showed that circulating melanoma cells in the blood of xenografted mice and metastatic nodules had higher levels of ROS than primary subcutaneous tumors (112). Metastasizing melanomas underwent reversible metabolic changes that increased their capacity to withstand oxidative stress, including increased dependence on NADPH-generating enzymes in the folate pathway. The GSH/GSSG ratio was lower in metastatic nodules than in subcutaneous tumors, suggesting that metastatic cells use antioxidants, including GSH, to buffer the higher ROS level present in these cells. Oxidative stress is a barrier to distant metastasis in these melanomas, as treatment with the antioxidant, NAC, increases the frequency of circulating melanoma cells in the blood and metastatic disease burden, without significantly affecting the growth of primary subcutaneous tumors. In contrast, folate pathway inhibition by low-dose methotrexate, ALDH1L2 knockdown, or MTHFD1 knockdown inhibited distant metastasis without significantly affecting the growth of subcutaneous tumors in the same mice (112). Similarly, treatment with NAC or the vitamin E analog, trolox, promoted distant metastasis without affecting the growth of subcutaneous tumors in an endogenous mouse model of malignant melanoma (80). Both NAC and trolox increased the GSH/GSSG ratio in melanoma cells and lymph nodes. Metastasis and increased tumor-cell migration depended on new GSH synthesis.
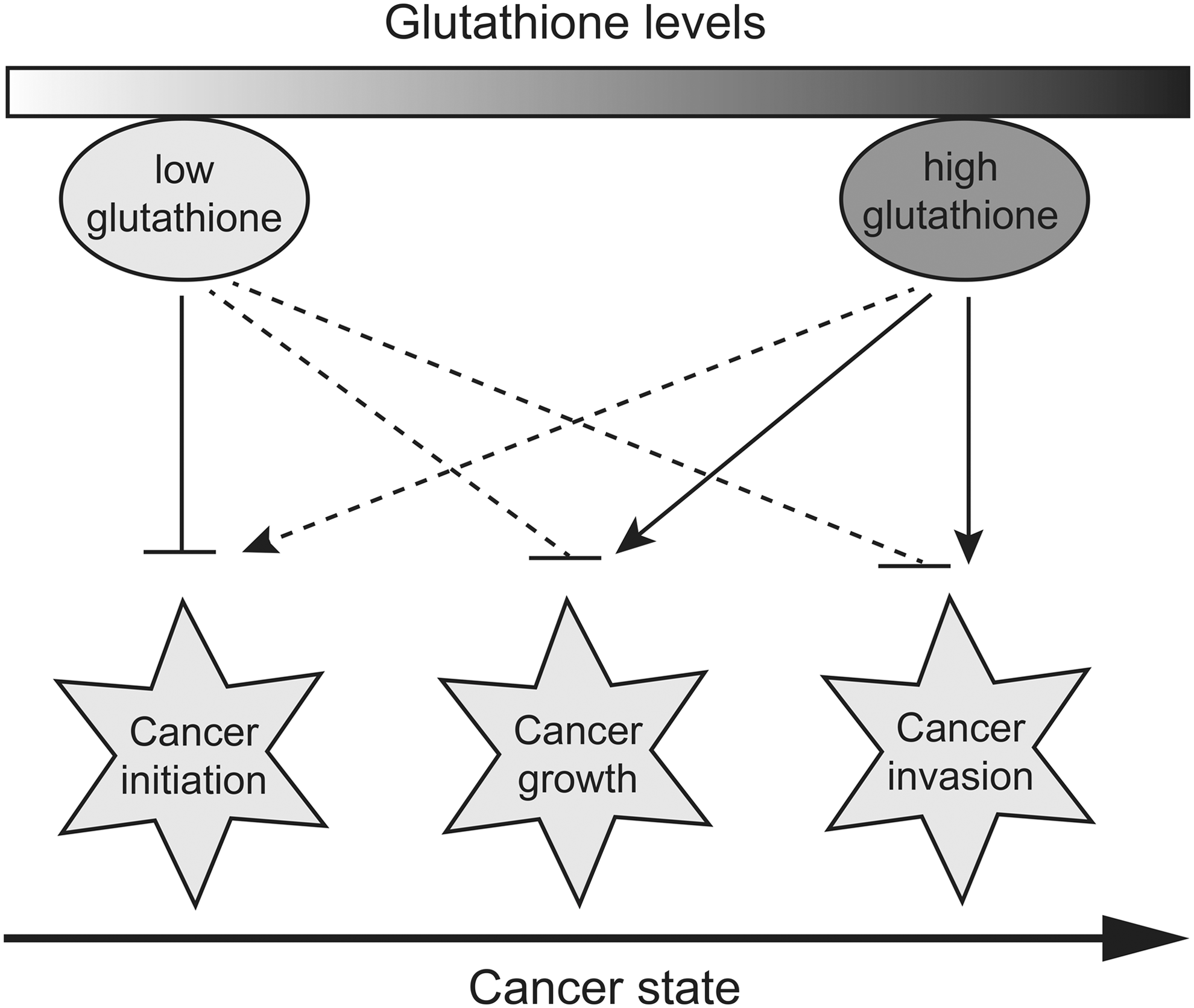
ROS act as a double-edged sword in oncogenesis: ROS promote cancer initiation by inducing mutagenesis and perhaps activating signaling pathways that promote proliferation, survival, and stress resistance. ROS also limit cancer growth by causing lethal oxidative stress to many cancer cells. Therefore, antioxidants, including GSH, may exert dual effects through ROS scavenging and/or regulation of the redox signaling pathway: reducing cancer risk in certain genetic backgrounds, but promoting cancer initiation, progression, and metastasis under many other circumstances, as shown by increasing evidence (Fig. 2). Indeed, glutathione and glutathione-based systems could be pieces of a larger puzzle in the vicious cycle driving carcinogenesis. There is increasing awareness that aberrant oncogenic signals, metabolic plasticity, and redox perturbations/adaptations are closely linked in cancer cells. For example, c-Myc has been shown to regulate glucose, glutamine, and serine/glycine metabolism, which are central hubs of cancer metabolism and constitute a source for precursors of GSH synthesis (137, 139). Likewise, cells with KRAS-activating mutations that promote lung cancer exhibit a glycolytic switch coupled to increased channeling of glucose-derived metabolites into the tricarboxylic acid cycle and GSH biosynthesis, resulting in enhanced GSH-mediated detoxification (72). These mechanistic complexities may explain why dietary supplementation with antioxidants or the use of antioxidants as adjuvant therapy in the treatment of cancer has often proved ineffective or even detrimental in clinical trials (46).
Aberrant Nuclear Factor Erythroid 2-Related Factor 2 Activity Promotes Glutathione Synthesis and Regeneration in Cancer Cells
The transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) is a master transcriptional regulator of cellular antioxidant systems (135). Nrf2 is primarily regulated by Kelch-like ECH-associated protein 1 (Keap1), a substrate adaptor of Cullin 3 (Cul3)-containing E3 ubiquitin ligase. Under basal conditions, Nrf2 is complexed with Keap1 via direct protein–protein interactions (Fig. 3). The Keap1-Cul3-E3 ubiquitin ligase complex tightly regulates Nrf2 protein levels by directing it to ubiquitin-dependent proteasomal degradation. Keap1 is modified by oxidation or adduction of cysteine residues in response to oxidative or electrophilic stresses, causing the release of Nrf2, its stabilization, and translocation to the nucleus where it heterodimerizes with small Maf proteins or other bZIP family members to activate genes containing an antioxidant-response element (ARE) within their regulatory regions (143). Nrf2 drives the transcription of a wide array of genes, including antioxidant, anti-inflammatory, and detoxification enzymes, as well as proteins that assist in the repair or removal of damaged macromolecules. Among these genes are those involved in GSH biosynthesis, utilization, and recycling (Fig. 3): (i) GCLC and GCLM, which encode the catalytic and modifier subunits of GCL, respectively; (ii) SLC7A11, which encodes the light chain (xCT) of cystine/glutamate antiporter Xc −, which imports cystine into the cells in exchange for glutamate (123); (iii) GR, which encodes GR, which reduces GSSG into GSH; (iv) genes that encode four principal NADPH-generating enzymes, including malic enzyme 1, isocitrate dehydrogenase 1, glucose-6-phosphate dehydrogenase, and 6-phosphogluconate dehydrogenase (32); and (v) genes that encode GPx2 and several GSTs (96, 152).
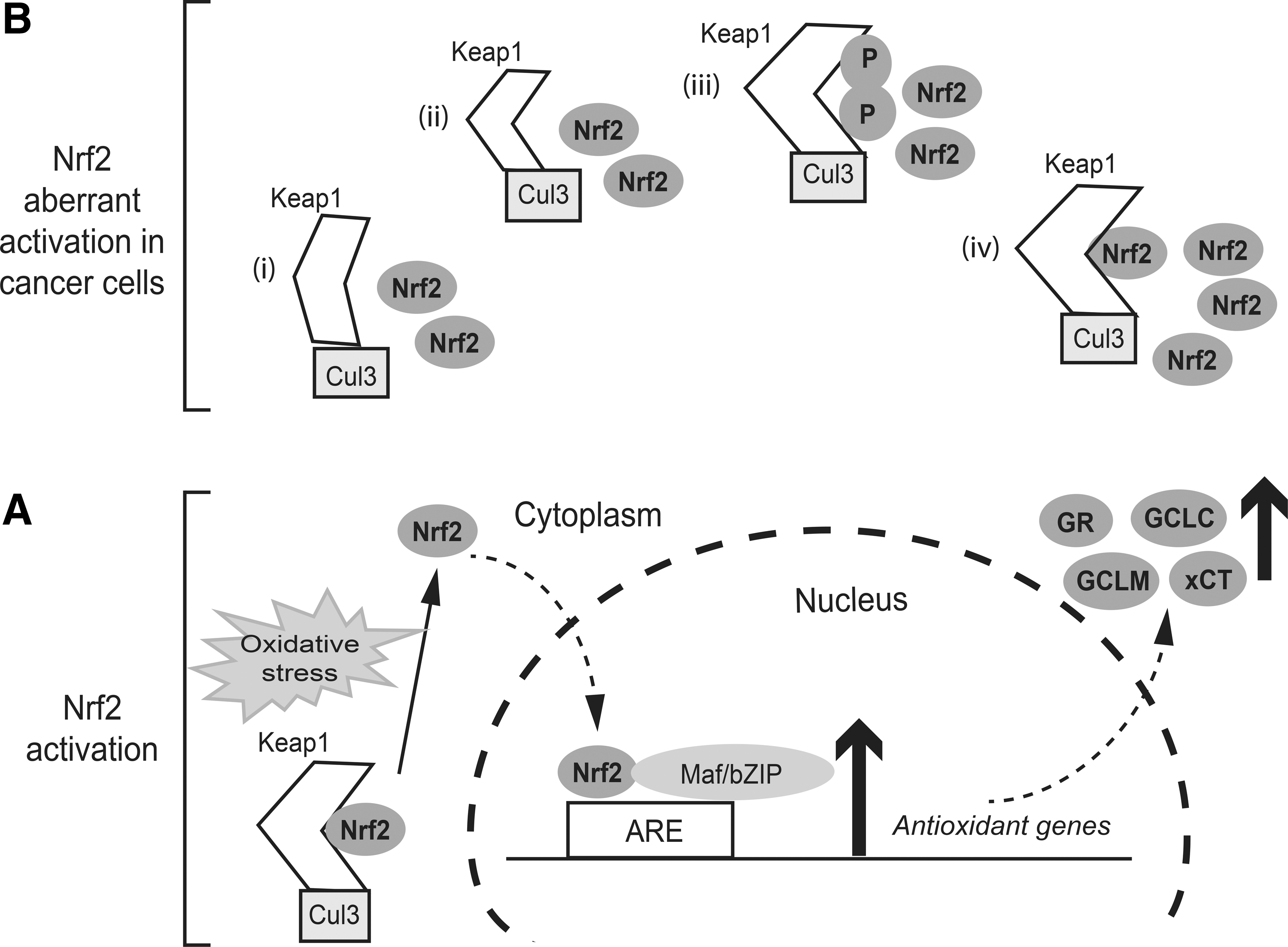
The ability of Nrf2 to activate cytoprotective genes gives it tumor suppressor properties, as has been demonstrated in Nrf2 knockout (Nrf2−/−) mouse models. Nrf2−/− mice are highly susceptible to carcinogenic agents and readily develop tumors on exposure to chemical carcinogens, or they have higher tumor burdens than their wild-type littermates (106, 115, 170). Further, reduced tumor incidence was observed in mouse models on co-administration of Nrf2 activators along with a carcinogen (170). Indeed, it is highly possible that the beneficial effects of many foods and traditional medicines could be partially due to their properties to activate Nrf2. On the other hand, many studies have revealed the “dark side of Nrf2.” Aberrant activation of Nrf2 in cancer cells has been associated with progression, metastatic invasion, angiogenesis, and intrinsic and acquired drug resistance in tumors and is indicative of poor prognosis (69). The causes of the constitutive activation of Nrf2 in cancer cells have been thoroughly reviewed recently (69, 97); they may include at least one of the following mechanisms (Fig. 3): (i) somatic mutations of KEAP1, NRF2, or the CUL3 gene, leading to impaired binding to Keap1; (ii) epigenetic silencing of the KEAP1 gene that inhibits its expression, resulting in the accumulation of Nrf2; (iii) aberrant accumulation of proteins, such as p21, DJ-1, which compete with Nrf2 for Keap1 binding, also leading to an increase in the stability of Nrf2 and the activation of Nrf2 target genes; and (iv) oncogene-mediated transcriptional upregulation of Nrf2. For example, KRAS and MYC have been shown to stabilize Nrf2 and promote the Nrf2-mediated antioxidant response. The precise mechanisms by which these oncogenes upregulate the transcription of Nrf2 are currently unknown.
Aberrant activation of Nrf2 could lead to deregulation of a large set of Nrf2 target genes, including those involved in GSH biosynthesis, utilization, and recycling (see the first paragraph of this section), thereby contributing to cancer malignancy. It is tempting to speculate that the role of glutathione and glutathione-based systems in cancer initiation, progression, and metastasis may also be intimately linked to Nrf2 activity. Recent studies provide evidence that Nrf2 may act as a tumor-suppressor or tumor-promotor, depending on the stage of carcinogenesis. Using the urethane-induced lung cancer mouse model, Nrf2−/− mice exhibited more tumor foci by 8 weeks after urethane administration than wild-type mice. However, after 16 weeks, tumors in the Nrf2-null mice showed less advanced malignancy than controls (125). Urethane-induced tumors were significantly smaller and less frequent in Keap1−/− mice, which express high levels of Nrf2, than in wild-type mice. In contrast, tumor cells derived from Keap1−/− mice and transplanted into nude mice exhibited higher tumorigenicity than cells derived from wild-type mice. These results consistently support the view that Nrf2 prevents the initiation of cancer but accelerates its progression (124).
In addition to Nrf2, the tumor suppressor p53 also contributes to the regulation of glutathione production and utilization and other antioxidant pathways. TIGAR (TP53-induced glycolysis and apoptosis regulator) is one of the targets regulated by p53 that has a pro-oxidant function (13). TIGAR has fructose-2,6-bisphosphatase activity, thus dampening the glycolytic pathway and promoting the shuttling of metabolites to the PPP. By upregulating TIGAR, p53 amplifies PPP-dependent NADPH and glutathione production (164, 176). Glutaminase 2 (GLS2) is also a p53 target gene with antioxidant activity (63, 140). There are two predominant human isozymes of glutaminase, GLS1 and GLS2, which exhibit distinct tissue distribution and are very differently regulated. Tight control of GLS expression is essential for GSH synthesis, as it converts glutamine to glutamate, which is, subsequently, used for GSH synthesis. The contribution of altered p53 activity to redox deregulation and glutathione-mediated drug resistance in cancer cells is still poorly understood.
Elevated Glutathione Levels and Increased GST Activity Enhance Drug Detoxification
The major contribution of glutathione and GSTs in the detoxification of xenobiotics predicts their important roles in drug resistance. The role of GST is to catalyze the conjugation of GSH with a wide variety of compounds, resulting in the formation of the corresponding GSH conjugates. Seven classes of cytosolic GSTs have been identified in human cells and are designated by Roman capitals: A, M, P, S, T, Z, and O (14, 134, 151). GSTs are involved in many different detoxification reactions (Fig. 4). Several conventional chemotherapeutic agents, including cisplatin, melphalan, chlorambucil, doxorubicin, paclitaxel, and cyclophosphamide, are substrates for GSTs (30, 60). An early study found that the expression of GSTs was upregulated in a panel of 60 human tumor cell lines, with GSTP1 being the major GST isozyme found in these cancer cells (149). Malignant gliomas also have higher expression of GSTP1 than benign gliomas (138). In addition, GSTP1 has been reported to be overexpressed in a variety of other malignancies, including leukemia, lung, colon, kidney, ovary, esophagus, and stomach cancers (45, 51, 64, 133, 144, 181). Taken together, the overexpression of GSTs, together with increased glutathione levels, drives cellular biotransformation of electrophilic compounds, leading to increased detoxification of anticancer agents and the development of drug resistance in cancer cells. GSH-conjugated xenobiotics and GSH-conjugated metabolites must be exported out of cells before they can be eliminated from the body. This efflux is often mediated by the MRP1 transporter, which is known for its role in protecting normal cells from toxic insults and in conferring drug resistance to cancer cells (22, 84) (Figs. 1 and 4). MRP1 also exports GSH and GSSG, and it may thus have a role in cellular responses to oxidative stress (22).
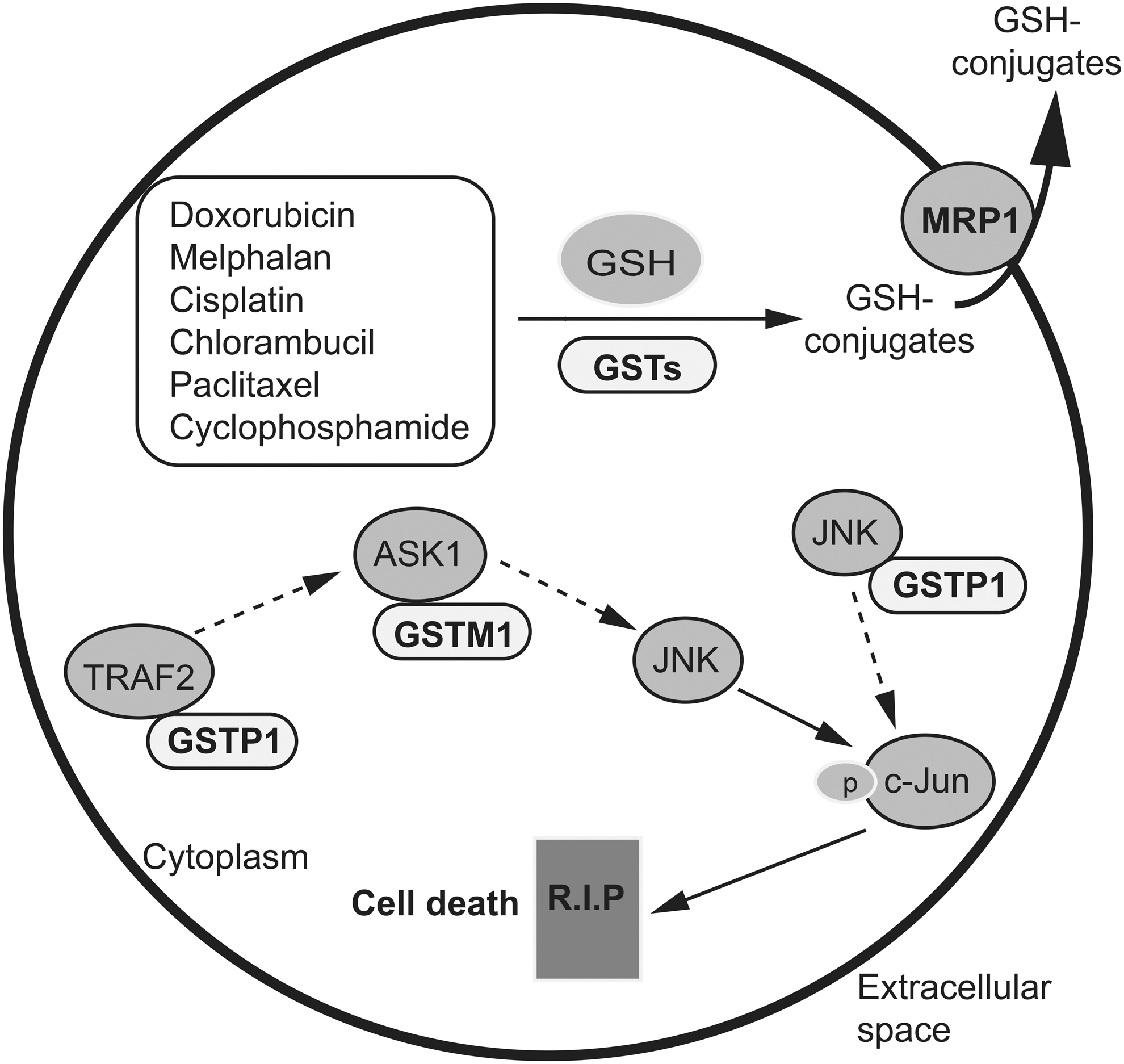
In addition to contributing to direct detoxification, several GST isoforms play roles in the regulation of signaling pathways (107, 150) (Fig. 4). For example, GSTP1 is involved in protecting cells against apoptotic signals by inhibiting c-jun N-terminal kinase 1 (JNK1) of the mitogen-activated protein kinase (MAPK) pathway via protein–protein interactions (1). Oxidative stress could cause the dissociation of GSTP1 from JNK1, allowing JNK1 to phosphorylate and activate c-Jun, which, in turn, activates the genes that are involved in apoptotic cell death pathways (15). JNK1 activation contributes to anticancer drug-induced cytotoxicity. It has been demonstrated that inhibition of GSTP1 is sufficient to cause JNK1 activation and it leads to apoptosis in cisplatin-sensitive and -resistant human osteosarcoma cell lines (126). Further, GSTP1 binds to tumor necrosis factor (TNF)-receptor-associated factor 2 (TRAF2), thus blocking its interaction with apoptosis signal-regulating kinase 1 (ASK1) and TRAF2-ASK1-induced apoptosis (167). GSTP1-mediated regulation of the MAPK signaling pathway provides a concrete example of GST-linked drug resistance mechanisms that do not involve direct detoxification by GSTs. In addition, GSTM1 binds to ASK1 and inhibits its ability to activate the JNK and p38 signaling pathways (20). The mechanism is similar to that proposed for GSTP1-JNK1. Altogether, these findings add a new dimension to the role of GSTs in drug resistance. Strategies to disrupt GST-JNK interactions could have a therapeutic impact, as the incidence of high GST levels is common in most major tumor types.
Increased GGT Activity and xCT Overexpression Enhance Glutathione Generation and Drug Resistance
GGT catalyzes the hydrolysis of the γ-glutamyl bond of extracellular GSH, GSSG, and GSH conjugates, thus releasing glutamate (57, 166) (Fig. 1). The cysteinylglycine or cystinylglycine dipeptide, resulting from GGT activity, can be cleaved by several dipeptidases on the surface of the cell, releasing cysteine (cystine) and glycine. Cysteine forms a key redox couple on its own with cystine. Glutamate and glycine can be transported into the cell by their respective amino acid transporters. Cysteine, which is the rate-limiting substrate for GSH synthesis, is taken up via alanine-cysteine-serine transporters, whereas cystine is taken up by the cystine/glutamate antiporter Xc −, composed of the xCT subunit credited with the transport activity and a heavy chain (4F2hc) (81). Once inside the cell, cystine is reduced to cysteine by GSH or TrxR1. Hence, the resulting amino acids are taken up by the cell and used for intracellular re-synthesis of GSH or other needs, providing the basis for the recycling of excreted GSH and GSSG in the salvage cycle. The cleavage of GSH conjugates and other γ-glutamyl compounds by GGT also plays an important role in the metabolism of leukotriene C4, prostaglandins, and estrogen. The removal of the γ-glutamyl moiety from GSH conjugates is often an initial step in the conversion of xenobiotic compounds, such as carcinogens and anticancer drugs, in detoxification processes (14).
Deregulation of GGT expression has been detected in several cancer types (114). In addition, the induction of GGT during cancer treatment has been observed in drug-resistant cancers (82). The expression of GGT provides cancer cells with an additional source of cysteine and cystine, allowing GGT-positive tumors to maintain higher levels of intracellular glutathione and thereby enhancing their resistance to pro-oxidant anticancer therapy. Several clinical studies have shown a strong correlation between GGT expression in tumors and poor patient survival (50).
The replenishment of intracellular glutathione depends largely on the availability of cysteine or cystine. Therefore, xCT has an important role in cellular cysteine and glutathione homeostasis. Elevated xCT expression is associated with the chemoresistance of cancer cells. For example, xCT expression levels negatively correlate with drug potency across the NCI-60 cancer cell lines (65), and the overexpression of xCT is commonly observed in drug-resistant cancers and considered a marker of poor patient survival (70, 75, 121). The regulatory mechanism of the human SLC7A11 gene, encoding xCT, is still poorly understood. The SLC7A11 gene is at least under the coordinate control of Nrf2 and activating transcription factor 4 (ATF4) (123, 174). Cancer stem-like cells (CSCs) often display high ROS-scavenging capacity and lower cellular ROS levels than the corresponding non-tumorigenic cells (111, 182). CD44 is expressed in CSCs and is a major CSC marker. Interestingly, a CD44 variant (CD44v) interacts with and stabilizes xCT in gastrointestinal cancer cells, resulting in increased cystine uptake (68, 81). The blocking of cystine uptake attenuates the metastatic potential and drug resistance of CD44v-expressing cells (68, 178). There are no reports on the co-regulation of xCT and the expression of GGT. Both can increase the supply of cysteine to cells, helping to maintain intracellular GSH levels and increasing resistance to anticancer drugs.
Cross-Talk Between the Glutathione and Trx Pathways Contributes to Drug Resistance
Similar to glutathione, the deregulation of the Trx pathway is highly involved in cancer biology and drug resistance. A complex interplay and compensatory activity between Trx- and glutathione-dependent pathways has been documented in different organisms, and in mammalian cells and cancer cells (19, 59, 119, 148). For example, a deficiency in GSH biosynthesis in keratinocytes is efficiently compensated by the cysteine/cystine and Trx systems (148). At physiological concentrations, GSH, together with GR and NADPH, can reduce Trx1 in vitro, and this reaction is strongly stimulated by Grx1 (34), indicating a backup function of the glutathione-dependent pathway in the reduction of Trx1 in cells lacking Trx reductase (TrxR1) activity. Similarly, TrxR1 deficiency results in compensatory upregulation of glutathione-metabolizing enzymes. The survival and growth of TrxR1-deficient tumors rely on a functional glutathione-dependent pathway (59). Reciprocally, glutathione-depleted cells and tumors upregulate the Trx pathway to support tumorigenesis. Another example of the functional interplay between the glutathione and Trx pathways is the rescue of cell growth by xCT overexpression in cells that are completely deficient in GSH production, which is highly dependent on the activity of the cytosolic Trx/TrxR1 system, presumably to reduce the excess of intracellular cystine (92). Functional redundancy between the glutathione and Trx pathways is the biological basis for therapeutic rationales that target both redox systems to achieve synergistic anticancer effects (12, 47, 156).
Elevated or Sustained Glutathione Levels Prevent Activation and Progression of Cell Death
The signaling pathways that regulate cell death have been extensively studied and characterized (43). Although oxidative stress and ROS/RNS formation have long been believed to be major players in the regulation of cell death, an important and direct role of glutathione/glutathione-based systems in the protection of cells against different forms of cell death, including apoptosis, necrosis, ferroptosis, and autophagy, has been increasingly recognized. Apoptosis mediated by extrinsic (receptors) and intrinsic (mitochondrial) pathways is a highly organized program that is characterized by the progressive activation of apoptotic signaling cascades. Necrotic cell death is a morphologically distinct form of cell death due to bioenergetic failure and oxidative damage. Although initially believed to be passive, it is now evident that the execution of necrotic cell death can also be finely regulated by specific signaling pathways and catabolic processes (necroptosis) (18). Autophagy is a major catabolic pathway by which eukaryotic cells degrade and recycle macromolecules and organelles. Ferroptosis is an oxidative, iron-dependent form of cell death that is distinct from apoptosis, classic necrosis, autophagy, and other forms of cell death. These cell death pathways are generally classified by biochemical and morphological criteria, whereas there are numerous examples of cell death displaying mixed characteristics.
Glutathione is a prominent protagonist in the extensive network that governs the decision between life and death of the cell. The links between glutathione and cellular apoptotic processes have been the most widely studied (21, 39). Glutathione depletion is a common early event preceding cell death, sensitizing cells to further stress insults, whereas glutathione replenishment confers drug resistance. Glutathione depletion during apoptosis can result from distinct mechanisms: (i) increased use of GSH by its oxidation to GSSG by ROS/RNS; (ii) export of cellular glutathione into the extracellular space, an important event that either initiates apoptotic signaling or promotes apoptotic progression; (iii) increased use of GSH in protein S-glutathionylation; or (iv) impairment of de novo GSH synthesis, as GCL could be a direct target of caspase 3, preventing GSH replenishment (41). In addition, the conjugation of glutathione with xenobiotics and electrophiles, and their subsequent extrusion by specific plasma membrane transporters, could also contribute to glutathione depletion during apoptosis.
Numerous studies using different cellular models have shown that glutathione depletion can affect apoptosis at multiple levels (Fig. 5). Glutathione depletion modulates the permeability transition pore (PTP) of the mitochondria, a channel complex that spans outer and inner mitochondrial membranes at points of contact (7, 9, 38). PTP is a major player in initiating both apoptotic and necrotic cell death, and a target of oxidative stress in mitochondria. In addition, glutathione depletion triggers mitochondrial B cell lymphoma 2 (Bcl-2)-associated X protein (Bax) translocation and cytochrome c (Cyt c) release (53). Released Cyt c requires depletion of cytosolic glutathione for its proapoptotic acitivity (109). On the other hand, glutathione content has been shown to be linked to the antiapoptotic role of Bcl-2 (85). For example, Bcl-2 regulates mitochondrial glutathione (mGSH) content by the direct interaction of the BH3 groove with GSH (183). This physical interaction contributes to the ability of Bcl-2 to modulate ROS levels through, at least partially, regulation of the mGSH pool, providing a survival advantage for Bcl-2-overexpressing cells. In addition, Bcl-2 overexpression reduces glutathione efflux from the cells (104, 105). Depletion of intracellular glutathione overcomes Bcl-2-mediated resistance to apoptosis (9, 122, 177). Glutathione depletion may also be a prerequisite for oxidative stress and the activation of cell death pathways (Fig. 5). Glutathione depletion occurs at earlier stages of the cell death program and is followed by secondary ROS accumulation (26, 40, 53, 77). In some cases, cellular glutathione content, rather than an excess of ROS and oxidative stress, regulates apoptosis (40). As already discussed, post-translational modification of redox-sensitive cysteines through S-glutathionylation results in glutathione depletion. Increasingly, the specific activation/deactivation of proteins via S-glutathionylation is increasingly considered an important regulator of cellular signaling pathways, including those involved in the apoptotic cascade (3). For example, TNF-induced apoptosis is accompanied by a significant increase in GRX activity and deglutathionylation of caspase-3. Deglutathionylated caspase-3 is a better substrate for caspase-8 and can be quickly cleaved by it (66, 108). The degree to which apoptotic components are affected by S-glutathionylation, specific cysteine residues that are susceptible to this post-translational modification, and the functional consequences are yet to be determined. Finally, cellular glutathione is implicated in MAPK-induced apoptosis. The Trx1/ASK1 and GSTP1/JNK complexes in the MAPK pathway function as redox switches that can be turned on or off by ROS. It is conceivable that cellular glutathione is a modulator of MAPK pathways, since glutathione is a key determinant of intracellular redox homeostasis and a major antioxidant. Indeed, a GSH/GSSG redox imbalance has been shown to activate MAPK signaling and exacerbate apoptosis in several cell models. Further, GSH-dependent S-glutathionylation of proteins with redox active cysteines has been implicated in MAPK signaling, although the mechanism is not well understood.
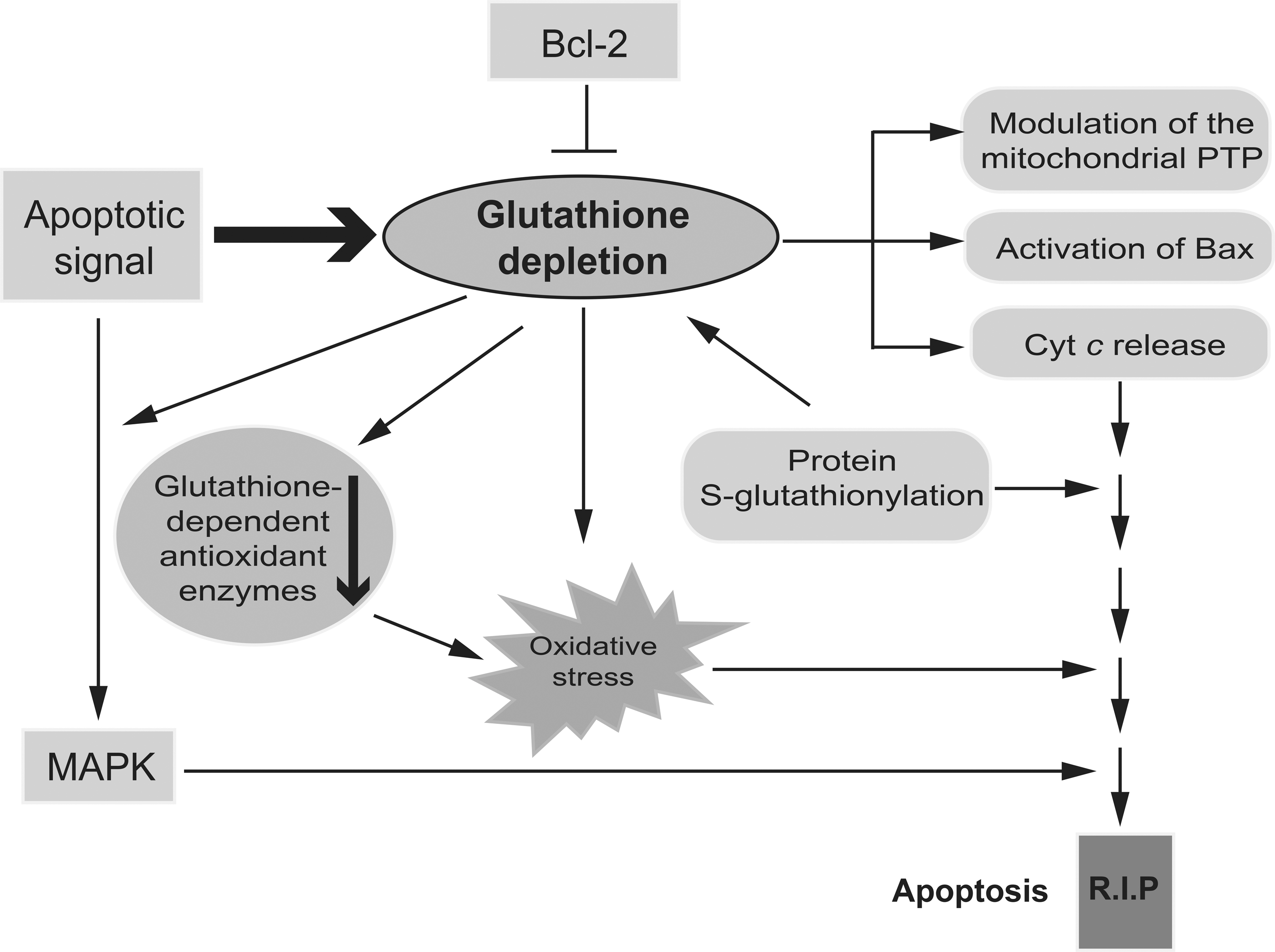
The precise contribution of the cytosolic versus mGSH pool in apoptosis is not well characterized. The role of mGSH is of particular relevance due to the pivotal role of mitochondria in apoptosis and ROS generation (94). Similar to the cytoplasmic GSH pool, reduced mGSH serves as a source of reductive power for certain antioxidant proteins, such as GPx. mGSH, mitochondrial membrane-bound GST, which has both glutathione transferase and peroxidase activities, and specific mitochondrial GPx, such as GPx4, provide the primary defense against oxidative damage to mitochondrial membranes by reducing H2O2 present on phospholipids and other lipid peroxides (5, 23, 129). GPx4 plays a role in preventing apoptosis and the maintenance of oxidative phosphorylation complexes in gut epithelial cells. Similary, ectopic GPx4 expression prevents TNF-α-induced ROS formation, phospholipid peroxidation, mitochondrial damage, and apoptotic death in Jurkat cells (79). Conversely, GPx4 knockdown enhances phospholipid peroxidation and increases TNF-α-dependent apoptosis (79). The anti-apoptotic effect of mGSH is also reflected by the protective role of mGSH in cardiolipin, an anionic phospholipid of the mitochondrial inner membrane that is prone to free radical oxidation (175). Cardiolipin regulates apoptosis by playing an important role in Cyt c release from mitochondria. After an apoptotic stimulus, mitochondrial ROS can oxidize cardiolipin, decreasing its affinity for Cyt c, and facilitate Cyt c detachment from the mitochondrial inner membrane (100, 131). As mitochondrial ROS are controlled by glutathione and other antioxidants, mGSH appears to modulate apoptotic cell death by indirectly regulating the redox state of cardiolipin. Globally speaking, the mechanistic details of how changes in compartmental glutathione redox status regulate signaling pathways that affect cell survival or death are yet to be resolved.
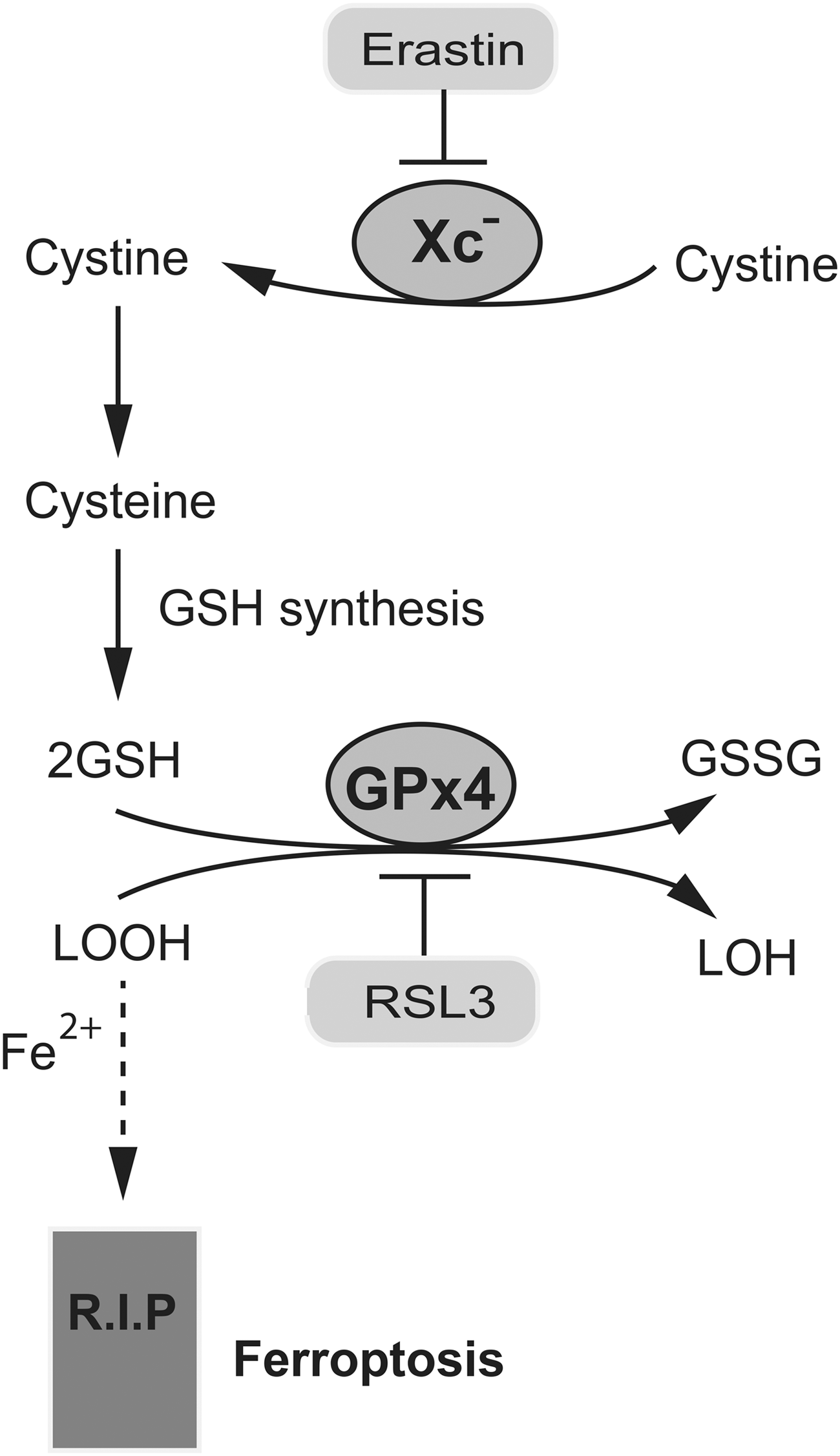
Recent studies have also revealed a protective role of glutathione in ferroptosis, an oxidative, iron-dependent form of cell death that is distinct from other forms (17, 169). The recognition of ferroptosis as a unique form of regulated cell death initially emerged from characterizing the lethal mechanism of action of the small molecules erastin and RSL3 (33) (Fig. 6). Erastin triggers ferroptosis by inhibiting the cystine/glutamate antiporter Xc −, causing significant intracellular glutathione depletion, and RSL3 targets the GPx, GPx4, during the initiation of ferroptosis (172). GPx4 peroxidase activity is directed toward lipid-based ROS, particularly lipid hydroperoxides (L-OOH), and uses the reducing power of glutathione. Erastin and RSL3 share a common cell death execution mechanism, although their initial targets are different. GPx4 is a central regulator of ferroptotic cell death, akin to Bcl-2 in apoptosis. Glutathione depletion or GPx4 inactivation causes iron-dependent accumulation of lipid-based ROS and the depletion of polyunsaturated fatty acids (PUFAs), leading to ferroptotic cell death. The molecular events that occur downstream of the oxidative fragmentation of PUFA to cause irreversible cell death are not fully defined. Further, although glutathione depletion can trigger either apoptosis or ferroptosis, how the decision between these two mechanisms is made is unknown.
A protective role of glutathione in other cell death processes has also been reported. For example, NAC prevents ROS-induced formation of autophagosomes and the subsequent degradation of proteins during starvation-induced autophagy (130). Lipopolysaccharide-induced autophagy is associated with ROS formation and glutathione depletion, and it is also prevented by NAC (179). Excessive glutathione depletion and oxidative stress have been reported to switch apoptosis to necrotic cell death (27, 158, 159). Ceramide is implicated as a secondary messenger for TNF-α-induced cell necrosis, and NAC or glutathione-monoethyl ester can delay the onset of ceramide-induced necrosis (27). Acute pancreatitis is an inflammatory process of the pancreatic gland that may eventually lead to a severe systemic inflammatory response and necrotic cell death. Pancreatic glutathione depletion is a hallmark of this disease that occurs in its initial phase. Administration of glutathione-monoethyl ester in mice increases pancreatic glutathione levels and has beneficial effects in acute pancreatitis, whereas the chemical inhibition of GSH synthesis by BSO worsens pancreatic necrosis (110).
Targeting Glutathione and Glutathione-Based Systems as a Rational Anticancer Strategy
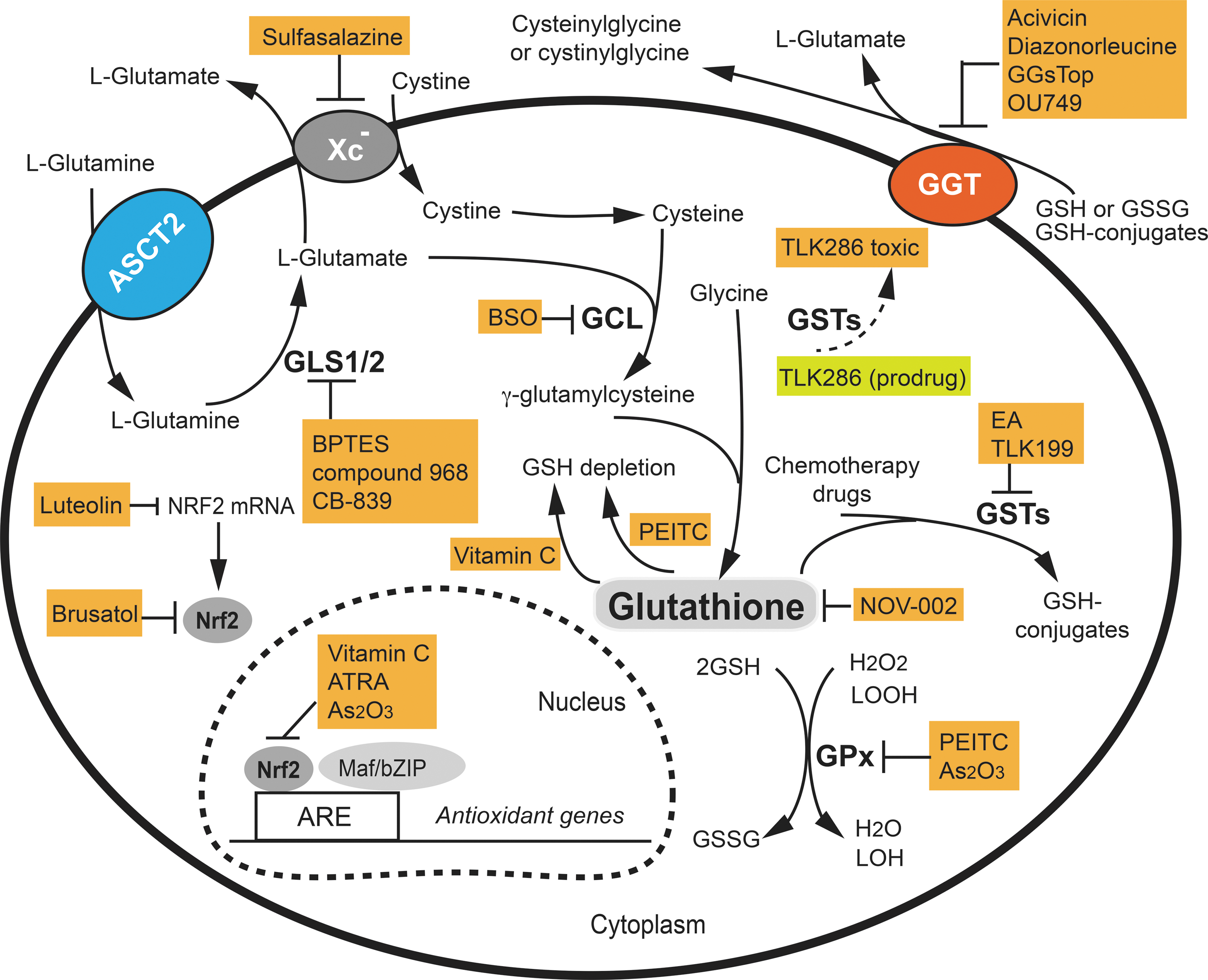
We have highlighted several key pathways underlying the pivotal role of glutathione and glutathione-based systems in resistance to the induction of cell death and anticancer drugs. The modulation of glutathione and glutathione systems, thus, represents a promising therapeutic strategy. Such mechanism-based pharmacological approaches have been studied for more than a decade with encouraging results (Fig. 7).
Pharmacological modulation of Nrf2 activity
Nrf2 is considered a promising target in cancer therapy as it is a master regulator of oxidative stress responses and multiple glutathione-related genes. There are many compounds that activate Nrf2, but no specific Nrf2 inhibitors are currently available. Among the molecules that have shown an Nrf2 inhibitory effect, ascorbic acid (vitamin C) was found to restore imatinib sensitivity to the imatinib-resistant cell line KCL22/SR, at least in part via inhibition of Nrf2-mediated gene expression by suppression of Nrf2 binding to AREs (146). All-trans retinoic acid (ATRA) significantly reduces the binding of Nrf2 to AREs and the ability of Nrf2 to activate its target genes in both cell and mouse models (42, 163). In the presence of ATRA, retinoic acid receptor α (RARα) forms a complex with Nrf2, preventing its binding to ARE (163). Brusatol, purified from extracts of the plant Brucea javanica, is able to enhance the depletion of Nrf2 protein in a Keap1-independent manner and to inhibit the Nrf2-mediated stress response and tumor growth, both in vitro and in vivo (103, 120). However, brusatol is cytotoxic, and its specificity and the precise mechanism by which it depletes Nrf2 are not yet understood. The flavonoid, luteolin, found at high concentrations in celery, green pepper, parsley, and perilla leaf, is a selective Nrf2 inhibitor. Non-small-cell lung cancer A549 cells possess constitutively active Nrf2. Luteolin treatment of A549 cells markedly reduced both Nrf2 mRNA and protein levels, leading to decreased Nrf2 binding to AREs, downregulation of ARE-driven genes, and glutathione depletion. The activity of luteolin appears to be related to its role in accelerating the turnover of Nrf2 mRNA (145). However, the antitumor properties of luteolin have also been attributed to other mechanisms (73, 87). The Nrf2-drived pathway represents one of the most important cellular defense mechanisms against oxidative stress and xenobiotic damage and plays a pivotal role in drug resistance. High-resolution structural studies of the Keap1-Nrf2 interaction should provide the necessary information for the rational design of inhibitors in the future.
Drugs that target glutathione metabolism and the redox state
Possible strategies for the depletion of cytosolic and mGSH that could sensitize cancer cells to chemo- and radiotherapy include inhibiting GSH synthesis, increasing the efflux of cytosolic glutathione, creating a shortage of cysteine, glutamate, and glycine, and inhibiting glutathione transport into mitochondria. As the rate-limiting enzyme in GSH synthesis, GCL has been an anticancer drug target for more than 30 years. The classical drug used to inhibit GCL activity is BSO (49), which has been routinely used to diminish intracellular GSH levels both in vitro and in vivo (Fig. 7). For example, BSO triggers apoptosis as a single agent and in combination with arsenic trioxide (As2O3) in solid tumors and acute promyelocytic leukemia (APL) cells (28, 90). In a phase I clinical study, continuous infusion of BSO produced consistent and profound glutathione depletion in tumors (<10% of pretreatment value) in patients with various types of cancer (ovarian, lung, breast, and colon cancer; melanoma) without significant BSO-related toxicity. A recent clinical pilot study showed that BSO-mediated glutathione depletion in combination with melphalan had therapeutic activity against recurrent or refractory high-risk neuroblastoma in children (4).
GSH depletion has also been demonstrated to be a part of the mechanism responsible for the anticancer activity of high doses of vitamin C (180). Vitamin C is oxidized to dehydroascorbate (DHA) in cell culture media and, subsequently, imported into cells by the glucose transporter GLUT1. High doses of vitamin C and the increased uptake of DHA into KRAS or BRAF mutant cancer cells, which express high levels of GLUT1, led to the accumulation of intracellular DHA. GSH is used as a reducing agent for the conversion of DHA into vitamin C. Thus, high doses of vitamin C cause the depletion of GSH and NADPH, and ultimately ATP, inducing an energetic crisis (180).
Another approach that is used for altering intracellular glutathione levels is based on manipulating the efflux of GSH conjugates out of the cell. This strategy takes advantage of the detoxifying role of glutathione toward xenobiotics, such as β-phenethyl isothiocyanate (PEITC). PEITC is a natural compound with chemopreventive and anticancer activity and it is found in consumable cruciferous vegetables (55). One of the molecular mechanisms underlying the anticancer properties of PEITC is the formation and efflux of GSH conjugates, disabling the GSH antioxidant system through depletion of the cellular glutathione pool and inhibition of GPx enzymes (55, 157) (Fig. 7). Cellular sensitivity toward PEITC correlates with constitutively high cellular glutathione levels (141). This may also explain the selective toxicity of PEITC for cancer cells, which generally have high glutathione levels. Available data from pre-clinical and clinical studies suggest PEITC to be among the promising anticancer agents available from natural sources (55).
GSH synthesis can also be modulated through the regulation of intracellular cysteine levels. The xCT subunit of cystine/glutamate antiporter Xc − is responsible for the export of glutamate in exchange for cystine (81). Sulfasalazine, an FDA-approved salicylate-based anti-inflammatory and immune-modulatory drug, has been shown to be a potent inhibitor of xCT (48) (Fig. 7). Several studies have shown that sulfasalazine used alone, or in combination with anticancer drugs, inhibits the growth of a large spectrum of human cancer cells in cell culture and/or mouse models (83, 89, 101, 173). Thus, sulfasalazine could be used as a sensitizer of chemotherapy and radiotherapies in cancer patients. More recently, SLC7A11, which encodes xCT, was shown to be specifically overexpressed in a panel of mouse and human triple-negative breast cancer cell-derived CSC-enriched tumorspheres (78). DNA vaccination-based immunotargeting of xCT in mice delayed established subcutaneous tumor growth, suppressed metastasis formation, and increased CSC chemosensitivity to doxorubicin.
Similarly, GGT cleaves extracellular GSH and GSSG, providing cells with an additional source of cysteine and cystine and increasing intracellular glutathione levels. The commonly used potent GGT inhibitors are glutamate analogs (Fig. 7). Acivicin and diazonorleucine are perfectly adapted for experimental research (2, 147), but they are too toxic for clinical use (35, 62). A new group of glutamate analogs and irreversible GGT inhibitors have been reported (56). The lead compound, GGsTop, inhibited renal GGT, showing a reno-protective effect in a rat ischemia/reperfusion-induced renal injury model (171), and inhibited lung lining fluid GGT activity in a mouse asthma model (160). The effects of this compound on the GGT of cancer cells have not been reported. Finally, another class of non-competitive inhibitors of GGT, OU749 and its analogs, has been reported (74, 165). These compounds are considered less toxic than glutamate analogs and have a large therapeutic window. Again, their effects on the GGT of cancer cells require further assessment.
The targeting of glutamate represents another strategy to alter intracellular glutathione levels. Glutamate is required for de novo GSH synthesis and for cells to acquire cystine through the Xc − antiporter. Glutamine is the precursor of glutamate in a reaction that is catalyzed by GLSs (95). In addition, glutamine metabolism has become a hot topic in cancer therapy in recent years as it is one of the key energy sources for many cancers (29, 61, 161). Thus, the targeting of GLSs and glutamine would have direct effects on glutamate synthesis and lower cellular glutathione levels (Fig. 7). Indeed, pharmacological inhibition of GLS by Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), CB-839, and compound 968 slowed the proliferation of several types of cancer cells in vitro and in xenograft models (52, 136, 168). Administration of BPTES slowed cancer cell proliferation in an immune-competent Myc-mediated mouse model of hepatocellular carcinoma through the inhibition of GLS, and it prolonged survival with no apparent toxicity (168).
Another potentially promising anticancer drug that affects glutathione metabolism is NOV-002, a GSSG mimetic. NOV-002 consists of GSSG formulated with cisplatin at an ∼1000:1 ratio (155). NOV-002 affects cellular redox homeostasis and exerts pleiotropic effects on oxidative signaling (54, 154). Administration of NOV-002 in combination with adjuvant chemotherapy to patients with HER2-negative breast cancers resulted in a favorable response rate and the mitigation of side effects compared with adjuvant chemotherapy alone (99).
Drugs that target GSTs
GSTs are involved in cellular biotransformation of electrophilic compounds, leading to increased detoxification of anticancer agents and the development of drug resistance. GST inhibitors are, therefore, potential cancer therapeutic targets. Among the various GST isoenzymes, GSTP1 has received more attention because it is usually overexpressed in cancer and has been associated with tumor drug resistance (134). Several early studies focused on the FDA-approved diuretic agent, ethacrynic acid (EA), which inhibits GST activity by binding to both GST and GSH (Fig. 7). EA was found to sensitize cancer cells to several anticancer drugs in vitro, but the lack of GST isoenzyme specificity and diuretic side effects limit its clinical use (44). Another class of GST inhibitors are GSH analogs that decrease GST catalytic efficiency. As a selective GSTP1 inhibitor, TLK199 (Ezatiostat HCl) potentiates the toxicity of several anticancer agents. An important feature of TLK199 is its capacity to activate JNK, thus promoting the growth and maturation of hematopoietic progenitor stem cells. This activity makes TLK199 a novel therapeutic agent for patients with myelodysplastic syndromes (91, 117, 118). Finally, another approach targeting GST has been to design GST-activated prodrugs, converting an inactive prodrug into a cytotoxic species. TLK286 (Canfosfamide HCl) is the lead candidate from this class of drugs (116, 150). TLK286 has been shown to act synergistically with a variety of chemotherapeutic agents, confirming that the cytotoxic potency of the drug increases in cells that overexpress GSTP1 (116).
Drug combination strategies to target both glutathione and other pathways
Rational drug combination directed against multiple targets is an effective anticancer strategy, as it decreases the risk of cancer drug resistance and allows the use of lower therapeutic doses. It is possible to maximally exploit redox mechanisms as a therapeutic strategy by conceiving different types of drug combinations: (i) combining one drug that targets the glutathione pathway with a drug targeting the Trx pathway; (ii) combining one drug that induces ROS generation with a drug that inhibits the cellular antioxidant capacity; and (iii) combining one drug that targets redox pathways with a standard chemotherapeutic drug or radiotherapy. Examples have accumulated that demonstrate the synergistic effect of rational drug combinations. This approach might be particularly useful in cancer cells that have become adapted to stress and are resistant to anticancer agents. These strategies are beyond the scope of this review, but readers are invited to consult recent comprehensive reviews on this important topic (12, 47, 156). Some redox modulating drugs, in fact, target multiple pathways. For example, As2O3 is a highly efficient pro-oxidant drug for the treatment of APL. In addition to its effect on reducing the binding of Nrf2 to ARE (163) (Fig. 7), As2O3 also inhibits GPxs and mitochondrial respiration, thereby causing increased ROS leakage, contributing to its antileukemia activity (113). Further, irreversible inhibition of TrxR is a key mechanism in As2O3-induced breast cancer cell apoptosis (86).
Conclusions and Perspectives
A large body of evidence reviewed here underlines the important roles that glutathione and glutathione-based systems play in carcinogenesis and anticancer drug resistance. High glutathione levels favor cancer initiation, progression, and metastasis in several types of cancer. In established tumors, aberrant Nrf2 activity, GGT activity, and xCT overexpression promote GSH synthesis/regeneration and inhibit cell death, whereas elevated GST activity and the complementary role of the Trx pathway further contribute to drug resistance. It will be important to uncover distinct mechanistic details regarding the role of glutathione in cancer initiation, progression, invasion, cell death, and drug resistance for a specific type of cancer. A detailed examination of the cross-talk between metabolic alterations and glutathione homeostasis will be helpful for dissecting the mechanisms leading to elevated or sustained glutathione levels in cancer cells. The broad application of state-of-the-art technologies, including the genetically encoded fluorescent redox-sensitive sensors, should lead to new insights into glutathione dynamics in subcellular compartments. One aspect that is frequently ignored relates to the redox-independent function of glutathione and should be explored in the context of carcinogenesis and anticancer drug resistance. Phamarcological interventions that target glutathione pathways should focus on mechanism-based rational drug combinations that are directed against multiple redox targets. A better understanding of the mechanisms of redox regulation in cancer cells and the development of effective, specific, and clinically safe inhibitors should offer new perspectives and successful strategies for cancer treatment.
Footnotes
Acknowledgments
Research in the authors' laboratory is funded by the French Centre National de la Recherche Scientifique (CNRS), the Institut Curie, and la Fondation ARC grant (PJA 20151203330) to M.E.H. E.H. is supported by the Ligue Nationale Contre le Cancer, and N.E.B. is supported by the Université Paris-Saclay.