
Abstract
Significance:
The number of people suffering from diabetes worldwide is steadily rising. Complications from diabetes, including cardiovascular and renal disease, contribute to the high morbidity and mortality associated with this disease.
Recent Advances:
Hyperglycemia promotes tissue damage through diverse mechanisms involving increased production of reactive oxygen species. Increased oxidative stress drives changes in chromatin structure that mediate gene expression changes leading to the upregulation of proinflammatory and profibrotic mediators. The epigenetic contribution to diabetes-induced changes in gene expression is increasingly recognized as a key factor in the development and progression of vascular diabetic complications.
Critical Issues:
The mechanisms through which stimuli from the diabetic milieu promote epigenetic changes remain poorly understood. In addition, glycemic control constitutes an important factor influencing epigenetic states in diabetes, and the phenomenon of hyperglycemic memory warrants further research.
Future Directions:
Knowledge of the molecular mechanisms underlying epigenetic changes in diabetes may allow the design of novel therapeutic strategies to reduce the burden of diabetic complications. Furthermore, certain epigenetic markers are detected early during the onset of diabetes and its complications and may prove useful as biomarkers for disease risk prediction.
Introduction
T
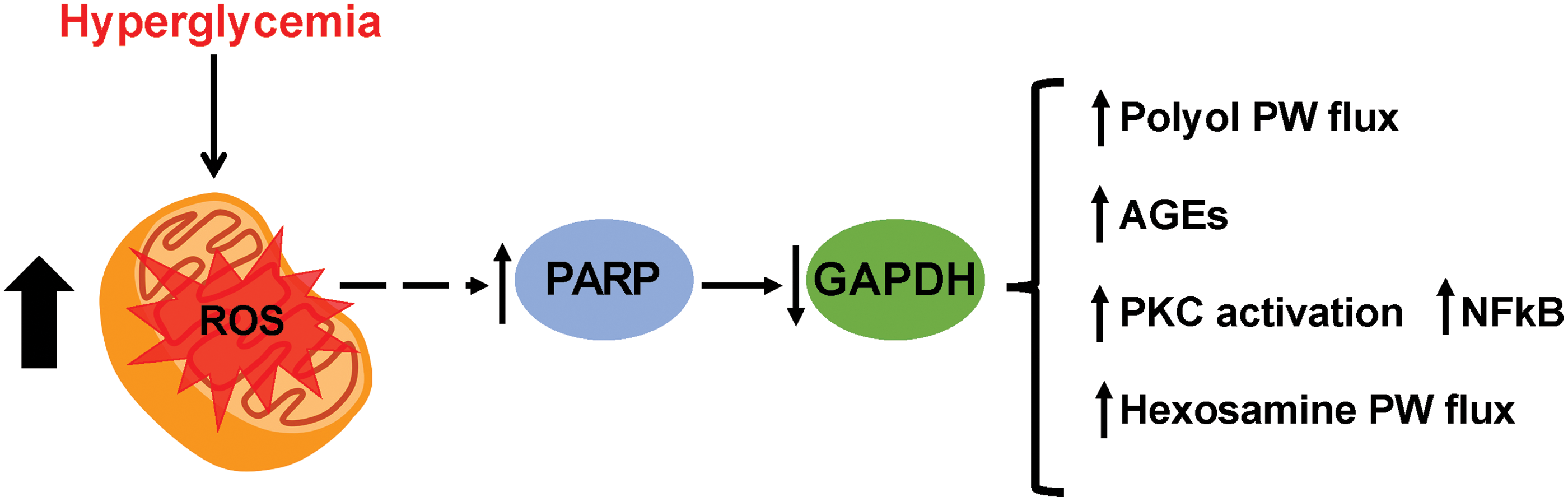
Long-term complications of diabetes include retinopathy (retinal damage and visual impairment), nephropathy (renal damage), and neuropathy (nerve damage) as well as cardiovascular disease. Retinopathy, nephropathy, and neuropathy are considered microvascular complications of diabetes, whereas cardiovascular events associated with the formation of atherosclerosis are considered macrovascular complications (51). The current paradigm for the development of diabetic complications involves the overproduction of reactive oxygen species (ROS) (21). According to the unifying mechanism for the pathobiology of diabetic complications, or Brownlee hypothesis, an increase in intracellular glucose results in overproduction of mitochondrial ROS that induce DNA damage, this, in turn, activates poly(ADP-ribose) polymerase (PARP). The resulting accumulation of ADP-ribose polymers on the enzyme glyceraldehyde-3 phosphate dehydrogenase (GAPDH) inhibits its activity, leading to the accumulation of all metabolites upstream in the glycolytic pathway. This process initiated by ROS overproduction causes cell damage via four major pathways: (i) increased flux through the polyol pathway, (ii) increased formation of advanced glycation end products (AGEs), (iii) activation of protein kinase C (PKC), and (iv) increased flux through the hexosamine pathway (Fig. 1) (21).
Together with hyperglycemia, other factors that characterize the diabetic state also contribute to the development and progression of diabetic complications. Hypertension, dyslipidemia, and hemostatic alterations are all associated with increased risk of cardiovascular disease in diabetic patients, including atherosclerosis, myocardial infarction, and stroke (10). This implies that, to significantly reduce the risk of vascular complications, therapeutic approaches should address all these risk factors in combination. Indeed, the STENO-2 trial showed that a multifactorial intervention targeting hypertension, hyperglycemia, platelet dysfunction, and lifestyle risk factors reduced the incidence of cardiovascular events and mortality due to cardiovascular disease in patients with type 2 diabetes (T2D) (59). However, despite a risk reduction for retinopathy, nephropathy, and neuropathy after intensive treatment, high rates of progression for one or more complications were observed (58). In addition, glucose-lowering treatment using the sodium–glucose cotransporter (SGLT) 2 inhibitor empagliflozin (EMPA-REG trial) resulted in a reduction in mortality due to cardiovascular events in patients with T2D and established cardiovascular diseases (199). This risk reduction, however, was likely due to hemodynamic improvement (heart failure hospitalization was reduced by 35%) and not an effect of empagliflozin on atherosclerosis progression (168). The outcomes of these trials highlight the importance of developing novel strategies to reduce the burden of vascular diabetic complications.
Epigenetic Gene Regulation
Epigenetic modifications are a critical component in the maintenance of normal gene expression patterns and in the development of disease. Epigenetic events such as DNA methylation and histone protein modifications have been associated with pathological gene expression changes during the development of diabetes and its complications. The effects of such modifications in gene expression are briefly explained in this section.
DNA methylation
DNA methylation in mammalian cells involves the addition of a methyl group to carbon 5 (C5) of cytosines in CpG dinucleotides. Although it was initially regarded as a silencing mark, we know DNA methylation is associated with both transcriptional repression and activation, depending on the genomic context (transcriptional start site, enhancers, and gene body) (71).
DNA methylation patterns are maintained through cell division by the action of enzymes called DNA methyltransferases. These enzymes recognize methylated sites in the parental DNA strand and add a methyl group to the corresponding site on the newly synthesized strand during DNA replication (38).
Histone modifications
Post-translational modifications to the tails of histone proteins can modify chromatin structure as well as mediate the recruitment of coregulatory proteins, modulating the accessibility of the transcriptional machinery to DNA. The most common histone modifications are acetylation and methylation, although phosphorylation, ubiquitination, and sumoylation have also been described. The enzymes responsible for writing or erasing such modifications have a high substrate and product specificity and their activity is closely linked to transcriptional activation or repression. The close relationship between specific histone tail modifications and their effect on transcription led to the proposal of the “histone code” hypothesis. This hypothesis states that histone modifications complement the genetic code by serving as binding sites for effector proteins and influencing chromatin accessibility and transcription factor binding (70).
Histone acetylation involves the addition of an acetyl group to the ɛ-amino group of lysine residues near the N-terminus of the protein. It was the first modification of histones to be discovered and it is associated with transcriptional activation. Lysine residues in histone tails subject to acetylation include lysine 9 (K9), K14, K18, K23, and K27 on histone H3 and K5, K8, K12, and K16 on histone H4 (82).
The enzymes responsible for adding the acetylation mark onto histone tails are called histone acetyltransferases (HATs) and the removal of such marks is mediated by histone deacetylases (HDACs) (33). Histone acetylation is highly dynamic and both HATs and HDACs are enriched at the promoters of actively transcribed genes (174). In addition to directly modulating chromatin accessibility, acetylated lysine residues on histone tails also serve as binding sites for bromodomain proteins that can further recruit transcriptional regulators (49).
Methyl groups can also be added to residues on the tails of histone proteins. Histone lysine methylation is carried out by histone methyltransferases (HMTs) and removed by lysine demethylases (LSDs) such as lysine demethylase 1 (LSD1) (150). Several histone lysine residues are subject to methylation, and their location and type of reaction (mono-, di-, or trimethylation) determine their effect in transcription. For example, methylation of lysines K9 and K27 on histone H3, and K20 and K59 on histone H4 is associated with transcriptional repression, whereas methylation of H3K4, H3K36, and H3K79 is characteristic of transcriptional activation (100).
Activating lysine methylation marks are differentially observed between gene regulatory elements. For example, monomethylated H3K4 (H3K4me1) is preferentially enriched at gene enhancers, whereas trimethylated H3K4 (H3K4me3) is enriched at gene promoters (64). Arginine residues within histone tails are also subject to methylation and protein arginine methyltransferases are responsible for this modification. They transfer the methyl group from the cofactor S-adenosylmethionine (SAM) to the guanidine group of arginine in histone and nonhistone protein substrates (82).
The tails of histone proteins are subject to a wider range of modifications that have an impact in transcription. Serine and threonine residues on histones H2A, H2B, and H4 are targets for β-N-acetylglucosamine (O-GlcNAc) transferase, suggesting that O-GlcNAcylation can be considered part of the histone code (139). Histones can also be ADP-ribosylated by enzymes of the PARP family and this reversible modification has been associated with promoting chromatin relaxation (62). Ubiquitination, particularly monoubiquitination, of histone proteins appears to play a diverse role in gene transcription by mediating interactions with histone-modifying enzymes (87, 170, 171). Similarly, small ubiquitin-related modifier proteins can modify sites on histone H4. The function of histone sumoylation is not completely understood but it is thought to be associated with transcriptional repression (151).
Noncoding RNAs and RNA modifications
Noncoding RNAs are considered part of the epigenetic machinery regulating gene expression as they can form RNA scaffolds that recruit DNA and histone-modifying proteins to sites of transcription (67). Furthermore, increasing evidence suggests that covalent modifications to RNA molecules themselves can directly regulate gene expression. The most common messenger RNA modification is N6-methyladenosine (m6A), a reversible modification akin to those on DNA and histone tails, that has been associated with transcript stability (52).
Histone Modifications as a Metabolic Sensing Mechanism
Epigenetic modifications of DNA and histones constitute a mechanism through which environmental changes influence cellular gene expression. Enzymes that catalyze methylation/demethylation as well as acetylation/deacetylation reactions require cofactors that derive from intermediary metabolism pathways. The cofactor SAM is required as a methyl donor for all DNA and histone methylation reactions. SAM is produced as part of the one-carbon metabolism cycle by condensation of ATP and methionine (5). In fact, methionine metabolism is directly linked to tissue histone methylation levels by modulating SAM concentrations (103). Demethylation reactions are also influenced by metabolic products. LSD1 activity is dependent on FAD+ levels within the cells, and the JmjC family of demethylases requires α-ketoglutarate as well as iron (FeII) and oxygen to catalyze the removal of methyl groups from histones (150).
Histone acetylation is strongly associated with cellular acetyl-CoA levels. Mammalian cells derive most of their acetyl-CoA from glucose-derived citrate by action of the enzyme ATP citrate lyase (41, 176). Acetyl-CoA can also be derived from lipids by fatty acid β oxidation. In this case, the gene expression changes induced favor lipid metabolism (65). This demonstrates that the availability of nutrients promotes gene expression programs characterized by the activation of genes involved in their metabolism by inducing epigenetic changes. HDACs of the sirtuin family (SIRT) are NAD+ dependent, thus their activity is influenced by cellular conditions that affect NAD+ levels such as changes in redox states (68, 141).
Serine and threonine residues in proteins, including H2A, H2B, and H4 histones, are subject to O-GlcNAc modifications. UDP-N-acetylglucosamine (UDP-GlcNAc) addition or removal from proteins is catalyzed by O-GlcNAc transferase and O-GlcNAcase, respectively (22, 139). UDP-GlcNAc derives from the hexosamine pathway of glucose metabolism and high glucose levels increase protein O-GlcNAcylation (61). Increased O-GlcNAc in β cells as a result of high glucose promotes histone modification changes near the insulin gene promoter that activates its transcription (37). This suggests that this modification acts as a sensor for rising glucose levels to promote insulin secretion.
Nuclear–cytoplasmic transport and epigenetic regulation
Many histone-modifying proteins are localized in both nuclear and cytoplasmic compartments. In this sense, environmental stimuli that promote gene expression changes through mediating histone modifications would need to promote changes in the subcellular localization of these enzymes. Indeed, many histone-modifying proteins alter their subcellular localization according to environmental stimuli. Hyperglycemia promotes the nuclear translocation of the Set7 lysine methyltransferase in cultured endothelial cells (122). Similarly, transforming growth factor β1 (TGFβ1) leads to increased nuclear Set7 in renal mesangial cells (143). In C2C12 cells, the HDAC SIRT1 translocates to the nucleus in response to oxidative stress where it leads to a reduction in histone H3 acetylation levels (161). In rodent models of heart failure, SIRT1 is mainly localized in the nucleus of cardiomyocytes, where it promotes the expression of manganese superoxide dismutase (MnSOD), increasing resistance to oxidative stress (160).
Excess production of ROS and increased oxidative stress are characteristic of diabetes. Many proteins, especially those involved in antioxidant responses, translocate into the cellular nucleus in response to oxidant stimuli. For example, rising ROS levels promote the nuclear localization of the NF-E2-related factor 2 (NRF2), where it activates the expression of genes that participate in the antioxidant response. Nuclear import of NRF2 requires transport by importins proteins, specifically importin α5/importin β1 (165). Diabetes markedly increases the expression of importin isoforms, including importin α5, in rat kidneys (80). This suggests that nuclear transport could be increased in diabetes and this may constitute another mechanism through which gene expression changes occur in the diabetic kidney. Growth factor signaling contributes to the abnormal tissue proliferation that characterizes diabetic complications, partly by promoting the nuclear translocation of histone-modifying proteins. Nerve growth receptor signaling promotes nuclear localization of the p300/CREB-binding protein-associated factor and hGCN5 acetyltransferases, increasing the expression of cell cycle genes during cell differentiation (179).
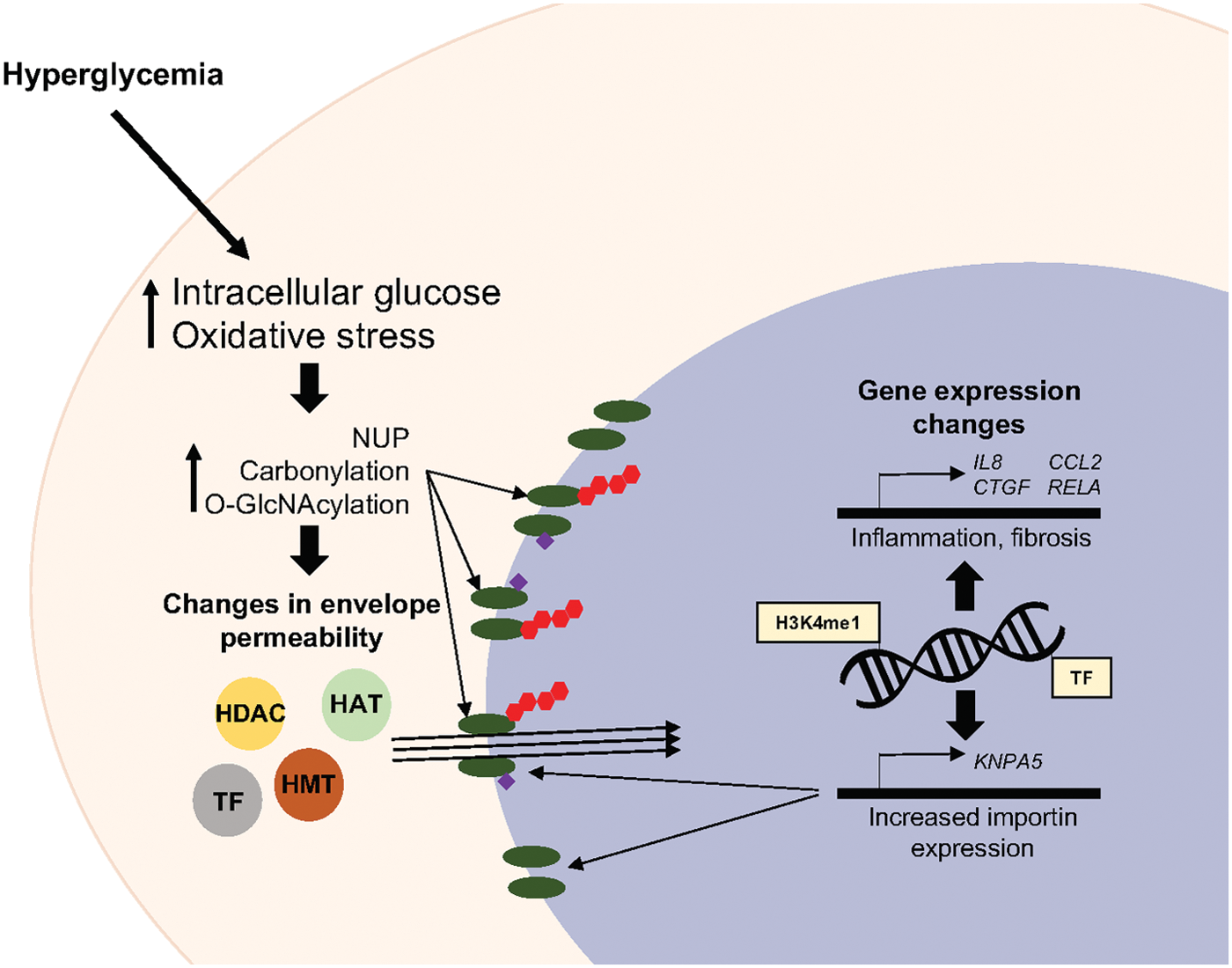
Hyperglycemia and oxidative stress result in increased protein O-GlcNAc and many nuclear pore proteins have been shown to acquire this modification (89). Glycosylation of nucleoporins affects their stability and ability to interact with other proteins (60, 89, 110). These changes affect nuclear pore specificity and are thought to translate into changes in nuclear envelope permeability (85). ROS can also directly affect the function of nuclear pore components. Oxidative stress induces carbonylation of proteins including nucleoporins, and this is associated with increased permeability of the nuclear envelope (32). The events that contribute to changes in nuclear envelope permeability in response to diabetic stimuli are summarized in Figure 2.
Epigenetic Mechanisms Underlying Diabetic Complications
The generation of mitochondrial ROS is behind the dysregulation of several pathways, ultimately leading to the development of diabetic complications. Indeed, persistent ROS signaling is thought to be a key mechanism for the establishment of hyperglycemic memory, discussed hereunder. Increased levels of intracellular glucose in endothelial cells promote the recruitment of the Set7 lysine methyltransferase to the p65 gene (RELA) promoter, resulting in the increase of H3K4me1 levels in an ROS-dependent mechanism (39). Furthermore, mice that are deficient in antioxidant uncoupling protein 2 and glyoxalase (GLO1), important antioxidant enzymes, have increased levels of H3K4me1 at the RelA promoter in response to hyperglycemia (39). P66Shc is a mitochondrial adaptor protein that is upregulated in endothelial cells in response to hyperglycemia and is implicated in the generation of ROS. This upregulation is maintained despite normalization of glucose levels and this is associated with increased histone acetylation and decreased DNA methylation at its gene promoter (127, 198). Similarly, p66Shc expression is persistently upregulated in blood monocytes from T2D patients despite intensive glycemic control (28). This redox protein is also induced by diabetes in the kidney, particularly in podocytes, where it contributes to the increase in oxidative stress and glomerular damage (18). These findings highlight another potential epigenetic mechanism underlying the development of diabetic complications and hyperglycemic memory in different tissues.
Postprandial increases in blood glucose observed in subjects with T2D stimulate ROS production even more than sustained hyperglycemia (111). In fact, minimizing glucose fluctuations in diabetic patients is associated with a decrease in circulating markers of inflammation and oxidative stress (138). This observation, together with persistent epigenetic changes that occur in response to hyperglycemia, highlights the importance of effective diabetes management treatments that focus not only on HbA1c but also on target glucose fluctuations.
Diabetic macrovascular disease
The association between diabetes and macrovascular disease has been recognized for >30 years with the observation that people with diabetes have a significantly higher risk of death from cardiovascular events (36, 86, 133). In fact, cardiovascular disease is a major cause of death in people with both type 1 diabetes (T1D) and T2D, accounting for a large proportion of healthcare expenditure in people with diabetes (114). The major pathological process behind cardiovascular disease is atherosclerosis, a progressive narrowing of arterial walls.
Inflammation is a key process in the development of atherosclerosis. Histone modifications mediate changes in the gene expression profile of vascular endothelial cells in response to high glucose levels, this is characterized by increased expression of proinflammatory genes (116, 130). In this context, Set7 is a key mediator of cellular responses to hyperglycemia. This enzyme promotes the methylation of histone and nonhistone proteins to activate the expression of proinflammatory genes (79, 90, 122). Set7 may act as a hyperglycemic sensor, translocating into the nucleus in response to high glucose levels to influence gene expression (78, 122). High glucose stimulation of endothelial cells induces the recruitment of Set7 to the RELA gene promoter and subsequent increase in the levels of H3K4me1. Importantly, this effect is maintained after the cells are returned to normal glucose conditions (39, 122). Persistent activation of nuclear factor κB (NFκB) was also described in vivo using a mouse model of hyperglycemic memory where transient hyperglycemia resulted in increased expression of NFκB target genes Hmox1 and Cxcl2 (IL-8) for up to 7 days (122). The role of Set7 as an activator of NFκB was also evidenced in cultured monocytes where knockdown of the enzyme resulted in a decrease in monocyte adhesion and reduced expression of 25% of all proinflammatory genes induced by tumor necrosis factor α (90).
NFκB activation results in increased expression of cellular adhesion molecules that contribute to the recruitment of inflammatory cells. Once monocytes adhere to the arterial wall, they differentiate into macrophages and migrate to the intima layer of the vessel, where they take up oxidized low-density lipoprotein (oxLDL) particles and become foam cells (69, 91). Cultured monocytes/macrophages exposed to oxLDL adopt a persistent proatherogenic gene expression profile mediated by changes in chromatin accessibility that promote the expression of proinflammatory genes (11, 136). These changes are characterized by increased levels of activating H3K4me3 at the promoters (11) and H3K27Ac at enhancers (136) of genes regulated by oxLDL.
Other chromatin remodeling events that occur at the RELA promoter and are associated with increased proinflammatory gene expression include the recruitment of the histone demethylase LSD1 and decreased binding of SUV39H1, which lead to a reduction in repressive histone methylation marks (19, 170). NFκB also promotes the expression of its proinflammatory targets by mediating the recruitment of HATs (105). In fact, high glucose treatment of vascular endothelial cells confers a specific hyperacetylation pattern that is directly correlated with transcriptional activation (130). Furthermore, dysregulation of HDAC expression in endothelial cells is associated with cardiovascular disease. A HDAC9 gene variant has been implicated in large vessel ischemic stroke by genome-wide association studies (GWASs) (14). Consistent with this association, increased HDAC9 expression is observed in carotid, aortic, and femoral plaques (98). In contrast, HDAC9 was significantly downregulated in aortas from diabetic db/db mice in association with increased markers of vascular calcification (15). Moreover, global levels of lysine acetylation were elevated in mouse aortas exposed to high glucose and palmitate and aortas from diabetic Goto-Kakizaki rats (24). This suggests that HDAC9 expression may be regulated differently in diabetic conditions. Pharmacological HDAC inhibition in human aortic endothelial cells (HAECs) results in both increases and decreases in histone acetylation (135). This highlights the complexities of gene regulation by histone acetylation and emphasizes the need for further understanding of these processes in the vasculature.
NFκB activation is also increased in vascular smooth muscle cells (VSMCs) in response to high glucose (63). These cells play a key role in the progression of macrovascular disease as atherogenic stimuli induce their transdifferentiation into macrophage-like cells (43). Angiotensin II (AngII) has an atherogenic effect in VSMCs as it increases the production of ROS and upregulates the expression of proinflammatory mediators (20). In vitro experiments suggest that some of these actions of AngII are mediated by histone modifications as exposure of VSMCs to AngII results in increased levels of H3K4me3 and H3K36 (88). The expression of 24 long noncoding RNAs is also altered in VSMCs by AngII exposure, a novel transcript lnc-Ang362 was identified in association with microRNAs (miRNAs) miR-221 and miR-222 as a regulator of cell proliferation in this cell type (88).
The role of miRNAs in gene regulation associated with macro and microvascular complications of diabetes is increasingly recognized and has been extensively reviewed by Zhang et al. (191). miRNAs such as miR-146a and miR-155 are upregulated by diabetes in animal models and appear to contribute to endothelial dysfunction and the progression of atherosclerosis (46, 123, 158, 180). In contrast, miR-126 is reduced in patients with diabetes in association with increased micro and macrovascular complications (8, 189). Similarly, miR-492 and miR-1 have an atheroprotective effect by reducing insulin resistance and endothelial dysfunction (44, 186). miRNAs can target other epigenetic modifiers, evidencing the complexity of epigenetic regulation. For example, miR-195 is upregulated in endothelial cells after high glucose exposure, resulting in the upregulation of fibronectin expression via targeting the HDAC SIRT1 (113). Similarly, miR-125b targets the HMT SUV39H1 in VSMCs, leading to an increase in proinflammatory gene expression (169).
Diabetic nephropathy
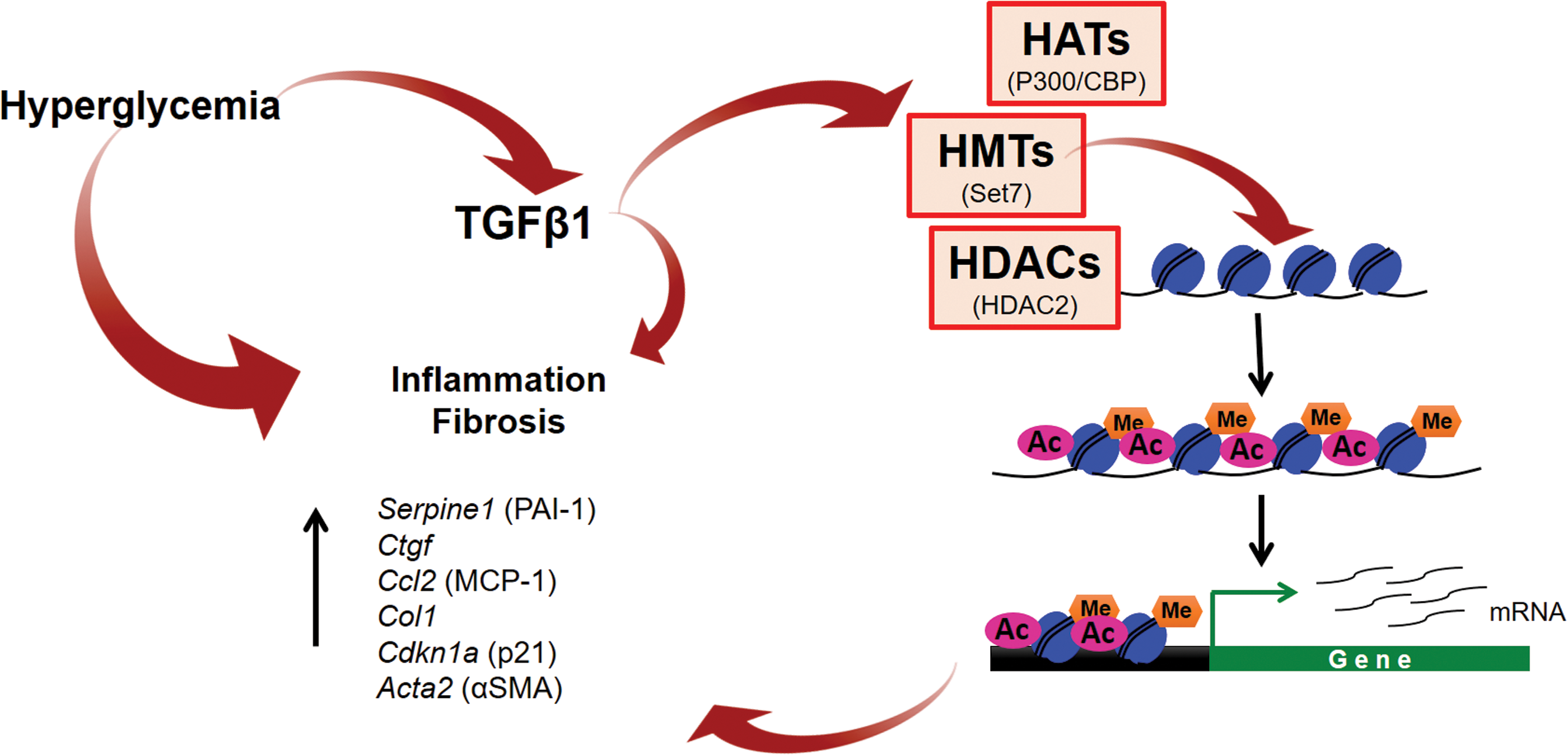
Chronic kidney disease can develop as a major complication of diabetes. In fact, diabetes constitutes the lead cause of end-stage renal disease (ESRD) (4). Kidney glomeruli comprise endothelial cells surrounded by podocytes that, together with the basement membrane, constitute the glomerular filtration barrier. Renal podocyte injury and loss are critical in the pathological progression of diabetic nephropathy and represent the mechanism through which proteins leak into the urinary compartment (125). The major pathological features of diabetic nephropathy are increase of glomerular basement membrane thickness and mesangial expansion, accumulation of extracellular matrix (ECM) components and hypertrophy of glomerular and tubular components (51, 148). Accumulating evidence indicates that pathological gene expression changes observed during the progression of diabetic nephropathy are mediated by histone modifications (Fig. 3). TGFβ1 is produced in the kidney in response to hyperglycemia and is a key mediator of kidney damage in diabetes by promoting the expression of profibrotic genes, partially by inducing changes in chromatin structure that facilitate transcriptional activation. TGFβ1 stimulates the expression of plasminogen activator inhibitor 1 by recruiting the p300/CBP acetylase and Set7 methyltranferase to its gene promoter (157, 188). Other TGFβ1 targets such as collagen 1 and connective tissue growth factor are also regulated by Set7-mediated histone methylation (157). Similarly, in hyperglycemic conditions, the myocardin-related transcription factor A recruits HATs and HMTs to its target genes, such as collagen, increasing histone acetylation and H3K4me3 levels at their promoters and promoting fibrotic gene expression in the kidney (181). Chronic hyperglycemia also leads to endoplasmic reticulum (ER) stress, which promotes the upregulation of MCP-1 in a histone methylation-dependent mechanism mediated by Set7 (25). During the progression of diabetes, there is an increase in production of the enzyme 12/15 lipooxygenase, which mediates lipid oxidation. Oxidized lipids such as 12(S)-hydroxyeicosatetraenoic acid cause kidney damage, partially through increasing the levels of Set7 in renal mesangial cells, resulting in the upregulation of profibrotic genes (187). Increased ROS production and TGFβ1 signaling also induce HDAC2 expression and this is associated with increased expression of α-smooth muscle actin and fibronectin in the diabetic kidney (119). Other HDACs are associated with renal damage in diabetic nephropathy. Increased renal expression levels of HDAC4 and HDAC9 have also been reported in diabetic nephropathy models, where they contributed to renal damage by promoting ECM accumulation and epithelial-to-mesenchymal transition (92, 119, 131, 172). In fact, HDAC inhibition mitigates diabetes-induced renal damage by attenuating oxidative stress, inflammation, and fibrosis in rat and mouse models of diabetes (1, 54, 119, 152). In contrast, the deacetylase SIRT1 plays a protective role in renal cells and its expression is reduced in diabetic nephropathy (182).
Increased TGFβ1 signaling is characteristic of other fibrotic pathologies, and epigenetic mechanisms are implicated. Set7 mediates the upregulation of profibrotic genes such as those encoding collagen 1 and fibronectin in a mouse model of unilateral ureteral obstruction (143). Furthermore, Set7 levels are directly correlated with the degree of fibrosis in human samples from IgA and membranous nephropathies (143). In addition, Set7-mediated histone methylation regulates TGFβ1 gene expression in the liver of rats after bile duct ligation (149). These observations strongly suggest that epigenetic modifications, particularly histone methylation by Set7, play a key role in mediating gene expression changes that drive the development of fibrosis in diverse disease environments.
Renal damage in diabetes is also mediated by miRNA-dependent gene repression, indeed miRNAs have been widely implicated in the development and progression of diabetic nephropathy. Several miRNAs, such as miR21, miR192, miR200-c, miR216a, and miR217, are highly expressed in diabetic kidney and promote the upregulation of ECM components contributing to glomerular hypertrophy characteristic of diabetic nephropathy (74 –77, 102, 128). In contrast, the expression of miR-1 and miR-146 is decreased by diabetes in association with increased expression of endothelin-1 and fibronectin (44, 46).
Diabetic retinopathy
Although epigenetic-mediated gene expression changes that contribute to vascular and renal disease have been well studied, epigenetic changes during the progression of diabetic retinopathy remain poorly understood. In a rat model of diabetic retinopathy, HDAC and HAT activities were dysregulated, resulting in a global decrease in histone acetylation in the retina and capillary cells (193). However, contrasting results have shown that retinal histone acetylation is increased in diabetes in association with increased proinflammatory gene expression (72). In this sense, understanding the role of histone acetylation in the development of diabetic retinopathy requires further investigation.
MnSOD protects retinal endothelial cells against oxidative stress-induced damage (84, 93). The gene encoding MnSOD, Sod2, is downregulated in the retina of diabetic rats in a process that involves increased expression of the H4K20 methyltransferase SUV420H2 and the LSD1. This results in increased H3K20me3 and decreased H3K4me1/me2 levels (194, 195). Diabetes-induced decrease in H3K9me2 and increase in H3K9ac at the matrix metalloproteinase 9 (MMP9) gene promoter activate its expression, contributing to capillary damage in the retina (196). A single nucleotide polymorphism in the gene encoding the HMT SUV39H2 was associated with diabetic retinopathy in patients with T1D (159). These observations suggest a role for histone methylation in the development of diabetic retinopathy.
The miRNA profile of the retina is altered by diabetes (83). Downregulation of miR200b is associated with the upregulation of its gene target, VEGF, a major driver of pathological angiogenesis during the progression of diabetic retinopathy (101). In contrast, upregulation of miR-29b in rat retinas during early diabetic nephropathy may have a protective effect against apoptosis of retinal ganglion cells (153). miR-126 and miR-320 may also be protective in this context by halting cell proliferation, angiogenesis, and tissue remodeling (45, 173, 184). NFκB activation also plays a pathological role in the development and progression of diabetic retinopathy. miR-146 and miR-146a have an anti-inflammatory effect in retinal endothelial cells by attenuating NFκB activation (191).
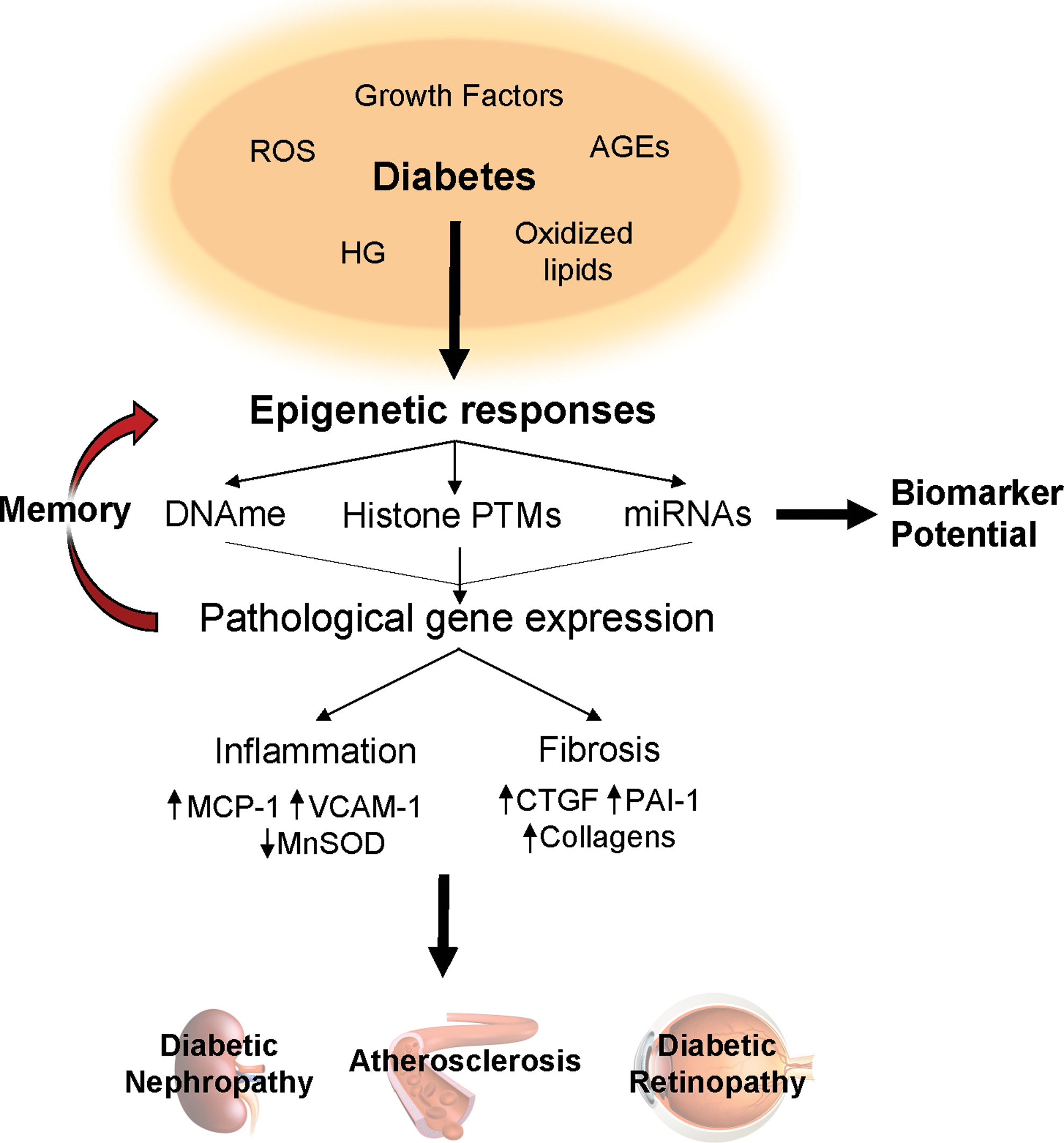
The mechanisms behind diabetes-induced epigenetic and gene expression changes that contribute to the development and progression of vascular diabetic complications are shown in Figure 4.
Hyperglycemic Memory
The Diabetes Control and Complications Trial (DCCT; 1983–1989) aimed to study the effect of intensive glycemic control compared with conventional treatment in patients with T1D. This seminal study showed that patients under intensive control had improved glycemic and HbA1c levels and this was associated with reduced incidence of albuminuria and risk of developing neuropathy and retinopathy (162). Given these outcomes, all participants were treated under the intensive control regime at the end of the study period. A follow-up study referred to as Epidemiology of Diabetes Interventions and Complications (EDIC) assessed the long-term effects of intensive control on the original DCCT cohort. During the course of the EDIC trial, all participants regardless of their treatment group during the DCCT achieved similar levels of HbA1c. Results showed that patients who had received intensive treatment during the DCCT and EDIC had improved kidney function and decreased risk of retinopathy progression and cardiovascular disease compared with those who had been on the conventional treatment arm (163, 164). The effect of tight glycemic control in patients with T2D was assessed during the UK Prospective Diabetes Study. The initial endpoint study (10-year treatment) revealed a significant reduction in the risk of developing microvascular but not macrovascular complications (167). However, a 10-year follow-up study demonstrated that intensive treatment also reduced the risk of developing macrovascular complications (66, 156). The conclusions from these studies suggested that previous periods of hyperglycemia cause long-lasting effects that result in the development of complications despite glycemic normalization. This phenomenon was termed hyperglycemic memory.
Several pathways have been implicated in the establishment of hyperglycemic memory. Hyperglycemia increases the generation of AGEs, which can lead to the covalent modification of proteins, lipids, and nucleic acids, resulting in sustained cellular damage (17). Intensive glycemic control in DCCT participants was associated with a reduction in glycated skin collagen (112) and with reduced incidence of complications during the follow-up EDIC study (53). These two studies suggest that AGE formation may underlie the phenomenon of hyperglycemic memory and propose the measurement of indicators of skin collagen glycation as a predictor of diabetic complications. Increased production of ROS causes direct damage to cellular proteins and organelles. Indeed, endothelial dysfunction is associated with increased ER stress and persistent activation of the unfolded protein response (9) and defective autophagy (47). In this sense, chronic activation of these pathways contributes to the maintenance of an inflammatory state in endothelial cells in diabetes.
Hyperglycemia itself, as well as ROS, can promote proinflammatory changes in gene expression that persist after the return to normoglycemia. Epigenetic modifications, particularly histone methylation, at the promoter of the p65 subunit mediate this process, resulting in persistent NFκB-dependent gene activation (39). It appears that chronic hyperglycemia creates an environment of low-grade inflammation characterized by increased protein glycation and oxidative stress that is maintained by a persistent increase in the expression of inflammatory mediators due to changes in epigenetic modifications.
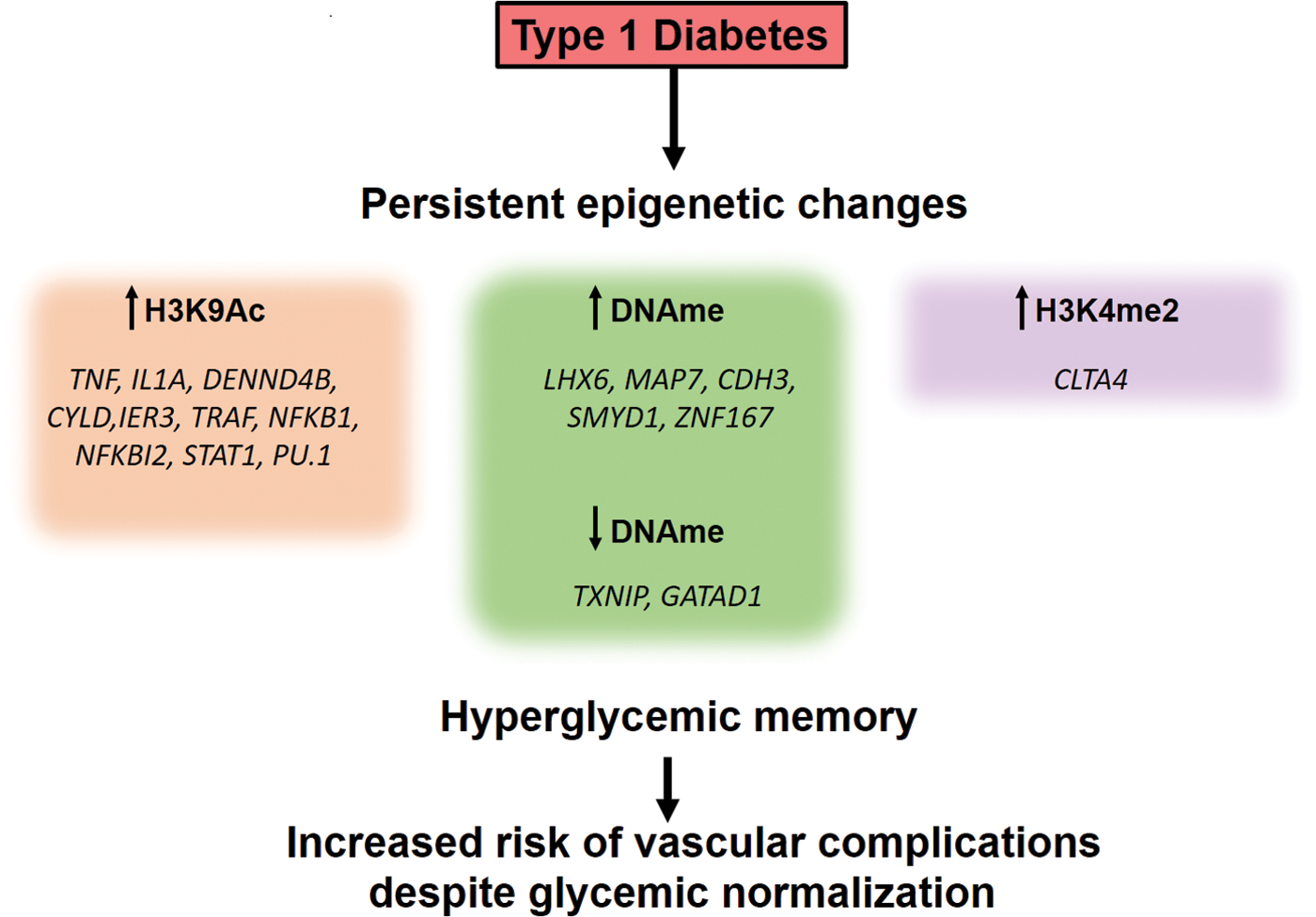
Indeed, recent studies using blood monocytes and lymphocytes isolated from DCCT participants showed there were specific differences in DNA methylation as well as histone acetylation and methylation at the sites of proinflammatory genes between patients on the intensive and conventional treatment groups (Fig. 5) (26, 104). Similarly, lymphocytes from subjects with T1D have higher levels of the H3K9me2 mark in genes associated with inflammation and autoimmunity than those from healthy individuals (106). Furthermore, the expression of the proinflammatory NFκB p65 subunit and some of its target genes is increased in mononuclear cells derived from T2D patients in a H3K4me1-dependent manner (126). In vitro experiments using HAECs also revealed persistent NFκB activation and proinflammatory gene expression after transient high glucose exposure in association with expression changes on miR-125b, miR-146a-5p, and miR-29a-3p (197). These observations implicate a role for epigenetic mechanisms mediating the phenomenon of hyperglycemic memory.
Epigenetic Modifications as Disease Predictors
We have described the role of epigenetic modifications in the development of tissue pathology in diabetes. As these changes occur early in disease pathology, there is potential to use them as diagnostic tools for the prediction of disease onset and/or progression.
Diabetes onset
Diabetes is estimated to affect >400 million people worldwide, making a significant contribution to global morbidity and mortality (121). With this estimate expected to increase, detecting people at risk of developing diabetes and its complications would represent a major public health advantage.
Diabetes is a heterogeneous disease and can be classified according to its etiology and clinical stage. T1D is an autoimmune disorder that causes the destruction of pancreatic β cells, resulting in a marked or absolute deficiency in insulin secretion. It is one of the most common autoimmune diseases and it is characterized by a selective immune T cell attack of β cells and, although its causes are not clear, it is thought to involve genetic susceptibility and an environmental trigger (31). In contrast, T2D, the most common form of the disease, refers to a defect in insulin action and/or secretion frequently related to insulin resistance. The onset of T2D is slow and noticeable. A state of glucose intolerance and subsequent hyperglycemia that develops during pregnancy are known as gestational diabetes (GD). It often resolves at the end of the pregnancy but can also progress to T2D (42).
Traditionally, the risk of developing diabetes was estimated based on biochemical parameters and other noninvasive measurements such as body mass index. The advent of new DNA sequencing technologies has resulted in widespread use of GWASs to detect genetic variants associated with diabetes risk (120). However, many variants detected do not map to coding genomic regions, making it difficult to assess their overall risk contribution. Improving technologies have allowed the interrogation of many genes and gene variants associated with diabetes risk for the presence of epigenetic modifications. For example, circulating levels of insulin-like growth factor binding protein 1 (IGFBP-1) have been associated with T1D and T2D. Recent studies have shown that DNA methylation at the IGFBP1 locus is inversely correlated with circulating IGFBP-1 in diabetic patients (55, 56). Similarly, polymorphisms in SLC30A8 confer increased T2D susceptibility, and DNA methylation of this gene is indeed increased in patients with T2D (147). A differentially methylated region within the TXNIP gene is also associated with T2D (50).
The diagnosis of T1D usually occurs when β cell destruction is close to 90% (178). However, the onset of the disease is slow and the discovery of novel biomarkers with strong predictive value, together with traditional biomarkers such as islet autoantigens, would provide a useful window for treatment (57, 178). Detection of β cell death in vivo by assessing the levels of differentially methylated circulating free Ins (insulin) and Iapp (amylin) DNA has been successful in monitoring the onset of diabetes in mouse models of T1D and patients with recent onset of the disease (3, 124). The risk of developing T2D is often estimated using an algorithm that combines biochemical parameters measured in blood, anthropometric measurements, and lifestyle factors (144). This approach can predict individuals at risk of developing T2D within people who already display metabolic abnormalities. However, it cannot identify susceptible individuals in the general population (57). miRNAs are detectable in tissues and bodily fluids such as blood and urine and are highly stable, which is why they have been proposed as biomarkers for cancer and other conditions (109, 115, 175). Altered tissue and circulating miRNA profiles have been observed in T1D and T2D, supporting the notion of miRNA-based tests as predictive markers of diabetes onset (Table 1).
miRNA, microRNA; PBMCs, peripheral blood mononuclear cells; qPCR, quantitative polymerase chain reaction.
Diabetic complications
GWASs have identified noncoding genetic variants associated with risk factors predisposing to the development of metabolic disease and diabetic complications. These may affect gene expression through epigenetic regulation. For example, some variants affect local DNA methylation and are referred to as methylation quantitative trait loci (meQTL). Many gene variants identified from blood cells, liver, and adipose tissue and associated with circulating lipid levels are meQTL (34).
Genome-wide methylation studies have identified differentially methylated regions associated with the risk of development and progression of nephropathy in diabetic patients (12, 142, 154). One such region was located within the UNC13B gene, where a single nucleotide polymorphism was recently identified in association with diabetic nephropathy risk (166). Specific differentially methylated CpG sites have also been identified in blood of T1D patients with proliferative diabetic retinopathy (2). In addition, global DNA methylation levels have been associated with the progression of diabetic nephropathy (95) and retinopathy (94). These observations suggest that DNA methylation changes detectable in blood cells can be useful biomarkers for the prediction of microvascular complications in diabetic patients.
miRNAs are also proposed biomarkers in this context. Endothelial cell-enriched miR-126 is reduced in response to high glucose exposure, this loss has also been observed in T2D patients (189). Moreover, miR-126 is also downregulated in patients with coronary artery disease, further implicating it in vascular damage (48). miR-503 is upregulated under diabetic conditions and it is thought to contribute to endothelial dysfunction and impaired neovascularization (23). Other miRNAs, such as miR-133a, miR-133b, miR-499, miR-208a, and miR-208b, are associated with cardiovascular disease, including myocardial infarction and heart failure; however, their relevance as biomarkers in diabetes mellitus has not been assessed (30).
Circulating and urinary miRNA signatures may provide valuable information as biomarkers of nephropathy progression in T1D and T2D. Circulating levels of mir-16, miR-21, miR-155, miR-210, and miR-638 are inversely correlated with glomerular filtration rate in patients with nephropathy at different disease stages (117). In patients with T1D, miR-21-5p and let-7b-5p were significantly associated with increased risk of ESRD progression, whereas miR-29a-3p and let-7c-5p were associated with protection against rapid nephropathy progression (129). Urinary miRNA profiling in T2D patients revealed 16 miRNA species that are deregulated only in diabetic nephropathy and correlated with albuminuria levels (35). Such associations have also been reported in T1D patients (6).
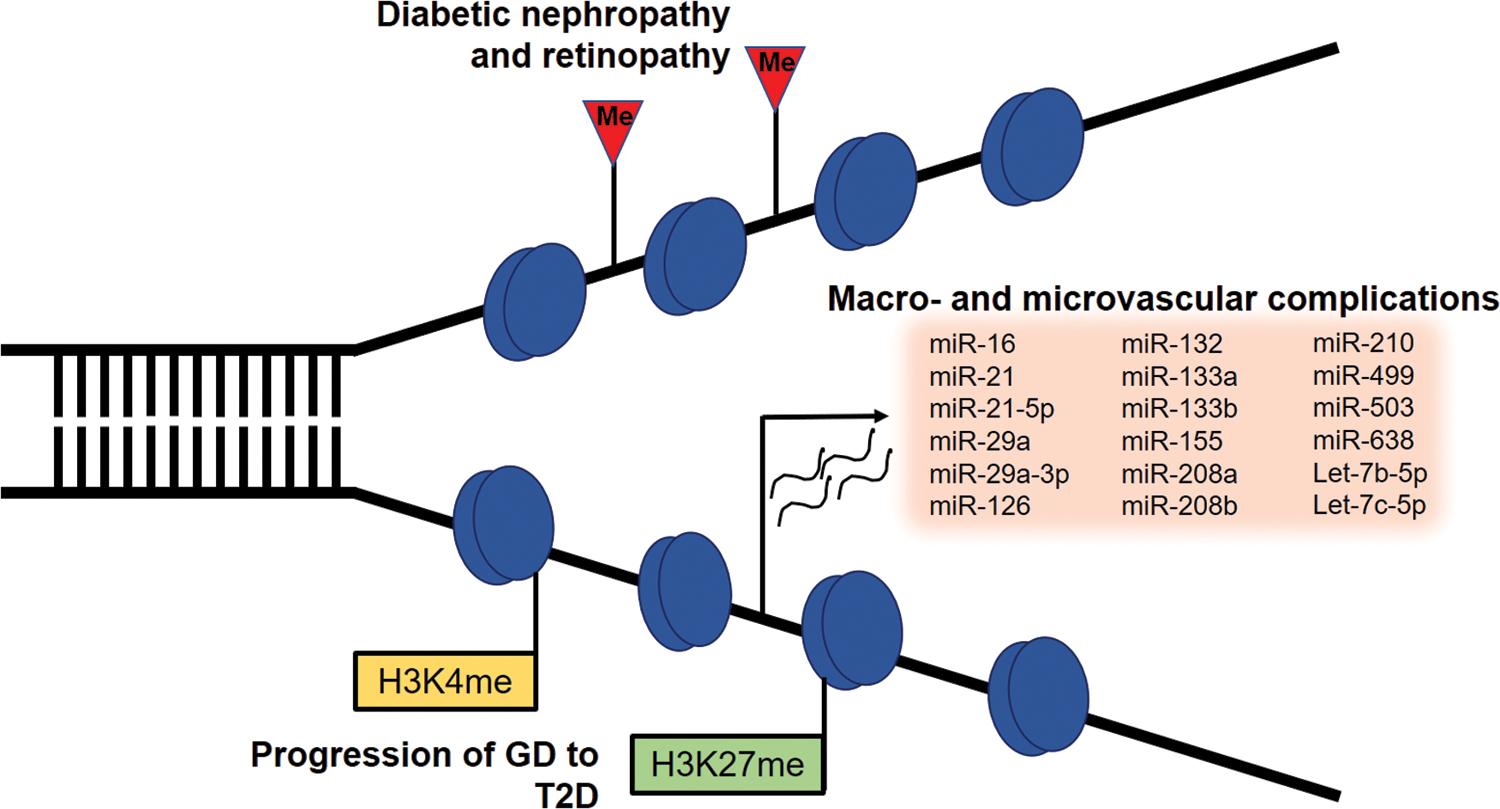
Serum miRNAs have also been proposed as candidate biomarkers for the development of GD. A signature characterized by aberrant expression of miR-222, miR-132, and miR-29a in serum of women in the second trimester of pregnancy precedes abnormalities in glucose tolerance and has predictive value for the development of GD (192). GD is associated with increased risk of T2D (13), and the availability of strong predictors for this risk would allow early intervention in women at risk. Global levels of dimethylated H3K27 and H3K4 were consistently reduced in white blood cells of women with GD who developed T2D, suggesting that these modifications could also provide a predictive tool for T2D (107). A summary of epigenetic events detected in circulating leukocytes and associated with complications of diabetes is shown in Figure 6.
Future Perspectives
Intensive glycemic control has failed to improve outcomes related to diabetic cardiomyopathy and heart failure in long-term randomized clinical trials. Persistent myocardial damage despite glycemic normalization was recently shown to be associated with persistent expression of diabetes-induced miRNAs that may be implicated in cellular damage in the heart (29). These findings highlight the need to further understand the mechanisms underlying hyperglycemic memory.
Recent trials show that treatment with the glucagon-like peptide 1 receptor (GLP-1R) agonist liraglutide in T2D patients with high risk for cardiovascular disease resulted in a reduction in the risk of death from cardiovascular disease as well as progression of kidney disease (96, 99). Because these patients already had a high risk of cardiovascular disease, these outcomes raise the possibility that liraglutide treatment can reverse hyperglycemic memory. Indeed, targeting the incretin pathway with GLP-1R agonists or dipeptidyl peptidase (DDP) 4 (an inhibitor of GLP-1 action) inhibitors attenuates AGE and ROS downstream pathways (including NFκB activation) that contribute to cellular damage in diabetes and are with the maintenance of hyperglycemic memory (16). Given the potential for GLP-1R agonists and DPP-4 inhibitors in preventing the progression of diabetic complications in at-risk groups by reversing changes associated with hyperglycemic memory, this hypothesis deserves further investigation.
Several therapeutic options that target the epigenome have been developed. miRNA-based approaches (miRNA mimics and antagonists) constitute a promising strategy to prevent or correct expression signatures associated with hyperglycemic memory and diabetic complications. A recent review by Prattichizzo et al. (132) explores the therapeutic potential for exosome and microvesicle delivery of miRNAs in this context.
We have discussed the accumulating research showing a strong association between epigenetic marks detected in blood cells and the risk of development and progression of diabetic complications. This information may provide a way to tailor therapy to patients based on their individual risk. In addition, gene polymorphisms associated with differential responses to antidiabetic drugs are also associated with specific epigenetic regulation. For example, cytochrome p450 (CYP450) genes are subject to regulation by DNA methylation and miRNAs, potentially accounting for the variability in drug response between individuals with the same CYP450 genotype (134). This knowledge may be applied to choose the therapeutic approach most likely to be effective.
Since the sequencing of the first human genome, high throughput sequencing (HTS) technologies have been developed and the time and cost of sequencing have decreased dramatically (137). Lower turnaround times and costs have led to the widespread use of HTS and the development of diverse applications that have helped understand the molecular mechanisms underlying common diseases such as cardiovascular and renal diseases (27, 108). These tools promise to be a valuable resource for further understanding epigenetic-mediated gene regulation in health and disease.
Concluding Remarks
Progress in the field of epigenetics has increased our understanding of gene regulation in health and disease. It is now apparent that knowledge of epigenetic modifications that drive pathological changes during the development and progression of diabetic complications may hold the key to developing novel and more effective treatment options. Epigenetic regulatory events may explain the disease risk associated with genomic variants, contributing to our ability to more accurately predict the onset or progression of diabetes. Furthermore, therapeutic options based on epigenetic regulation (i.e., miRNA delivery) may provide a way to restore physiological gene expression patterns and prevent disease progression.