
Abstract
Significance:
The precise role and impact of reactive oxygen species (ROS) in stem cells, which are essential for lifelong tissue homeostasis and regeneration, remain of significant interest to the field. The long-term regenerative potential of a stem cell compartment is determined by the delicate balance between quiescence, self-renewal, and differentiation, all of which can be influenced by ROS levels.
Recent Advances:
The past decade has seen a growing appreciation for the importance of ROS and redox homeostasis in various stem cell compartments, particularly those of hematopoietic, neural, and muscle tissues. In recent years, the importance of proteostasis and mitochondria in relation to stem cell biology and redox homeostasis has garnered considerable interest.
Critical Issues:
Here, we explore the reciprocal relationship between ROS and stem cells, with significant emphasis on mitochondria as a core component of redox homeostasis. We discuss how redox signaling, involving cell-fate determining protein kinases and transcription factors, can control stem cell function and fate. We also address the impact of oxidative stress on stem cells, especially oxidative damage of lipids, proteins, and nucleic acids. We further discuss ROS management in stem cells, and present recent evidence supporting the importance of mitochondrial activity and its modulation (via mitochondrial clearance, biogenesis, dynamics, and distribution [i.e., segregation and transfer]) in stem cell redox homeostasis.
Future Directions:
Therefore, elucidating the intricate links between mitochondria, cellular metabolism, and redox homeostasis is envisioned to be critical for our understanding of ROS in stem cell biology and its therapeutic relevance in regenerative medicine. Antioxid. Redox Signal. 29, 149–168.
Introduction
S
The regenerative potential of stem cells is intimately connected to intracellular reactive oxygen species (ROS) levels and cellular redox homeostasis (Fig. 1). At steady state, ROS are maintained at low basal levels in HSCs (relative to committed progenitors) and have been shown to increase with differentiation (48, 90). The low basal ROS levels serve to preserve stem cell potential by maintaining an appropriate balance between stem cell quiescence, differentiation, and self-renewal (57, 137, 141). Suppression of ROS in NSCs and HSCs below basal levels leads to significantly reduced regenerative potential, characterized by impaired proliferation (e.g., exit from quiescent G0 phase), differentiation, and self-renewal (60, 79, 141). Conversely, excess ROS have been associated with a decline in the function and regenerative potential of multiple stem cell compartments (10, 13, 25, 38, 53, 54, 67, 107, 108, 141). For instance, ROS accumulation in HSCs results in severely impaired long-term reconstitution potential, and exhaustion of the stem cell pool (53, 54, 141), which was attributed to p38-mitogen-activated protein kinase (MAPK)-mediated loss of quiescence and senescence induction (54). Notably, further elevation in ROS induces cell death in stem cells (24, 90). Therefore, physiological levels of ROS are required for stem cell function, and can greatly influence cell-fate decisions.
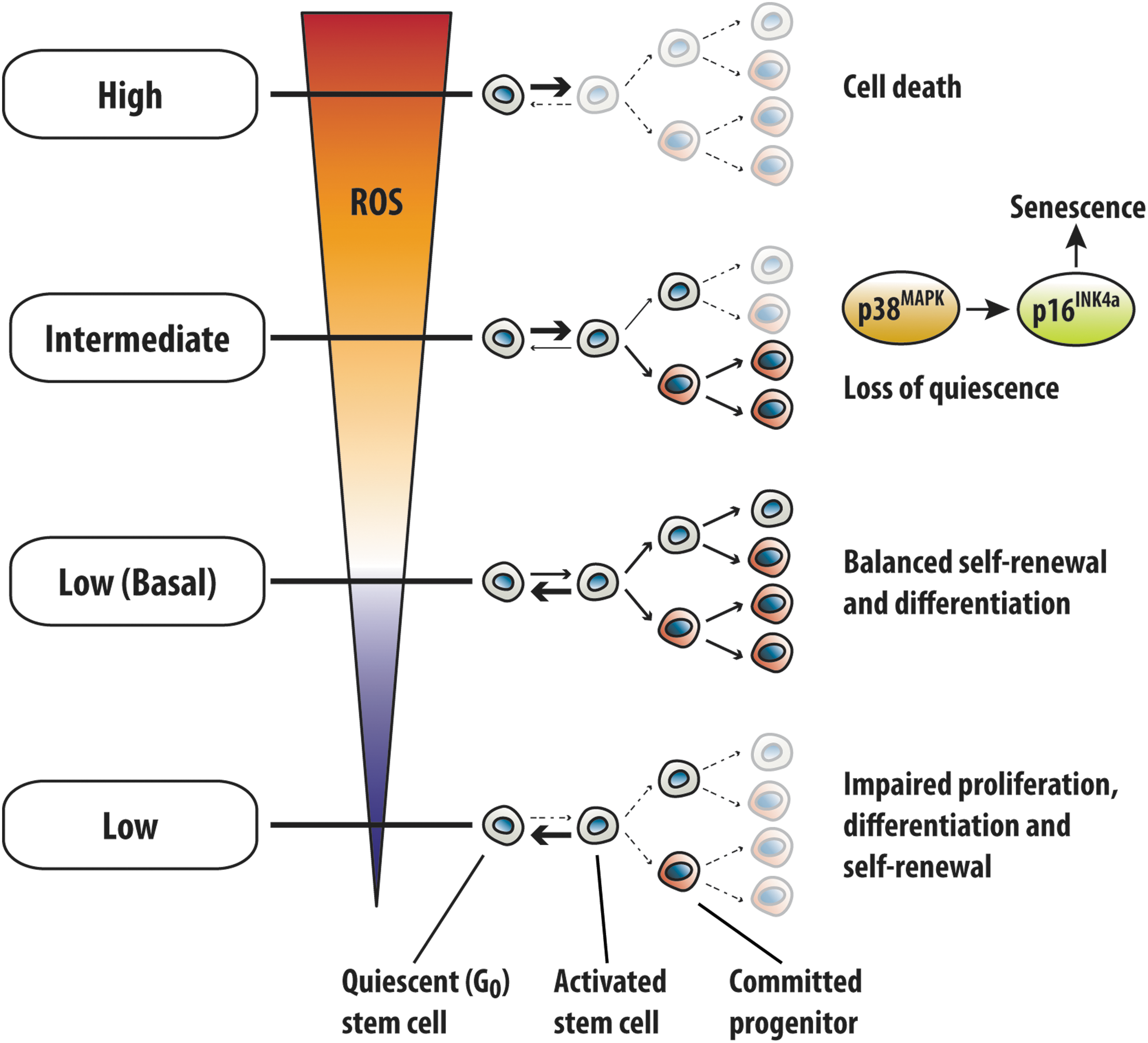
In this review, we explore the reciprocal relationship between ROS and tissue stem cells, with an emphasis on recent discoveries highlighting the importance of proteostasis (i.e., protein homeostasis) and mitochondria in stem cells. We begin with a brief overview of ROS generation in stem cells, followed by a discussion on the functional aspect of ROS in stem cells. We then review the impact of oxidative stress on stem cells, and further discuss how ROS are managed in stem cells. Finally, we examine a collection of recent studies on mitochondria and stem cells and discuss these in the context of redox homeostasis.
ROS in General
ROS encompass a diverse range of oxygen-containing entities that exhibit greater reactivity than molecular oxygen (O2). Collectively, ROS refer to both oxygen-free radicals and several nonradical species that easily give rise to radicals (43). Physiologically relevant ROS typically encountered intracellularly include superoxide radical anion (O2 •−), hydroxyl radical (OH•), and hydrogen peroxide (H2O2) (14). At steady state, these ROS exist at picomolar to nanomolar concentrations (14), with lifetimes ranging between nanoseconds to seconds, depending on the inherent reactivity and presence of antioxidants (30). Notably, the lifetime of H2O2 is greater than other intracellular ROS. In addition, H2O2 can easily diffuse across membranes (30). Thus, H2O2 is proposed to be the predominant ROS involved in cell function (e.g., redox signaling).
ROS are generated from the partial reduction of O2 to O2 •−, which occurs as a result of oxygen's preferential acceptance of one electron at a time during redox reactions. Mitochondria are a major source of ROS, a consequence of its role in energy (i.e., ATP) production via oxidative phosphorylation (OXPHOS). The processes underlying ROS generation in mitochondria have been reviewed by Murphy (103). In brief, OXPHOS involves sequential transfer of electrons along the electron transport chain (ETC), which predisposes O2 to one-electron reduction, resulting in O2 •− generation (103). The O2 •− generated is rapidly converted to H2O2 by mitochondrial superoxide dismutase 2 (SOD2) or cytosolic superoxide dismutase 1 (SOD1) (43). Thus, mitochondria are major sites of O2 •− and H2O2 generation, and serve as central modulators of redox homeostasis in stem cells (see the Mitochondria: The Epicenter of ROS Management in Stem Cells section).
Another major source of ROS is the NADPH oxidase (NOX) family of ROS-generating enzymes, which are potentially important in stem cells [reviewed by Skonieczna et al. (134)], although further studies are required to ascertain the role of NOX in stem cells. Other potential sources of ROS in stem cells include nitric oxide synthase (NOS) and cytochrome p450. However, further studies are needed to understand their role in stem cells.
The Need for ROS in Stem Cells
ROS have classically been viewed as detrimental to cellular function; however, there is a growing appreciation of the physiological utility of ROS in cell signaling (i.e., redox signaling). The mechanism underlying redox signaling entails ROS-mediated oxidation of target proteins (also known as redox sensors) at specific, redox-reactive amino acid residues that are critical for protein functionality. Specifically, the sulfur-containing amino acids cysteine (Cys) and methionine (Met) are known to be involved in redox signaling, of which the involvement of reactive Cys residues has been extensively documented and well established (48). Unlike other amino acid residues (including Met), Cys is inherently more sensitive to ROS-mediated oxidation because of its thiol group (−SH), which under physiological conditions predominantly exists as the strongly nucleophilic thiolate anion (−S−) (35). As such, exposure to physiological levels of ROS (e.g., nanomolar amounts of H2O2) readily oxidizes Cys thiolate anion (Cys−S−) residues to Cys sulfenic acid (Cys−SOH) residues (35). Similarly, the less reactive Met (Met−S−R′) residue can be oxidized to Met sulfoxide (Met−SO), although at a significantly slower rate than Cys, with similar rates for reactions involving highly reactive ROS (e.g., HOCl and OH•) (32). Importantly, both the Met−SO and Cys−SOH modifications are of lower oxidation states, and can easily be reversed by specific antioxidant defenses, thereby permitting reactive Met and Cys residues to serve as redox-dependent molecular switches. The reduction of Met−SO back to Met is catalyzed by methionine sulfoxide reductases [reviewed by Drazic and Winter (32)], whereas the reduction of Cys−SOH to Cys is dependent on several classes of proteins, including glutaredoxins, thioredoxins, and thioredoxin reductases. If not reversed, further exposure to high ROS levels results in hyperoxidation of Cys−SOH to Cys sulfinic acid (Cys−SO2H) followed by Cys sulfonic (Cys−SO3H) acid (35) and Met−SO to Met sulfone (Met−SO2) (32). Although sulfiredoxin can reduce Cys−SO2H back to Cys−SOH, this is restricted to the Cys−SO2H residues of peroxiredoxins (43). Thus, these high oxidation state modifications (Cys−SO2H, Cys−SO3H, and Met−SO2) are generally irreversible, resulting in permanent damage of the target protein. Therefore, a key aspect of redox signaling is the reversibility of ROS-modified reactive amino acid residues critical for target protein function.
Protein kinases
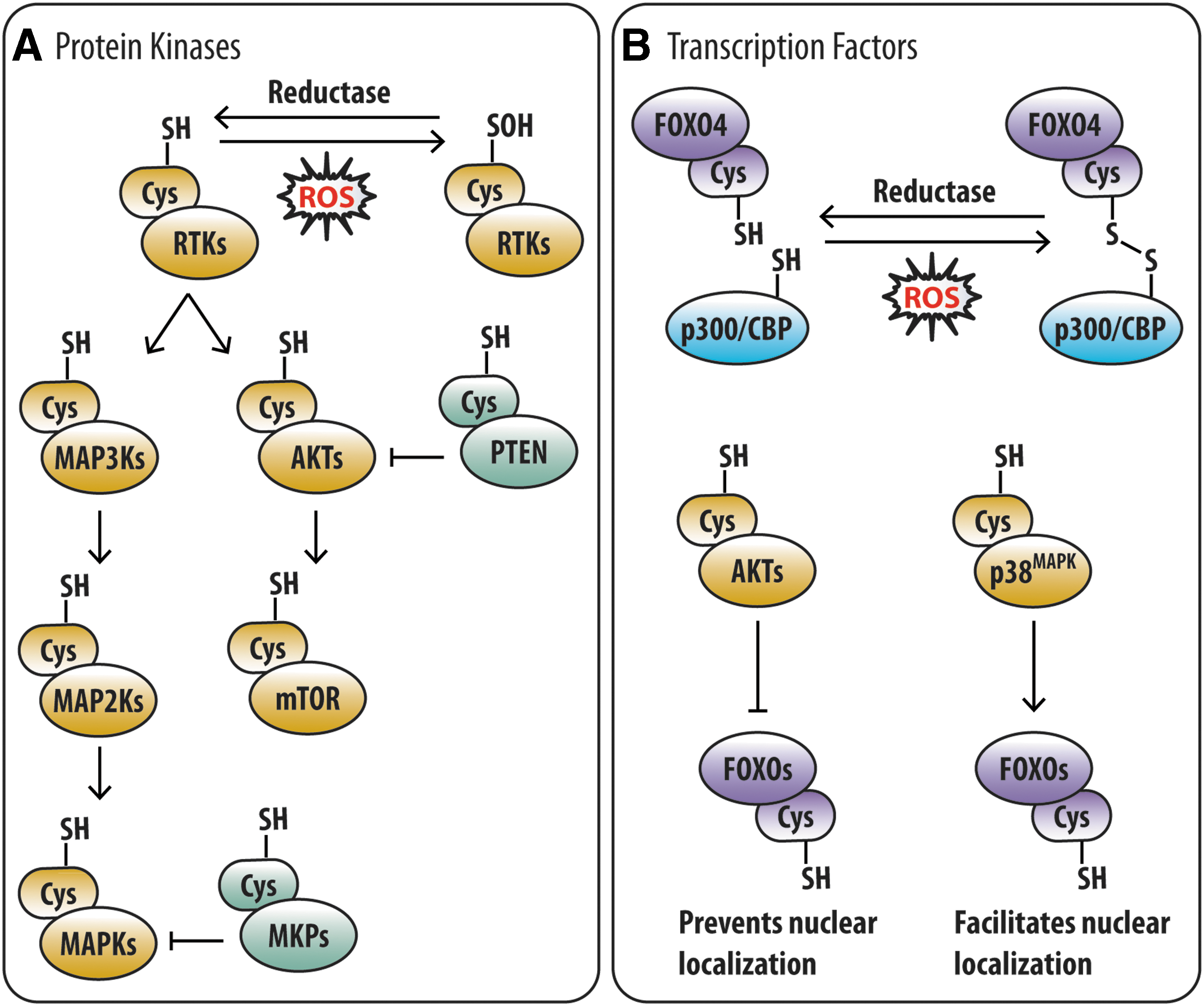
Diverse protein kinase families, which are important for stem cells, are amenable to redox modulation (8) (Fig. 2A). These include receptor tyrosine kinases (RTKs), AKTs, MAPKs, ataxia-telangiectasia mutated (ATM), and mechanistic target of rapamycin (mTOR). In fact, studies on growth factor stimulation of RTKs contributed to defining the principle of redox signaling. Initial indication of the importance of ROS in RTK signaling arose from observations that platelet-derived growth factor and epidermal growth factor (EGF) stimulation induced a sudden NOX-dependent spike in ROS (22, 109, 111), which was necessary for downstream signaling (5, 139). The spike in ROS directly modulates RTK activity via oxidative modification of a critical Cys residue within its active site, as demonstrated in the EGF receptor (111). In addition, the spike in ROS also modulates downstream RTK signaling by inactivating protein tyrosine phosphatases (PTPs) and dual specificity phosphatases (DUSPs) via oxidation of a conserved reactive Cys residue critical for phosphatase activity (77, 81, 123, 151). Thus, redox signaling encompasses oxidative modification of the kinase or its regulatory phosphatase.
The RTK c-KIT (CD117) is widely recognized as a marker used to identify hematopoietic stem/progenitor cells (HSPCs; Lineage-marker−Sca-1+c-Kit+), of which less than 10% are HSCs (116). Functionally, c-Kit is critical for HSC function (129, 145) and activates multiple downstream signaling pathways (AKT, MAPK, etc.) upon activation. Importantly, the magnitude of c-KIT signaling determines HSC reconstitution potential (131). Notably, the PTPs PTPN11/SHP2 and PTP4A2/PRL2, which positively regulate AKT and extracellular signal regulated kinase (ERK) activity downstream of c-KIT in HSCs, are required for stem cell maintenance (17, 73). In addition, phosphatase and tensin homologue (PTEN), a DUSP that negatively regulates AKT, is required for the maintenance of the HSC and muSC pool (167). Thus, ROS can direct stem cell fate by modulating the RTK signaling cascade via oxidative modification.
The AKT family of serine/threonine (Ser/Thr) kinases is also responsive to cellular redox state (i.e., redox responsive) and important for stem cells. AKT exhibits susceptibility to H2O2-mediated oxidation at two conserved Cys residues (Cys-297 and Cys-311), which predominantly exist in a reduced state (102). Exposure of AKT to H2O2 in vitro leads to disulfide bond formation between its two reactive Cys residues, recruitment of protein phosphatase 2A, AKT dephosphorylation, and suppression of AKT activity (102). Notably, ectopic expression of constitutively active AKT1 in HSPCs resulted in hyperproliferation, apoptosis, and eventual exhaustion of the stem cell compartment (68). Similarly, HSC exhaustion, involving loss of quiescence and hyperproliferation, resulted from inactivation of PTEN, a redox-responsive negative regulator of AKT (168). Very recently, the requirement for PTEN in muSC maintenance, by preventing spontaneous activation and premature differentiation, was demonstrated (167). Conversely, AKT1/2 double-deficient HSCs exhibited increased quiescence and impaired differentiation (60). Interestingly, these functional defects could be attributed to low ROS levels (60). Given that AKT signaling inhibits FOXO3 (12), which transcriptionally upregulates antioxidant genes to suppress ROS in HSCs (75, 93, 161), the redox responsiveness of AKT may serve as a feedback mechanism to regulate ROS in stem cells.
Another redox-responsive kinase family that regulates stem cell fate is the MAPK family of proline (Pro)-directed Ser/Thr kinases, comprising ERK, c-Jun N-terminal kinase (JNK), and p38-MAPK subfamilies. Like other kinase families, MAPK activity can be modulated by cellular redox state, both directly and indirectly. Treatment in vitro with a low H2O2 concentration (0.1 μM) was reported to oxidize ERK2 at Cys-38 and Cys-214, whereas significantly higher H2O2 concentrations (1 μM and 20 μM) were required for oxidative modification of reactive Cys residues in JNK2 and p38-MAPK (36). Mechanistically, the oxidative modification of ERK2 in LP07 cells was found to be required for its interaction with MEK1/2 (a mitogen-activated protein kinase kinase [MAP2Ks]) and nuclear translocation of the ERK2-MEK1/2 complex from the mitochondria (36). Likewise, direct oxidation of JNKs and p38-MAPKs has been proposed to modulate interaction with their cognate MAP2Ks (36), although this remains to be experimentally verified. Alternatively, modulation of MAPK activity can also occur indirectly via ROS-mediated inactivation of MAPK phosphatases (MKPs; Fig. 2A), which dephosphorylate and inactivate MAPKs. TNFα-induced ROS accumulation was shown to inactivate MKPs (MKP-1, -3, -5, and -7) in HeLa cells and fibroblasts via oxidative modification of a conserved Cys residue located within the catalytic pocket, resulting in sustained JNK activation (62). Sustained JNK activation was also observed in MIN6 cells upon oxidative modification of MKP-1 (49). Indirect modulation of MAPKs can also occur via redox regulation of upstream kinases (e.g., RTKs, MAP2Ks, and mitogen-activated protein kinase kinase kinase [MAP3Ks]; Fig. 2A). One such example is ASK1, an MAPK that is part of the JNK and p38-MAPK signaling cascade, which is inactive when associated with reduced thioredoxin (89). Upon ROS exposure, thioredoxin becomes oxidized and dissociates from ASK1, leading to the activation of ASK1 (89). Thus, redox modulation of MAPK signaling in stem cells can occur at multiple levels along the signaling cascade.
The p38-MAPK subfamily comprises p38α (MAPK13), p38β (MAPK11), p38γ (MAPK12), and p38δ (MAPK13), and is a key component of the cellular stress response in stem cells. In response to oxidative stress induced by glutathione depletion (in vitro), ATM-deficiency (in vivo) or thioredoxin-interacting protein-deficiency (in vivo) HSCs were shown to activate p38-MAPK, resulting in loss of quiescence and eventual exhaustion of the HSC pool (53, 59). Likewise, p38-MAPK activation in the HSC compartment was demonstrated upon aging and serial bone marrow transplantation (BMT), in response to elevated intracellular ROS levels, resulting in impaired HSC function (53, 59). Notably, ROS-induced activation of p38-MAPK in HSPCs was dependent on ASK1 (53). Further insight into p38-MAPK signaling in HSCs was recently provided, in which p38α was shown to initiate HSC proliferation and support HSC reconstitution capability during stress hematopoiesis (i.e., BMT and 5-fluorouracil treatment), by activating purine metabolism in a microphthalmia-associated transcription factor-dependent manner (64). In the context of muSCs (studied ex vivo), p38α/β was shown to be necessary for activation of quiescent muSCs and induction of MYOD (58), a marker of myogenic commitment and progenitor proliferation (127). Remarkably, asymmetric division of activated muSCs was shown to involve asymmetric activation of p38α/β in daughter cells (147). The daughter cells containing activated p38α/β expressed MYOD, whereas those without activated p38α/β lacked MYOD expression, which reflects reversion to quiescence and retention of self-renewal capability (147). In agreement, a recent study showed that p38-MAPK activation in human muSCs was associated with muSC activation and differentiation, whereas p38-MAPK inhibition was associated with muSC expansion (20). Similarly, a study of muSCs in vivo demonstrated that p38α was involved in the regulation of muSC proliferation and myoblast commitment (11). Meanwhile, in vitro studies on NSCs and SSCs have reported that pharmacological inhibition of p38-MAPK diminishes proliferation and self-renewal (69, 106, 124). Therefore, redox modulation of protein kinases functions to regulate stem cells in multiple stem cell compartments.
Transcription factors
Several transcription factors important for stem cell function are redox responsive (8). These include p53, hypoxia inducible factors, nuclear factor erythroid-2-related factor (NRFs), and forkhead transcription factors of the O-class (FOXOs). In particular, the role of FOXOs (comprising FOXO1, FOXO3, FOXO4, and FOXO6) in stem cells has been extensively characterized (7, 39, 93, 107, 119, 120, 146, 161, 163). The major FOXO in SSCs and NSCs is FOXO1 and FOXO3, respectively. In contrast, the HSC compartment does not exhibit reliance on a single FOXO, at least under steady-state conditions (93, 146), because FOXO3 was demonstrated to be necessary for HSC maintenance during aging and stress (i.e., BMT and 5-fluorouracil treatment) (93). However, a subsequent study (161) reported high FOXO1 and FOXO3 expression (relative to FOXO4) in HSPCs, predominant nuclear localization of FOXO3, and reduced HSCs in FOXO3-deficient mice. These findings, in addition to the apoptotic phenotype observed in FOXO1 and FOXO3 double-deficient mice (146), suggest that FOXO1 and FOXO3 are likely to be functionally important in the HSC compartment. In all three stem cell compartments, FOXO deficiency results in the loss of stem cell quiescence and aberrant cell cycling, leading to attrition of the stem cell compartment and transient expansion of downstream progenitors. Thus, FOXOs are required for regulating self-renewal and/or differentiation in multiple stem cell compartments.
The transcriptional activity of FOXOs is contingent upon subcellular localization and interaction with transcriptional coregulators, both of which can be modulated by redox-dependent post-translational modifications (Fig. 2B). The transcriptional coactivator and lysine acetyltransferase p300/CBP, which has been implicated in HSC maintenance (18, 118), is a well-known FOXO binding partner known to modulate FOXO activity via acetylation (70). Notably, interaction between human FOXO4 and p300/CBP was shown to be dependent on ROS-induced formation of an intermolecular disulfide bond involving Cys-477, which is conserved across humans and murine FOXOs (29). Interestingly, FOXO4 contains four other potentially reactive Cys residues that appear to be involved in redox-mediated disulfide-dependent interactions with partners other than p300/CBP (29). Thus, changes in cellular redox state can directly modulate the activity of redox-responsive transcription factors to determine stem cell fate.
In addition to direct modulation, ROS-mediated modulation of FOXO activity can also occur indirectly via the action of redox-responsive kinases (Fig. 2B). FOXOs can be phosphorylated at several Ser/Thr residues by multiple kinases across different kinase groups (70), several of which participate in redox signaling (26). For instance, AKT phosphorylation sites have been reported in all FOXOs, whereas p38MAPK and ERK phosphorylation sites have been reported in FOXO1 and FOXO3 (70). AKT-mediated phosphorylation of FOXOs generally suppresses FOXO activity by preventing nuclear localization (70). In contrast, p38MAPK-mediated phosphorylation is proposed to enhance FOXO activity by facilitating nuclear localization (FOXO3) or interaction with other transcription factors (FOXO1) (4, 46). ERK-mediated phosphorylation may enhance FOXO activity by facilitating interaction with other transcription factors (4) or suppress FOXO activity by promoting its degradation (162). In addition, since AKT and MAPKs are located downstream of the RTK signaling cascade, ROS-mediated modulation of RTK activity can also alter FOXO activity. As such, the transcriptional activity of FOXOs, and other transcription factors regulated by redox-responsive kinases, can be altered by a change in cellular redox state.
The Need to Regulate ROS in Stem Cells
Stem cells experience oxidative stress when ROS levels exceed available antioxidant resources. This occurs when antioxidant defenses are impaired. For instance, germline genetic variation (e.g., single nucleotide polymorphisms) or mutations can impair the function of antioxidant defense components. Several studies [reviewed by Yu and Huang (165)] have reported an association between male infertility (i.e., defective spermatogenesis) and genetic variation/mutation of antioxidant genes, including NRF2, glutathione S-transferase, SOD, catalase, and glutathione peroxidase. Notably, oxidative stress in FOXO1/3/4 triple-deficient HSCs and NSCs has been attributed to downregulation of several ROS-detoxifying enzymes, including catalase and SOD (107, 146). Genetic variation in NRF2, which transcriptionally upregulates many antioxidants, has also been associated with respiratory, cardiovascular, gastrointestinal, and neurodegenerative diseases [reviewed in Cho et al. (23)]. Similarly, genetic variation in NRF2-binding cis-acting antioxidant response elements is associated with gastrointestinal and neurodegenerative diseases (78). Interestingly, deficiency in dietary antioxidants or essential constituents of antioxidant proteins (e.g., sulfur-containing amino acids) may also cause oxidative stress (43).
Conversely, oxidative stress in stem cells occurs under circumstances wherein ROS generation is increased. For instance, ROS levels increase in stem cells exposed to ambient air (87), chemotherapeutic agents (e.g., 5-fluorouracil), and ionizing radiation (24, 128). In fact, NOS-dependent ROS generation occurs within seconds of irradiation, lasting several minutes thereafter (80). Interestingly, irradiation-induced ROS were greater in HSPCs than in lineage-committed progenitors (128), thereby suggesting selective ROS generation in stem cells. Activation of resting stem cells in response to tissue injury or exposure to inflammatory stimuli (e.g., during infection) also results in oxidative stress because of increased ROS generation from elevated mitochondrial activity (154).
Oxidative stress generally leads to a decline in stem cell function. The association between impaired stem cell function and oxidative stress was initially observed in the HSC compartment of ATM-deficient mice (53), and subsequently in many other loss-of-function models. Oxidative stress induced directly by buthionine sulfoximine treatment (depletes intracellular glutathione) also reduced HSC reconstitution ability in a dose-dependent manner without affecting progenitor differentiation (54), thereby suggesting that oxidative stress selectively impairs the stem cell compartment. Similarly, serial BMT resulted in increasing oxidative stress and progressive decline in reconstitution ability (54). Conversely, reducing oxidative stress in human HSPCs via p38α inhibition enhanced reconstitution ability (6). In addition, oxidative stress also compromised the function of muSCs (38), NSCs (107), and MSCs (10, 13, 25, 108).
The outcome of oxidative stress varies depending on the stem cell type and degree of stress (i.e., magnitude and exposure time) encountered, but can result in one or a combination of the following: proliferation, adaptive responses, oxidative damage, senescence, and/or death (43).
Proliferation
Proliferation is a common response of stem cells to oxidative stress. As demonstrated in HSCs in vivo (21, 54, 74, 87, 88, 93, 117, 128, 140, 141, 146, 149, 161, 169), increased ROS are typically associated with loss of quiescence and increased proliferation, ultimately leading to stem cell exhaustion. Importantly, these defects were rescued by antioxidant administration (21, 54, 74, 88, 117, 146, 169), thus establishing oxidative stress as a cause of stem cell proliferation. Unlike HSCs, increased ROS were associated with aberrant proliferation of NSCs (69, 107). Interestingly, antioxidant treatment failed to rescue the proliferative defect (107), thus excluding ROS as cause. Therefore, oxidative stress can activate stem cells to proliferate, potentially leading to stem cell exhaustion, although this does not apply to all stem cell compartments.
Oxidative damage
Oxidative damage can have catastrophic consequences on the stem cell compartment. In stem cells, ROS can damage biomolecules such as lipids, proteins, and nucleic acids (Fig. 3). Notably, the oxidative damage sustained can be irreversible, resulting in permanent cellular injury and loss of stem cell function.
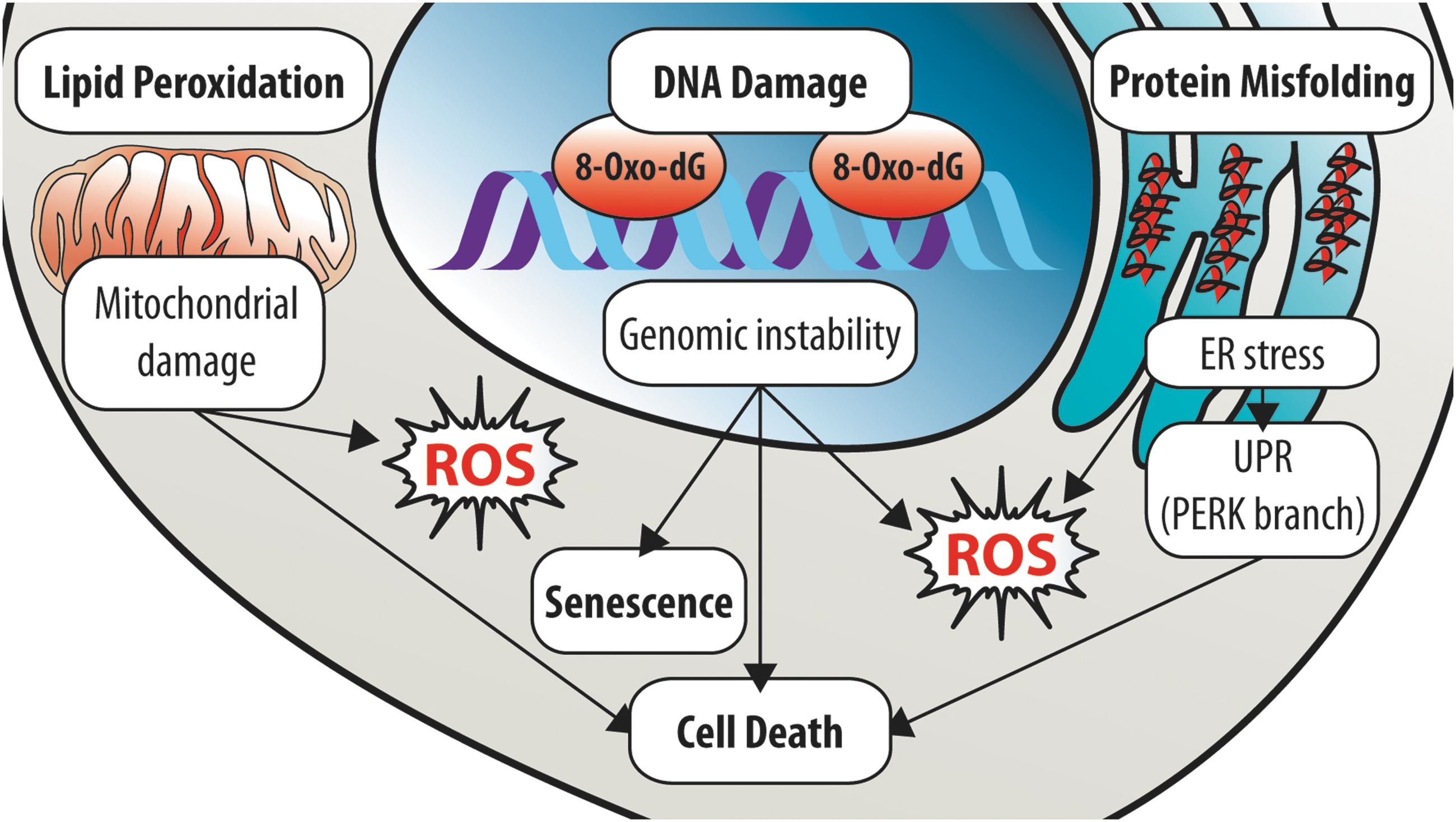
Lipid peroxidation
Lipids are functionally essential biomolecules; yet, the impact of lipid peroxidation on stem cells has not been extensively characterized. Lipids encompass diverse classes of hydrophobic or amphipathic small molecules, such as fatty acyls, glycerophospholipids, and sphingolipids. (33). Functionally, lipids can serve as structural components of cell and organelle membranes, metabolites, and cell signaling molecules. Membranes contain significant amounts of lipids composed of fatty acyl side chains with multiple carbon–carbon double bonds (i.e., polyunsaturated fatty acid [PUFA] chains), which are particularly susceptible to oxidation (43). A notable example is the lipid cardiolipin, which is composed of four PUFA side chains and located at the inner mitochondrial membrane (IMM) (164), thus rendering it highly susceptible to peroxidation. Cardiolipin peroxidation has been implicated in apoptosis and mitochondria dysfunction (164). Furthermore, as a major component of mitochondrial membranes, cardiolipin, as well as other mitochondrial lipids, can influence mitochondrial dynamics (see the Mitochondrial dynamics section) (158). Therefore, stem cells are potentially vulnerable to peroxidation of functionally critical lipids, particularly those regulating mitochondria function.
Lipid catabolism involves fatty acid oxidation (FAO), which predominantly occurs in the mitochondria, and in the peroxisomes to a lesser extent. Mitochondrial FAO was detected in HSPCs but not in differentiated (i.e., lineage-negative) cells, and was shown to be important for maintenance of the HSC compartment (52). Similarly, mitochondrial FAO was shown to be important in neural stem/progenitor cells (NSPCs) for proliferation (136), whereas fatty acid synthase-dependent lipogenesis was shown to regulate NSPC activity (72). Notably, an increase in ROS has been reported to inhibit FAO (125). Thus, oxidative stress may also impact stem cells by disrupting lipid metabolism.
Protein damage
Stem cell injury resulting from damage of proteins during oxidative stress can occur via multiple mechanisms. The integrity of proteins can be compromised directly by ROS, and also indirectly via ROS-induced DNA mutation or aberrant RNA translation (43), resulting in either dysfunctional or misfolded (i.e., nonfunctional) proteins. ROS-mediated protein damage can impair critical processes, for example, damage of DNA repair proteins affects DNA repair and causes DNA damage accumulation, which is highly detrimental to stem cells (see the DNA damage section). Alternatively, ROS-mediated protein damage can impair the function of organelles such as the endoplasmic reticulum (ER) and mitochondria (see the Mitochondria: The Epicenter of ROS Management in Stem Cells section). As the major site of protein folding, the ER lumen has an oxidizing environment, which is necessary for oxidative protein folding [i.e., stabilization of tertiary and quaternary structure by disulfide bond formation between Cys residues, reviewed by Cao and Kaufman (16)], a process that generates H2O2. Furthermore, the antioxidant defense capability of the ER appears limited (16). These factors render the ER highly vulnerable to oxidative stress, particularly under conditions of increased protein folding load. Oxidative stress disrupts oxidative protein folding and leads to accumulation of misfolded proteins within the ER, resulting in ER stress and ER stress-associated ROS generation. In addition, ER stress can further exacerbate oxidative stress via Ca2+release, which stimulates mitochondrial ROS (ROSmt) production (16). In turn, the ROSmt produced causes further release of Ca2+, resulting in a cumulative increase in ROS that ultimately triggers apoptosis (16). Meanwhile, ER stress leads to activation of the unfolded protein response (UPR). The UPR comprises three well-defined and distinct branches, which are defined by the ER transmembrane signal transducer (155): activating transcription factor 6 (ATF6), inositol requiring enzyme 1 (IRE1), and PRKR-like ER kinase (PERK). These three branches are activated sequentially (ATF6 and IRE1, followed by PERK) to increase ER protein folding capacity and relieve ER protein folding load, thus facilitating proteostasis and cell survival (155). Conversely, in response to severe chronic ER stress, the UPR induces apoptosis via the PERK branch (155). Notably, the UPR can also be activated by metal ions and lipid oxidation products. Therefore, oxidative damage can deplete the stem cell compartment via induction of ER stress and UPR activation.
The importance of proteome integrity and proteostasis in stem cells has become increasingly apparent of late. Several groups have recently demonstrated that in response to ER stress, the UPR is activated and induces apoptosis in both murine and human HSCs (15, 92, 150). Notably, the PERK branch was preferentially activated, and selectively induced apoptosis in human HSCs (150). In agreement, ER stress and UPR activation were associated with impaired engraftment (human HSCs) and long-term reconstitution ability (murine HSCs), which were rescued by reducing ER stress (92, 150). Most recently, the deleterious consequence of protein misfolding and ER stress on stem cells was also demonstrated in fetal liver HSCs, which exhibited greater protein synthesis than their adult counterparts (133). Intriguingly, fetal liver HSCs were shown to be dependent on maternal bile acids to suppress ER stress, reduce protein aggregation, and prevent ER stress-associated apoptosis (133). In further support, a recent study demonstrated that protein synthesis in HSCs is highly regulated (132). Protein synthesis in HSCs, at steady-state and under proliferative conditions, was significantly reduced compared with lineage-committed progenitors and differentiated hematopoietic cells (132). Remarkably, perturbation of HSC proteostasis by increasing or decreasing protein synthesis was associated with a decline in the long-term multilineage reconstitution ability (132). Similarly, perturbation of mitochondrial proteostasis in SIRT7−/− HSCs induced mitochondrial protein folding stress (PFSmt) and mitochondrial UPR (UPRmt) activation, leading to reduced quiescence, apoptosis, and impaired HSC maintenance (94). Therefore, perturbation of the proteome in stem cells and their mitochondria is a significant cause of stem cell decline under oxidative stress.
DNA damage
Among the ROS-susceptible biomolecules, oxidative damage involving DNA (i.e., chromosomal DNA) in stem cells is of particular concern. Stem cells are typically long-lived and can give rise to numerous progeny, whereas DNA is retained throughout a cell's lifetime and transmitted during cell division. Hence, mutations arising from ROS-mediated DNA damage can be rapidly propagated. Furthermore, DNA is the largest cellular macromolecule and is highly susceptible to ROS-induced damage. ROS can directly damage DNA via oxidative modification of the DNA backbone (i.e., 2-deoxyribose) and DNA bases, in addition to causing DNA strand breaks (43). Of the DNA bases, guanine is most vulnerable to oxidation because of its low redox potential (104). Consequently, 2-deoxyguanosine in DNA is frequently oxidized to 8-oxo-2′-deoxyguanosine [8-oxo-dG; 400–1500 lesions daily per cell (71)], which functionally mimics a “T,” resulting in G-to-T transversion mutations, if not corrected before DNA replication (104). Multiple studies have shown that elevated ROS levels are associated with increased 8-oxo-dG lesions and DNA strand breaks in both murine and human HSCs (7, 34, 154, 160). Importantly, the increase in DNA strand breaks could be rescued by antioxidant treatment (7, 34), thus confirming ROS as a cause of DNA damage in stem cells. Conversely, a study recently reported that DNA damage in HSPCs leads to mitochondrial BID-dependent ROS generation (144). Thus, oxidative DNA damage can further exacerbate oxidative stress in stem cells. Notably, the severe impact of DNA damage on stem cell function is well established (122, 144, 154, 160). Therefore, ROS-induced DNA damage can have potentially deleterious consequences on stem cell compartments.
Senescence
Cellular senescence is a stress-induced irreversible growth-arrested state, in which cells cease responding to mitogenic and oncogenic stimuli. Initial studies attributed senescence to telomere attrition (9, 44), which has been proposed to activate DNA damage response and p53 (101). Subsequently, diverse stressors have been recognized as triggers of senescence, including oncogene activation, UPR, DNA damage, and ROS (130). Although these triggers initiate signaling via different pathways, all signals eventually lead to the activation of cyclin-dependent kinase inhibitors (i.e., p16INK4a, p15INK4b, p21, and p27), typically via the activation of p53 (101). Notably, these triggers have all been associated with elevated ROS levels. For instance, ROS accumulation has been proposed to induce oncogene-induced senescence (27, 43). In addition, ROS has been implicated as both a cause and consequence of DNA damage and UPR (91). Furthermore, senescent cells are reported to have elevated oxidized DNA and proteins (43, 63). In fact, elevated ROS levels are thought to be more important than telomere attrition in triggering senescence (43). Thus, oxidative stress is a major trigger of cellular senescence.
Oxidative stress can trigger cellular senescence in stem cells. Experimentally, senescence is characterized in vivo using several biomarkers [reviewed by Sharpless and Sherr (130)], of which elevated β-galactosidase activity and p16INK4a upregulation are the most widely accepted. Sublethal irradiation of mice was found to induce p21, p16INK4a, and β-galactosidase expression specifically in the HSPCs, but not in lineage-committed progenitors (19, 128, 156). This HSPC-selective irradiation-induced senescence was subsequently found to be associated with increased ROS and independent of p16INK4a (128). Oxidative stress-induced p38-MAPK-mediated senescence of HSPCs, because of elevated mitochondrial ROS generation, was also demonstrated in P2Y14-deficienct mice in response to irradiation and other stressors (i.e., chemotherapy, BMT, and aging) (24). Notably, senescence in the HSPC compartment was associated with a decline in long-term reconstitution ability (24, 128, 156). Loss of BMI1 proto-oncogene, a member of the polycomb group of transcription factors known to regulate senescence, was reported to elevate ROSmt and activate DNA damage response in thymocytes (83). Interestingly, in both HSCs and NSCs, BMI1 is required for repression of senescence-associated genes and self-renewal of the stem cell compartment (95, 96, 110). Irradiation was also shown to trigger senescence in muSCs in vivo (19). In murine MSCs, TGF-β1-mediated increase in ROSmt and decrease in SOD2 expression were associated with TGF-β1-induced senescence in vitro (159). Similarly, treatment of human MSCs in vitro with sublethal concentrations of H2O2 was shown to trigger senescence (10, 13, 25) and involve DNA damage response and p38-MAPK activation (10). Therefore, oxidative stress can compromise the function of stem cell compartments via induction of cellular senescence.
Cell death
The ultimate response to oxidative stress is cell death, which is induced in response to severe oxidative damage. Apoptotic cell death, potentially resulting from DNA damage and/or mitochondrial dysfunction (83, 88, 99), has been observed in HSCs in response to oxidative stress (41, 74, 83, 88, 99, 117, 140, 146, 169). Notably, apoptosis via the intrinsic pathway requires cardiolipin peroxidation for cytochrome c release and subsequent caspase activation (164). Under extreme oxidative stress, caspases can be inactivated, leading to death by necrosis (43). Therefore, oxidative stress can adversely affect stem cell function and fate.
Management of ROS in Stem Cells
Given the deleterious effects of oxidative stress, stem cells possess several strategies to manage ROS accumulation and adapt to oxidative stress (Fig. 4). These strategies encompass resisting oxidative damage, limiting ROS generation, promoting ROS removal, and compartmentalization.
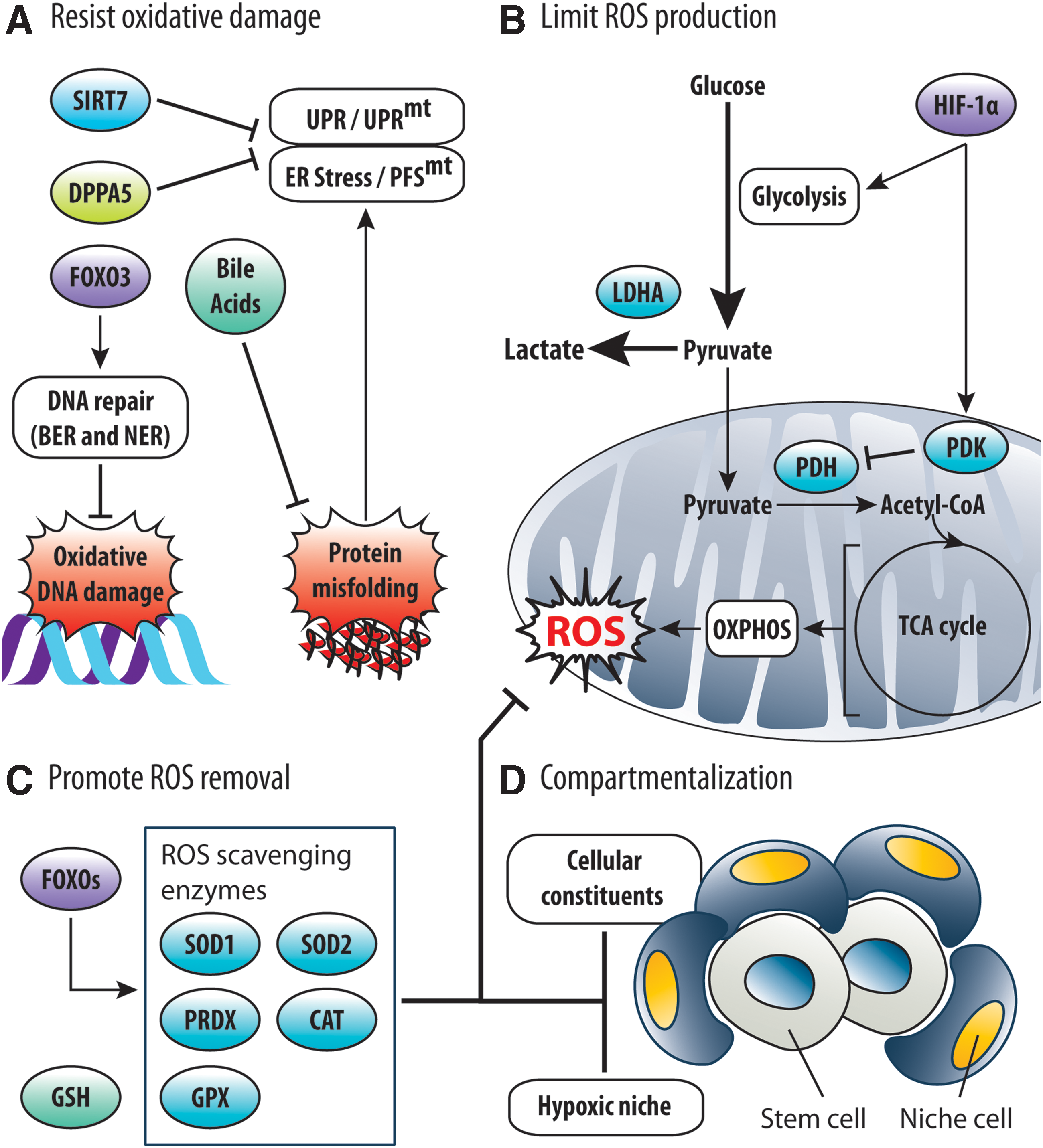
Stem cells have protective mechanisms to resist oxidative damage (Fig. 4A). For instance, oxidative protein damage in stem cells can elicit ER stress/PFSmt and consequent UPR/UPRmt activation (see the Protein damage section), leading to apoptosis and reduced stem cell potential. As a countermeasure, HSCs express DPPA5 to attenuate UPR, suppress ROS, and prevent apoptosis (92). HSCs also express SIRT7, which represses NRF1, to alleviate PFSmt and UPRmt activation (94). Similarly, fetal liver HSCs are supplied maternal bile acids to reduce protein misfolding/aggregation and ER stress (133). Very recently, FOXO3 expression in HSCs was reported to confer resistance to oxidative DNA damage by promoting expression of base excision repair and nucleotide excision repair genes (7). Therefore, stem cells are equipped to mitigate the impact of ROS-mediated damage.
Stem cells prevent ROS accumulation by limiting ROS generation (Fig. 4B). To do so, stem cells suppress metabolic activity and preferentially utilize glycolysis over OXPHOS [reviewed by Ito and Suda (55) and Perales-Clemente et al. (112)]. For example, selective expression of HIF-1α in HSCs promotes anaerobic glycolysis via the action of pyruvate dehydrogenase kinases (PDKs) (141), which reduces influx of glycolytic intermediates into the mitochondria to suppress ROS generation (142). Likewise, HSCs require lactate dehydrogenase A (LDHA) to facilitate glycolysis, limit OXPHOS, and suppress ROS production (157). Notably, the use of metabolic programs to regulate ROS generation has also been demonstrated in NSCs (163). Thus, stem cell metabolism is an important determinant of redox homeostasis and stem cell fate. Alternatively, ROS generation can also be controlled by modulating mitochondria (see the Mitochondria: The Epicenter of ROS Management in Stem Cells section).
Stem cells prevent ROS accumulation by removing ROS (Fig. 4C). Stem cells constitutively express a repertoire of ROS scavenging enzymes to remove ROS. SOD1 and SOD2 are responsible for converting O2 •− to H2O2, which is subsequently neutralized by catalase or peroxidases such as glutathione peroxidase and peroxiredoxin (43). Notably, expression of ROS scavenging enzymes in HSCs and NSCs was shown to be dependent on FOXOs (107, 161). In addition, the highly abundant and ubiquitous intracellular antioxidant glutathione may also support ROS removal in stem cells by scavenging ROS directly, or serving as a substrate of glutathione peroxidases (43). Therefore, ROS management in stem cells is mediated by antioxidants and ROS scavenging enzymes.
Stem cells are compartmentalized within a microenvironment (i.e., niche), which facilitates redox homeostasis (Fig. 4D). Residence within a hypoxic niche is known to suppress ROS generation, via a HIF-1α-directed metabolic program, and preserve stem cell function in HSCs, MSCs, and NSCs [reviewed by Ito and Suda (55)]. Very recently, the vascular architecture of the bone marrow (BM) niche was reported to be important for ROS suppression in HSCs (51). Notably, cellular constituents of the niche also contribute to redox regulation of stem cells. For example, α-smooth muscle actin-expressing monocytes/macrophages in the BM niche release prostaglandin E2 to inhibit AKT and suppress ROS generation in HSCs (86). In addition, BM stromal cells (BMSCs) can off-load ROS from HSCs under stress via Connexin-43 (143). Therefore, ROS management in stem cells involves cell-extrinsic and cell-intrinsic mechanisms.
Mitochondria: The Epicenter of ROS Management in Stem Cells
Mitochondria are involved in diverse processes (i.e., various metabolic processes, apoptosis, and calcium homeostasis) including energy and redox homeostasis. An evolutionarily conserved role (i.e., function) of mitochondria is the production of ATP, which is dependent on the electrochemical proton motive force (Δp) generated by OXPHOS (113). Since Δp is a sum of the mitochondrial pH gradient (ΔpH) and mitochondrial membrane potential (ΔΨm), ΔΨm is widely used experimentally as an indicator of mitochondrial activity (113). In healthy (i.e., functional) mitochondria, ΔΨm and ROS generation are positively correlated, especially at high ΔΨm (76, 148). As such, altered ΔΨm generally reflects a change in ROS generation. Therefore, ROS generation in stem cells can be regulated by modulating mitochondrial activity.
Mitochondrial activity
Mitochondrial activity can significantly influence the function and fate of stem cells (Fig. 5). Compared with committed progenitors, HSCs have low mitochondria activity as inferred from the significantly lower ΔΨm in HSCs (152). Within the HSC compartment, cells with lower ΔΨm were recently reported to possess significantly enhanced long-term reconstitution potential (138, 152). In striking contrast, phenotypic HSCs with high ΔΨm had barely any reconstitution potential (138, 152). In agreement, most (80%) ΔΨm-low HSCs expressed CD150, which marks HSCs with enhanced self-renewal and long-term reconstitution capability (98), whereas none of the ΔΨm-high HSCs expressed CD150 (138). Expression of CD41, which identifies cells with short-/intermediate-term reconstitution ability within the HSC compartment, was also absent in most (70%) ΔΨm-low HSCs (152). Consistent with these observations, increased ΔΨm in autophagy-deficient HSCs was associated with increased ROS, reduced self-renewal, and impaired long-term reconstitution potential (47). In further support, a recent landmark study (87), which addressed the impact of ambient air on HSCs during isolation, demonstrated that the increased mitochondrial activity in air-exposed HSCs (compared with hypoxia-isolated HCSs) was associated with increased HSC differentiation plus a reduction in the number of phenotypic and functional HSCs, a phenomenon termed extraphysiological oxygen shock/stress (EPHOSS) (87). Notably, the consequences associated with increased mitochondrial activity in the HSC compartment have been attributed to increased ROS generation (87, 138) and DNA damage (138). Interestingly, EPHOSS-induced ROS generation was linked to cyclophilin D-mediated opening of the mitochondrial permeability transition pore (87). Therefore, mitochondria serve as effectors of stem cell fate, where elevated mitochondrial activity corresponds with a decline in stem cell potential.
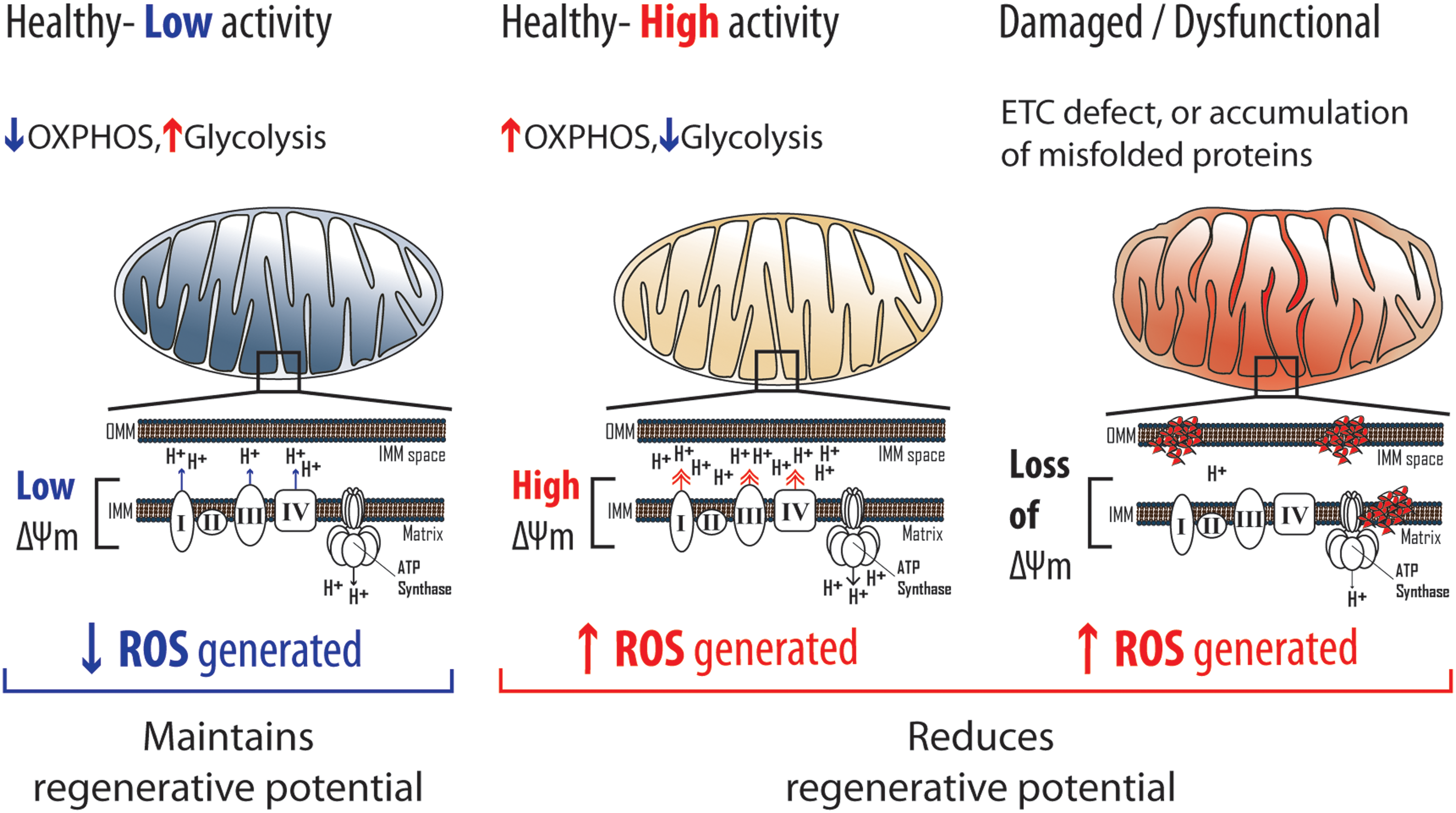
Conversely, loss of mitochondrial activity because of mitochondrial damage/dysfunction may be detrimental to stem cells (Fig. 5). Unlike healthy mitochondria, damaged mitochondria (i.e., dysfunctional) are unable to maintain ΔΨm because of ROS-mediated accumulation of misfolded proteins or mitochondrial ETC defects (105). For instance, antimycin A (ETC complex III inhibitor) or carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; an ETC uncoupling agent) treatment impairs mitochondrial function and reduces ΔΨm in NSCs (67). In turn, damaged mitochondria are thought to further stimulate ROS generation (24, 82). In agreement, MuSCs from aged mice, and autophagy-deficient MuSCs, were reported to have reduced ΔΨm, which was associated with senescence and impaired regenerative potential (i.e., MuSC activation and expansion) (38). Notably, the MuSC defects associated with reduced ΔΨm were attributed to increased ROS (38). Similarly, HSCs from aged mice exhibited reduced ΔΨm, which was associated with myeloid-biased differentiation and impaired self-renewal (47). However, unlike aged MuSCs, aged HSCs had lower ROS levels than their younger counterparts (47). In NSCs deficient for PINK1, which facilitates clearance of damaged mitochondria (see the Mitochondrial clearance section), ΔΨm was reduced and associated with impaired in vitro growth and differentiation (2). Taken together, the mentioned instances suggest that the loss of mitochondrial activity because of accumulation of damaged mitochondria is detrimental to stem cells.
Mitochondrial clearance
The mitochondrial activity in stem cells can be modulated by regulating the clearance of mitochondria (Fig. 6A). In stem cells, macroautophagy (hereafter referred to as autophagy) is the major process known to facilitate mitochondria clearance. Autophagy is an essential, housekeeping, and stress-responsive catabolic process, which is responsible for regulating organelle and macromolecule (i.e., proteins, lipids, carbohydrates, and iron) quality and quantity via lysosomal degradation (66). Several studies on HSCs (41, 47, 56, 99, 152), as well as MuSCs (38), have recently identified autophagy as critical for maintenance of the stem cell pool and regenerative potential, both at steady-state and under stress conditions. The requirement for autophagy in stem cells has been attributed to the clearance of damaged mitochondria via mitophagy (38, 56, 152), a mitochondria-selective form of autophagy that is dependent on the proteins PINK1 and PARKIN [reviewed in Nguyen et al. (105)]. In brief, PINK1 functions as a sensor of damaged mitochondria. Healthy mitochondria can import PINK1 into the IMM for processing, which promotes rapid degradation of PINK1 (105). Failure to import PINK1, because of compromised ΔΨm, leads to accumulation of PINK1 on the outer mitochondrial membrane (OMM) and recruitment of the E3 ubiquitin ligase PARKIN, which labels mitochondria for degradation by the autophagy machinery (105). Notably, Tie2+ HSCs (stem cell potential-enriched HSC fraction) at steady state were recently shown to preferentially express mitophagy-related genes, including Pink1 and Parkin, which were induced by the PPARdelta-FAO pathway (56). Moreover, PINK1 and PARKIN expression was shown to be critical for the reconstitution potential of Tie2+ HSCs (56). In addition, ΔΨm-low HSCs cultured in the presence of differentiation-inducing cytokines exhibited significantly improved long-term multilineage reconstitution ability if treated with FCCP (152). Therefore, stem cell potential is highly dependent on the clearance of damaged mitochondria via mitophagy.
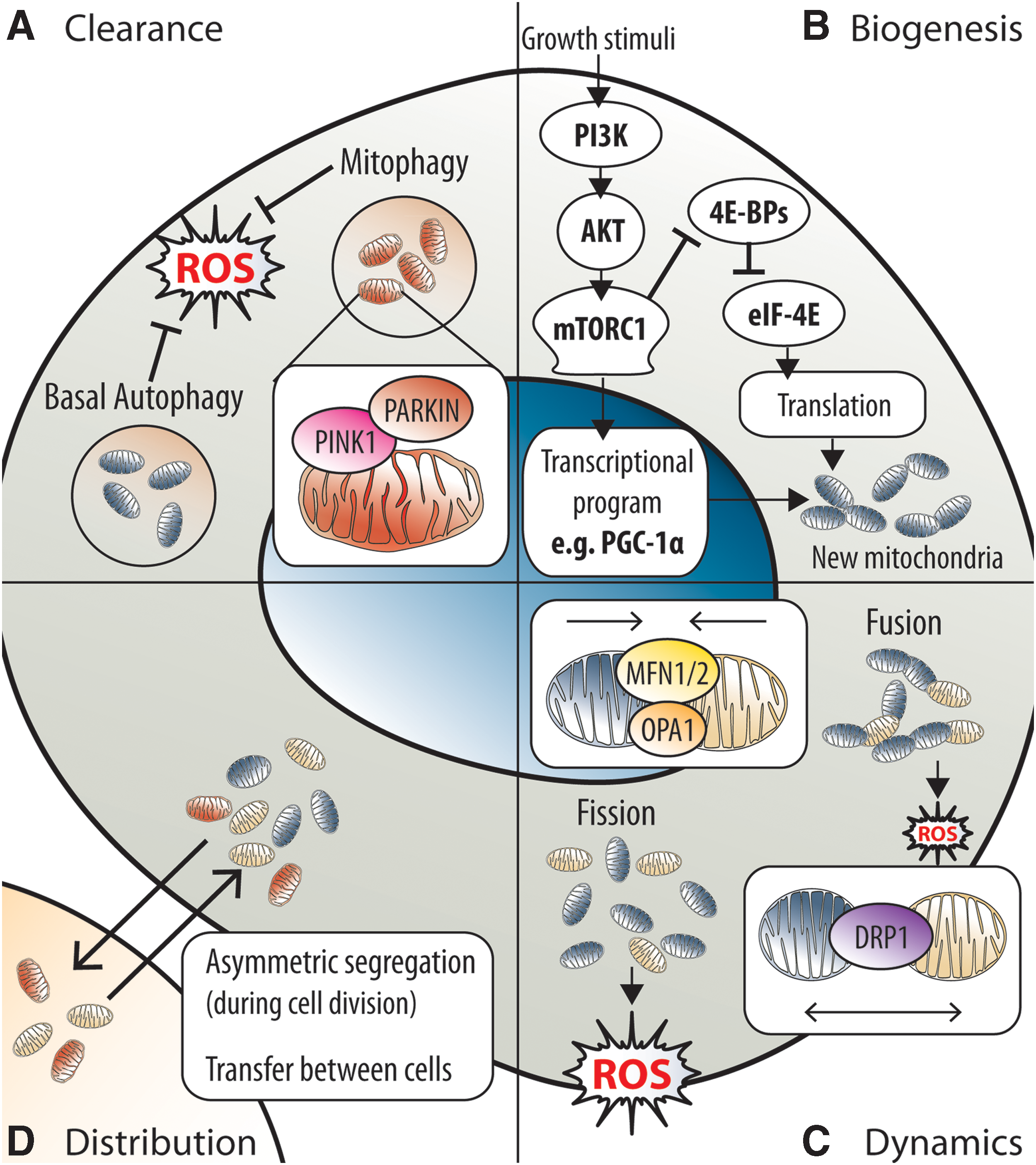
Apart from damaged mitochondria, the clearance of healthy mitochondria is likewise important for redox homeostasis in stem cells. Accumulation of healthy mitochondria and increased mitochondrial superoxide has been observed upon inhibition of autophagy in HSPCs (99). Most recently, autophagy was demonstrated to be important in HSCs for the preservation of stem cell potential, especially in the context of aging, by suppressing oxidative metabolism via the reduction of healthy mitochondria (47). Interestingly, the mechanism underlying autophagy-dependent clearance of healthy mitochondria appears to be distinct from mitophagy (47) and remains to be elucidated. In agreement, recent observations indicate that HSCs with lower mitochondrial mass and activity possess superior stem cell potential (138, 152). Thus, basal autophagy is necessary for the maintenance of stem cells.
Mitochondrial biogenesis
The mitochondrial activity in stem cells can be modulated by mTOR complex 1 (mTORC1)-mediated regulation of mitochondrial biogenesis (Fig. 6B). mTORC1 promotes mitochondrial biogenesis and activity by inhibiting eIF4E-binding proteins to stimulate synthesis of nucleus-encoded mitochondrial proteins (97) and promoting expression of the transcriptional coactivator PGC-1α (28), a key coordinator of mitochondrial biogenesis. Notably, mTORC1 is critical for HSC (21, 37, 61), NSC (45), and MuSC (121) compartments. Interestingly, mTORC1, which upregulates mitochondrial activity in stem cells (21, 37, 121), was reported to enhance the functional potential and facilitate activation of stem cells (121). In HSCs, hyperactivation of the PI3K-AKT-mTORC1 signaling cascade, with corresponding upregulation of PGC-1α, was recently reported to promote mitochondrial biogenesis (117). Similarly, hyperactivation of mTORC1 because of loss of Tsc1, a negative regulator of mTORC1, has also been implicated in promoting mitochondrial biogenesis in HSCs (21). The dysregulated mitochondrial biogenesis increased oxidative phosphorylation and elevated ROS levels, culminating in stem cell exhaustion (i.e., loss of quiescence and apoptosis) and impaired long-term reconstitution (21, 117). In agreement, similar defects in the HSC compartment were observed with constitutive activation of AKT1 or inhibition of PTEN (68, 168). Whereas AKT1/2 double deficiency, which presumptively reduces mitochondrial biogenesis, produced the opposite effect (i.e., decreased ROS and increased quiescence) in HSCs, resulting in impaired reconstitution (60). Notably, the effects of dysregulated mitochondrial biogenesis were confined to the HSPC compartment and not observed in lineage-marker positive cells (117). Furthermore, several miRNAs, which suppress multiple components of the PI3K-AKT-mTORC1 signaling cascade including the downstream target PGC-1α, were selectively expressed in the more primitive CD49lo HSC compartment (117). Therefore, dysregulated mitochondrial biogenesis and the resulting redox imbalance are selectively detrimental for stem cells.
Mitochondrial dynamics
Stem cell fate can also be controlled by modulating mitochondrial dynamics (Fig. 6C). The dynamic nature of mitochondria is a function of its constant fusion and fission, the balance of which determines their net morphology (153). Under physiological conditions, the morphology of mitochondria varies between fragmented and filamentous (i.e., elongated and fused) and closely reflects mitochondrial activity (153). Both HSCs and NSCs have longer mitochondria than their differentiated progeny (67, 85). In agreement, HSCs deficient in mitochondrial clearance accumulated elongated and fused mitochondria (47), thereby suggesting that mitochondria in stem cells are inclined to fuse. Interestingly, differentiation of stem cells to committed progenitors corresponds with shortening of mitochondria (67, 85) and a switch from glycolytic to oxidative metabolism, with increased O2 consumption (67, 166).
Mitochondrial fission is driven by DRP1, whereas fusion of IMM and OMM involves OPA1, and both MFN1 and MFN2, respectively (153). The importance of mitochondrial dynamics in stem cells has recently been demonstrated in HSCs (85), and most recently in NSCs (67). Functionally, mitochondria elongation/fusion induced by DRP1 deficiency improved stem cell potential (67). Conversely, mitochondria fragmentation (i.e., fission) induced by MFN1/2 deficiency or OPA1 deficiency caused aberrant differentiation and impaired stem cell maintenance (67, 85). Mechanistically, mitochondria fusion and fission have been associated with reduced and increased ROS generation, respectively (153). Correspondingly, increased ROS levels were observed in MFN1/2-deficient cells (100). In agreement, inducing mitochondrial elongation/fusion in NSCs reduced ROS and ROSmt levels, whereas mitochondrial fragmentation elevated ROS and ROSmt levels physiologically, without inducing oxidative damage (67). Furthermore, mitochondrial fragmentation is associated with reduced levels of SOD1 and complex I (67), a major site of ROSmt generation. Moreover, physiological elevation of ROS, induced by perturbing complex I, reduced NSC self-renewal (67). Importantly, impaired self-renewal in OPA1-deficient NSCs was restored by antioxidant treatment (67). Notably, the physiological ROS elevation generated by mitochondrial fragmentation was shown to function as a signaling messenger, activating transcriptional programs that inhibit self-renewal and promote differentiation (67). In contrast, mitochondrial fragmentation in MFN2-deficient HSCs did not alter ROS levels. This is consistent with the mitochondrial fusion-independent role of MFN2 in determining HSC fate by negatively regulating nuclear factor of activated T-cells (NFAT) transcriptional activity via intracellular Ca2+ buffering (85). Therefore, mitochondrial dynamics can influence stem cell fate, through the modulation of ROS or Ca2+ levels, to regulate cell fate-determining transcriptional programs.
Mitochondrial distribution
Regulation of mitochondrial activity by modulating mitochondria distribution between cells (Fig. 6D) is an emerging concept of potential relevance to stem cell biology and redox homeostasis. During cell division, mitochondria and other organelles are distributed between daughter cells. However, the functional relevance of mitochondria distribution in stem cells has only been revealed recently. A recent study (65) using stem-like mammary epithelial cells demonstrated that old mitochondria were segregated asymmetrically between daughter cells, unlike other organelles, which were distributed symmetrically. More specifically, one daughter cell obtained mostly young mitochondria, whereas the other obtained a mix of young and old mitochondria (65). Importantly, daughter cells that inherited fewer old mitochondria were more stem like, as determined by mammosphere forming capability (65). Interestingly, asymmetric mitochondrial segregation, and the consequent effect on daughter cell stemness, appears to be dependent on mitochondrial dynamics (65), although the precise mechanisms remain to be clarified. In addition, whether asymmetrical segregation of old or damaged mitochondria occurs in vivo, and in other tissue stem cells (HSCs, NSCs, etc.), remains unknown.
Mitochondria distribution can also occur via transfer of mitochondria between cells. Intercellular distribution of mitochondria (i.e., mitochondrial transfer) involving stem cells was first described more than a decade ago in vitro, in which mitochondria transfer from human MSCs to aerobic respiration-deficient A549 cells rescued aerobic respiration (135). Since then, several studies have reported unidirectional mitochondrial transfer from MSCs to various cell types in vitro (1, 84, 114, 115) and in vivo (3). Importantly, mitochondrial transfer was functionally beneficial for recipient cells (3, 84, 135), especially in the context of tissue injury, wherein mitochondrial transfer reduced recipient cell apoptosis (3, 84). Interestingly, mitochondrial transfer from MSCs occurred to a greater extent than other cells of mesenchymal origin (3), thereby suggesting that MSCs have a propensity to donate mitochondria. Notably, the reciprocal effect of mitochondrial transfer on MSCs has not been established. Thus, further studies are needed to determine the functional importance of mitochondrial transfer in stem cells.
The mechanisms underlying mitochondrial transfer, as well as its physiological relevance in other stem cell compartments, are not well understood. Factors reported to be involved in mitochondrial transfer include MIRO1 (3), which is part of the mitochondrial transport machinery, and Connexin-43 (50), a constituent of gap junctional channels. Interestingly, Connexin-43 is required for BMSC expression of CXCL12 (126), which is essential for HSC maintenance and function (31, 42, 126). In addition, Connexin-43 is also expressed in HSCs and facilitates the regenerative response of HSCs to myeloablation (143). Coincidentally, reduction of ROS accumulation in HSCs in vivo after myeloablation has been attributed to Connexin-43, which purportedly mediates transfer of ROS from HSCs to BMSCs (143). Alternatively, Connexin-43 may participate in redox homeostasis of HSCs by mediating transfer of ROS scavengers (e.g., glutathione), second messengers (e.g., Ca2+) or metabolites that alter ROS accumulation. Furthermore, given the importance of the BM niche in HSC maintenance, and the detrimental effects of mitochondria accumulation and increased ROS on HSCs, redox homeostasis in HSCs, particularly under stress conditions, may involve mitochondrial transfer that could be facilitated by Connexin-43. In fact, preliminary data on mitochondrial transfer between HSCs and BMSCs, potentially involving Connexin-43, have recently been presented (40). Therefore, the functional implications and mechanisms of mitochondrial transfer, in relation to redox homeostasis and stem cell fate, are of significant interest.
Concluding Remarks
The importance of ROS as a determinant of stem cell fate and function has become increasingly apparent. For instance, the concept of ROS in regulating stem cell function via redox signaling appears to be gaining acceptance. However, evidence regarding the occurrence and functional relevance of redox signaling in stem cells is still lacking. Nonetheless, it is possible that stem cells employ redox signaling to coordinate self-renewal and differentiation, or to regulate stem cell activity in response to stress. Unlike redox signaling, the detrimental impact of ROS on stem cells in the form of oxidative stress has been appreciated for well more than a decade. Notably, the detrimental effect of oxidative damage involving protein folding stress and dysregulated proteostasis has recently gained recognition. With regard to ROS management, the importance of mitochondria in stem cell function has garnered considerable interest in recent years; as such, our understanding of how mitochondria facilitate redox homeostasis in stem cells has increased considerably. At present, we propose that mitochondria, which are a major site of ROS generation and a node for diverse cell fate-determining processes, represent a key regulatory hub for ROS management in stem cells.
Footnotes
Acknowledgments
This work was supported by the Singapore Ministry of Health's National Medical Research Council STaR Investigator Award (NMRC/STaR/019/2014) and the European Commission under the 7th Framework Program (SyStemAge: FP7-HEALTH-2012-INNOVATION-1).