
Abstract
Significance:
Cancer cells accumulate high levels of iron and reactive oxygen species (ROS) to promote their high metabolic activity and proliferation rate. However, high levels of iron and ROS can also lead to enhanced oxidative stress and the activation of cell death pathways such as apoptosis and ferroptosis. This has led to the proposal that different drugs that target iron and/or ROS metabolism could be used as anticancer drugs. However, due to the complex role iron and ROS play in cells, the majority of these drugs yielded mixed results, highlighting a critical need to identify new players in the regulation of iron and ROS homeostasis in cancer cells.
Recent Advances:
NEET proteins belong to a newly discovered class of iron–sulfur proteins (2Fe-2S) required for the regulation of iron and ROS homeostasis in cells. Recent studies revealed that the NEET proteins NAF-1 (CISD2) and mitoNEET (CISD1) play a critical role in promoting the proliferation of cancer cells, supporting tumor growth and metastasis. Moreover, the function of NEET proteins in cancer cells was found to be dependent of the degree of lability of their 2Fe-2S clusters.
Critical Issues:
NEET proteins could represent a key regulatory link between the maintenance of high iron and ROS in cancer cells, the activation of cell death and survival pathways, and cellular proliferation.
Future Directions:
Because the function of NEET proteins depends on the lability of their clusters, drugs that target the 2Fe2S clusters of NEET proteins could be used as promising anticancer drugs.
Introduction
NEET proteins belong to a newly discovered class of iron–sulfur proteins (2Fe-2S). They are highly expressed in many cancers, supporting cellular proliferation and tumor growth. Recently, a key role was identified for the clusters of NEET proteins in regulating the levels of iron and reactive oxygen species (ROS) in cancer cells. In addition, NEET proteins were found to suppress the activation of autophagy and apoptosis in cancer cells. The central role NEET proteins play in cancer cells has led to the hypothesis that targeting these proteins, or their clusters, could be a viable strategy in the fight against cancer.
Iron and Cancer: the “Iron Addiction” Phenotype
A key phenotype of cancer cells discovered in the past decade is “iron addiction” (8, 23, 47, 49, 68, 69). Iron addiction describes the overaccumulation of iron by cancer cells and has been found, among others, in breast, lung, renal, liver, gastric, leukemia, lymphoma, myeloma, pancreas, skin, and prostate cancers (68, 69). Although the exact cause of “iron addiction” is largely unknown, it is proposed that cancer cells require excess iron to support their high metabolic rate, due to iron's key role as a cofactor for many different proteins involved in processes such as DNA and protein synthesis, glycolysis, and respiration (8, 47, 49, 68, 69). Consequently, treatments that target cancer cells' addiction to iron have been proposed and some are in clinical trials (7, 8, 36, 45, 51, 68, 69).
Alterations in a number of different cellular pathways involved in the regulation of iron homeostasis in cells were found to be associated with different cancers (8, 47, 49, 50, 68, 69). These include (I) the enhanced expression of the iron-uptake protein transferrin receptor-1 (TFR1) on the plasma membrane (PM) of cancer cells; (II) the suppressed accumulation of the iron-secretion protein ferroprotein (FPN), mediated in part by the local autocrine synthesis of hepcidin by some cancer cells; (III) the enhanced secretion of iron from endosomes through the divalent metal transporter DMT1 (mediated due to increased expression of the six transmembrane epithelial antigens of the prostate protein that reduces the ferric iron delivered by TFR1 to ferrous iron); (IV) the suppressed, or enhanced, expression of the iron storage protein ferritin (FT); and (V) the enhanced expression of the iron regulatory protein (IRP) 2 that is thought to control some of the mentioned responses (2, 68, 69, 73). The overall outcome of these alterations is an increase in the cytosolic labile iron pool (cLIP) in cancer cells. This iron is then used as a cofactor for the synthesis of different proteins, as well as transported into mitochondria via mitoferrin, endosomes, or other yet unknown mechanisms, and used for the synthesis of heme and/or iron–sulfur (Fe-S) moieties and proteins [Fig. 1 (47, 49, 57, 69)]. In addition to these established mechanisms, epidermal growth factor receptor, a known oncogenic factor, was recently found to bind to and regulate the subcellular distribution of TFR1 through its tyrosine kinase activity (71). Furthermore, the tumor microenvironment was also shown to support the “iron addiction” of cancer cells (50). Thus, tumor-associated macrophages (TAMs) and tumor-associated lymphocytes (TALs) have recently been shown to display an “iron donor” phenotype, with increased expression of FPN and FT, supporting iron secretion (50). The different alterations in cellular pathways that regulate iron homeostasis in cancer cells (Fig. 1) as well as the altered iron metabolism of TAMs and TALs in the tumor microenvironment (50) result, therefore, in high levels of labile iron in cancer cells.

Despite its presumed beneficial effects on promoting cellular proliferation, the overaccumulation of iron by cancer cells does not come without a drawback. Thus, labile iron can react with hydrogen peroxide and/or superoxide radicals to form the highly reactive hydroxyl radical [Fenton reaction (8, 9, 31, 60)] that can harm cells. The high levels of free iron can, therefore, promote cell death via ROS-mediated activation of different cell death pathways, such as apoptosis (8, 9, 60). Furthermore, high levels of iron and ROS can promote another type of cell death pathways termed ferroptosis (10, 19, 20, 24, 35, 46, 76, 78). Thus, in the absence of mechanisms for lipid peroxide scavenging (e.g., glutathione and glutathione peroxidase 4) and iron chelation (e.g., by heat shock protein β-1; HSPB1, and FT), ferroptosis can be induced and results in the death of cancer cells that are particularly sensitive to this type of cell death due to their high LIP (19, 20, 35, 76, 78). Indeed, a number of different therapy avenues have been proposed for the targeting of cancer cells based on the activation of ferroptosis (36, 47). These join other chelation (to lower iron and inhibit cellular proliferation) or iron overloading (to induce ROS and cell death of cancer cells) therapies previously proposed to target the “iron addiction” of cancer cells (7, 8, 45, 51, 68, 69). All of these approaches attempt, therefore, to take advantage of the fine, and apparently dangerous, balancing act cancer cells use to maintain their high metabolic rate via in part to the overaccumulation of iron. Because iron, ROS, and cellular proliferation are highly interlinked in cells (2, 7, 60), the role of ROS in cancer cells needs also to be further addressed.
ROS and Cancer: Playing a Dangerous Game with the Double-Edge Sword of Life
Like stem cells, and in addition to their “iron addiction,” cancer cells appear to also “suffer” from a form of “ROS addiction.” Thus, compared with normal cells, cancer cells accumulate ROS to a higher level and maintain a higher baseline of ROS [referred to as “the tumorigenic ROS level” (8, 18, 29, 52, 58, 60]. This state manifests itself in a shift in the cytosolic thiol redox balance to a more oxidized state that affects the activation of a number of signaling pathways and promotes cellular proliferation (55, 58). For example, overaccumulation of ROS can inactivate different phosphatases that alter the phosphorylation status of protein targets of MAPK-ERK and AKT-kinase and promote cellular proliferation (70). Low levels of ROS can also stabilize hypoxia-inducible factor 1-alpha (HIF1α) and promote cellular survival during hypoxia in a process known as pseudohypoxic HIF activation (58). Indeed, cancer cells were shown to generate ROS even under anaerobic conditions via electron “leakage” from the Qo site of complex III in the mitochondrial electron transfer chain (58). A number of other physiological adaptations of cancer cells were also proposed to result in the excess formation of ROS (reviewed in Refs. 8, 18, 29, 58, 60).
In addition to their role in promoting the survival and proliferation of cancer cells, ROS are also thought to be important for the tumor phenotype because they induce DNA damage, resulting in different types of mutations and overall genome instability (18, 58). These mutations are thought to be required for the “evolution” of cancer cells and their clonal formation (18, 58, 60). ROS-generated DNA mutations can be induced in the mitochondria (mtDNA), where some of the ROS is formed, or in genomic DNA, because ROS such as hydrogen peroxide can diffuse into the nuclei. The role of the abundant iron in cancer cells could of course be key for the generation of some of these mutations, because iron catalyzes the formation of hydroxyl radicals required for the direct oxidation of DNA (31). In addition to direct DNA oxidation by hydroxyl radicals, ROS-induced mutations may also be formed by the inhibition of DNA synthesis or repair enzymes by hydrogen peroxide (31, 58). It, therefore, appears as if cancer cells are using ROS to promote cellular proliferation on the one hand, and inducing DNA mutations on the other. This use is fundamentally different from the use of ROS by normal cells, in which ROS are used for proliferation and defense, but are maintained at a low level to prevent cellular damage (31). A number of different therapies have been proposed to take advantage of the high ROS levels of cancer cells (8, 9, 29). These include antioxidants to lower ROS and prevent proliferation, or pro-oxidants to “burn” cancer cells by excess ROS. However, due to the complex nature of ROS metabolism and its integration into almost all aspects of cellular life, the majority of these strategies have failed (58).
Because cancer cells contain high levels of iron and ROS, they are required to maintain a tight balance between these two dangerous compounds to survive and thrive. Identifying new proteins and pathways involved in maintaining this balance in cancer cells could shed new light on cancer cell's iron/ROS metabolism, as well as highlight new therapy avenues. The newly discovered family of NEET proteins could be one such group of proteins.
NEET Proteins: A New Class of Fe-S Proteins with Unique Features
NEET proteins belong to a novel family of iron–sulfur proteins (2Fe-2S) defined by a unique CDGSH amino acid sequence in their Fe-S cluster-binding domain (15, 53, 54, 64, 75). They are unique because their 2Fe-2S cluster is both labile and redox active, owing to its coordination structure of 3Cys:1His, in which the His is solvent accessible and can be easily protonated (63, 64, 79). In humans, three different genes encode for NEET proteins (Table 1): (i) CISD1 encodes for mitoNEET (mNT), localized to the outer mitochondrial membrane (OMM); (ii) CISD2 encodes for NAF-1, localized to the endoplasmic reticulum (ER), OMM, and the membranes that connect the ER and the mitochondria (mitochondria-associated membranes [MAMs]); and (iii) CISD3 that encodes for Miner 2, localized to the mitochondria. MitoNEET and NAF-1 are characterized by a unique homodimeric structure, in which two protomers, each containing one 2Fe-2S cluster, intertwine to form the unique “NEET fold.” In contrast, Miner 2 has a monomeric structure that contains two 2Fe-2S clusters (64). MitoNEET was the first member of this family to be discovered as a target for the antidiabetic drug pioglitazone (14). After the crystallization and characterization of mitoNEET, NAF-1, and other NEET proteins, it was found that they are highly conserved and can be found in many different unicellular and multicellular organisms (34, 64).
Because mitoNEET and NAF-1 were found to donate their 2Fe-2S clusters to apo-acceptor proteins, a putative role for NEET proteins was proposed in mediating the transfer of Fe/Fe-S clusters into or out of the mitochondria (22, 39, 42, 64). This putative role was then demonstrated using loss-of-function analysis in plant and animal cells (53, 61), as well as a gain-of-function approach in mice (17, 39), highlighting a conserved role for NEET proteins in cellular iron and Fe-S management (34, 64). Endeavors to identify possible NEET interactors and potential endogenous apo-acceptors have identified several different cellular partners of these proteins. Thus, mitoNEET was found to interact with cytosolic aconitase/IRP1 (22) and anamorsin (42), two Fe-S proteins involved in iron–sulfur biogenesis and iron regulation; with glutathione reductase (GR) (40), a redox regulator; with glutamate dehydrogenase 1 (GLDH) (56), a key metabolic enzyme and an insulin regulator; and with the RING-between-RING E3 ligase Parkin protein (PARKIN) (38). NAF-1 was found to interact with BCL-2, a key protein involved in the regulation of apoptosis and autophagy (11, 65), as well as with the calpain-2 catalytic subunit (CAPN2), a Ca2+-activated protease involved in apoptosis activation (44), and anamorsin (42). In addition, mNT was recently found to interact with NAF-1 and donate its cluster to it, suggesting that mNT and NAF-1 could function as part of an 2Fe-2S cluster relay that mobilizes 2Fe-2S clusters from the mitochondria to the cytosol [Fig. 2 (37)].
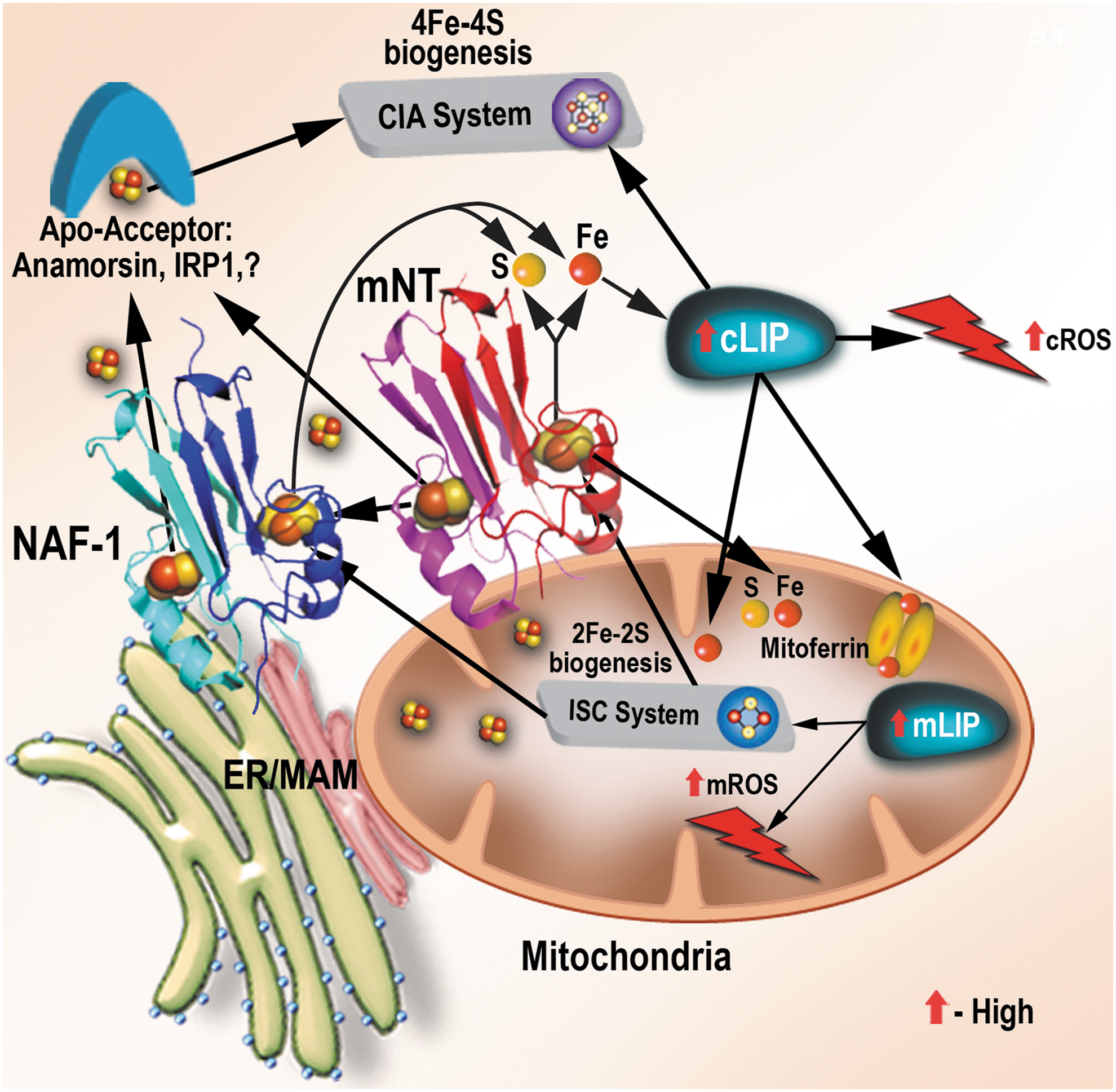
The interactions of the OMM-localized mitoNEET protein with key cytosolic IRPs such as aconitase/IRP1 and anamorsin could suggest that mitoNEET may link between the iron and/or Fe-S cluster status of the mitochondria and the regulation of iron homeostasis in the cytosol (Fig. 2). The interactions of mitoNEET with the redox and metabolic enzymes GR and GLDH may suggest that mitoNEET functions as a link between the metabolic and/or redox state of the cell and iron homeostasis (64). MitoNEET may, therefore, play a key role linking Fe-S/redox/metabolic changes with iron metabolism regulated through aconitase/IRP1 (Fig. 2). The interaction of mitoNEET with PARKIN, a protein involved in the activation of mitophagic pathways (39), could further suggest that mitoNEET is important in regulating mitochondrial stability. In support of the proposed role of mitoNEET in regulating iron homeostasis and mitochondrial stability, shRNA suppression of mitoNEET in different systems resulted in the overaccumulation of iron and ROS in mitochondria and the activation of autophagy (16, 61), and overexpression of mitoNEET protected mitochondria from overaccumulation of iron and ROS (39), as well as cells from ferroptosis (80), and promoted cellular proliferation (59).
In contrast to mitoNEET, NAF-1 that is localized to the ER, mitochondria, and MAMs interacts with BCL-2 and CAPN2 and could be involved in linking between the state of Fe-S/Fe in the mitochondria and the activation of apoptosis and/or autophagy. Because both mitoNEET and NAF-1 can donate their clusters or transfer electrons (via oxidation/reduction) to their interacting partners (22, 26, 28, 40 –42, 53, 63, 64, 67, 74, 81), and because such changes in mitoNEET or NAF-1 could induce conformational alterations (4, 5), it was proposed that the binding of NEET proteins to some of their partners could be regulated by the presence or absence of the cluster, and/or its redox state (11, 64). Thus, for example, holo-NAF-1 was proposed to regulate autophagy via binding to BCL-2 at the ER and stabilizing the binding of BCL-2 to BECLIN 1, therefore, inhibiting autophagy activation (11). In contrast, apo-NAF-1, which lost its cluster, due, for example, to nutrient stress, was proposed to lose its ability to bind to BCL-2 and stabilize its interaction with BECLIN 1, therefore, initiating autophagy (11). Such a link between cluster availability, the holo/apo state of NEET proteins, and the activation of cell death/autophagy pathways could be at the heart of NEET protein's mode-of-action in cells (64). In support of the role of NAF-1 in the activation of autophagy and apoptosis, suppression of NAF-1 expression was shown to activate autophagy and apoptosis, presumably because the lower levels of NAF-1 protein could not interact with BCL-2 and prevent the activation of these pathways (33, 61). It should be noted that the binding of NAF-1 to BCL-2 was shown to be mediated at the same sites as BH3-containg proteins (65), and that in addition to stabilizing BCL-2 interactions with BECLIN 1, NAF-1 could compete for BCL-2 binding with BH3-containing pro-apoptotic proteins. In addition, NAF-1 could activate apoptosis via its interactions with calpain-2 [CAPN2; (44)]. The proposed link NEET proteins establish between Fe/Fe-S availability and the regulation of cell death via apoptosis/ferroptosis and/or autophagy highlights a putative key role NEET proteins could play in many human pathologies. Considering that autophagy can also act as a survival pathway, NAF-1 could be controlling survival or death pathways in cells (11, 33, 61).
NEET Proteins and Human Pathologies
Several studies have implicated NEET proteins in the progression of different human pathologies and aging. These include diabetes, obesity, neurodegeneration, myocardial injury, cystic fibrosis (CF), and aging (1, 13, 16, 21, 30, 32, 38, 39, 62, 64, 74). The involvement of NAF-1 in key metabolic and regulatory processes was revealed in null mice (12, 74), as well as in the human genetic disease type 2 Wolfram Syndrome (T2-WFS) that results from a recessive mutation in CISD2, the gene encoding NAF-1 (1, 16). T2-WFS2 manifests in pancreatic β-cell degeneration resulting from cellular stress and apoptosis that leads to severe insulin deficiency mimicking type 1 diabetes (16). In addition, it is accompanied by neurodegeneration leading to sight and hearing loss, and a deficiency in blood clotting leading to severe bleedings and ulcers (1, 74). Null CISD2 mice show similar phenotypes that include early aging, blindness, skeletal muscle distortions, excessive bleeding, and neural degeneration (13, 74). In agreement with the effects described previously on mitochondria and autophagy activation in cells with suppressed NAF-1 expression (61), the mitochondria of null NAF-1 mice exhibited increased oxidative damage, abnormal shape, and dysfunction that were accompanied by enhanced autophagy (74). In contrast to the shortened lifespan of null NAF-1 mice or T2-WFS patients (1, 16), mice that overexpressed NAF-1 had delayed aging (64). In addition to these, several studies conducted on NAF-1-deficient cells revealed dysregulated calcium signaling and ER stress (16, 44, 74). Interestingly, some of the abnormalities attributed to NAF-1 deficiency could be corrected by iron chelators such as deferiprone (16, 61) and antioxidants such as NAC (74). Some of the additional commonalities between studies on NAF-1 deficiency conducted in cells and null CISD2 mice include the activation of apoptosis, although it is unknown whether this activation results from the lack of NAF-1 interaction with BCL-2, CAPN2, or other cellular proteins (33). Overall, the studies already described point to a key role for NAF-1 in regulating autophagy, apoptosis, ER calcium signaling, and mitochondrial iron and ROS homeostasis, which could be involved, among others, in diabetes, neurodegeneration, and aging.
MitoNEET that was originally identified in cross-linking experiments as a target of thiazolidinediones (TZDs), a class of type 2 diabetes drugs, has been implicated in obesity, diabetes, CF, and neurodegenerative diseases (64). Overexpression of mitoNEET in adipocytes of transgenic mice enhanced lipid uptake and storage resulting in an expansion of adipose tissue mass without a loss of insulin sensitivity (38). This was accompanied by a reduction in ectopic lipid accumulation, and an increase in adipocyte-produced adiponectin. Overexpression of mitoNEET also resulted in reduced mitochondrial iron levels, a decrease in electron transport chain activity, and a lower rate of β-oxidation, resulting in a marked decrease in mitochondrial ROS production (39). The level of mNT expression was further found to markedly affect the dynamics of cellular and whole body lipid (white/brown fat) homeostasis, and the protection of β-cells from oxidative state (38). In addition to the studies already described, mitoNEET was also implicated in neurodegeneration based on its interaction with PARKIN, a causal gene associated with autosomal recessive early onset parkinsonism, as well as its involvement in protecting neural cells during spinal cord injury (32). A protective role against hypoxia and reoxygenation-derived oxidative stress was also proposed for mitoNEET in cardiomyocytes (30). In addition, mitoNEET was proposed to be linked to CF, based on its expression association with the key CF chloride channel, CF transmembrane conductance regulator (64). The interaction of mitoNEET with PARKIN was recently shown to also affect pancreatic α- and β-cell survival and cross talk via a PARKIN-dependent mitophagic pathway (38). Because mitochondrial dysfunction is at the heart of many of the pathologies already described, it could be speculated that one of mitoNEET's roles in cells is to protect the mitochondria from stress, may be even in a similar manner to how NAF-1 protects cells from mitochondrial and/or ER stress. This function could also be linked to mitoNEET's (and NAF-1's) role in mediating iron and ROS homeostasis in cells (64). Indeed, mitoNEET was recently shown to protect cells from ferroptosis (80).
The iron addiction phenotype of cancer cells (9, 47, 49, 68, 69) and the enhanced levels of ROS cancer cells generate (8, 18, 29, 58, 60) could suggest that NEET proteins are involved in the protection of mitochondria from iron and ROS overaccumulation in tumors. Hereunder we describe recent studies identifying a newly discovered key role for NEET proteins in cancer cells.
NEET Proteins and Cancer: New Players in the Regulation of Iron and ROS
A survey of the C-bio database reveals that a large number of different tumors are accompanied by alterations in NEET protein expression and/or in mutations in NEET proteins (Fig. 3; Table 2). Furthermore, although newly discovered (64), the role of NEET proteins in promoting cellular proliferation, supporting cancer cell survival, and enhancing metastasis has already been described in a number of publications (12, 17, 25, 33, 43, 48, 59, 61, 72, 77, 79). NEET proteins were first implicated in cancer through studies on breast cancer cells. Thus, Salem et al. (59) found that overexpression of mitoNEET in triple-negative MDA-MB-231 cells resulted in increased abundance of mitochondrial oxidative phosphorylation (OXPHOS) proteins, enhanced resistance to autophagy under starvation, and increased xenograft tumor growth by up to approximately threefold (Fig. 4). Suppression of mitoNEET or NAF-1 in MDA-MB-231 and MCF-7 was subsequently reported by Sohn et al. (61) to result in the overaccumulation of iron and ROS in mitochondria, enhanced autophagy under normal conditions, and reduced xenograft tumor growth by up to 90% [Fig. 4 (61)]. Interestingly, mitoNEET or NAF-1 could not compensate for the deficiency in each other, suggesting that they may be functioning in the same pathway to regulate mitochondrial iron and ROS metabolism in cancer cells [Fig. 2 (37, 61)].
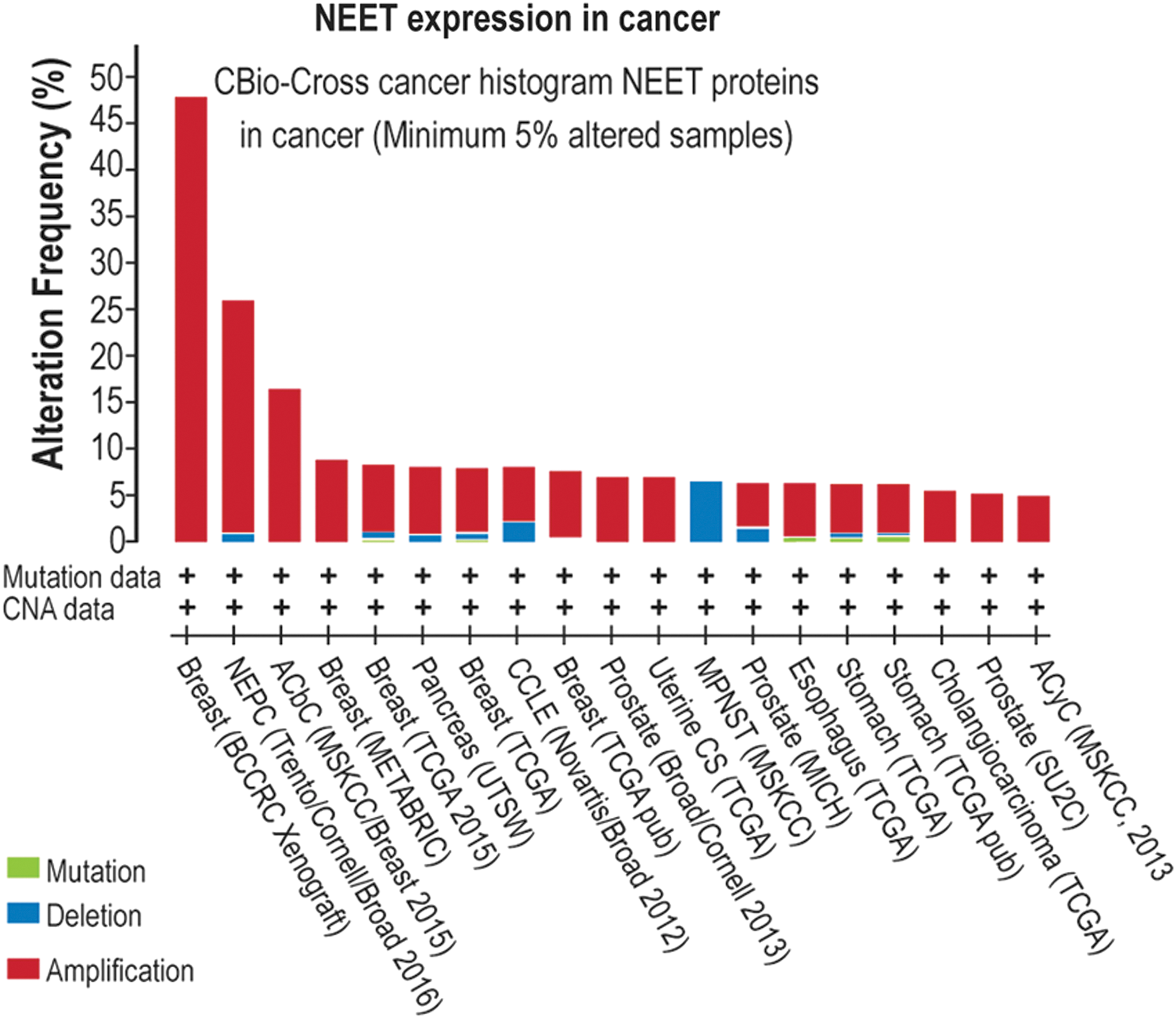

EMT, epithelial–mesenchymal transition; HIF1α, hypoxia-inducible factor 1-alpha; PSA, prostate-specific antigen.
The role of NAF-1 in regulating breast cancer cell metabolism and cell death was further addressed by Holt et al. (33), who discovered that cancer cells and tumors with suppressed expression of NAF-1 display enhanced activation of apoptosis (Fig. 4). Suppression of NAF-1 in breast cancer cells was also shown by Holt et al. to result in an increased uptake of iron into cells and mitochondria, a metabolic shift that enhances oxygenic glycolysis, and the activation of cellular stress pathways associated with HIF1α stabilization and mTOR inactivation (33). NAF-1 was, therefore, proposed to support mitochondrial iron and ROS metabolism, promoting cancer cell survival via stabilizing of HIF1α and suppression of apoptosis. The role of NAF-1 in promoting cancer cell proliferation was further examined by Darash-Yahana et al., (17) who discovered that overexpression of NAF-1 in xenograft breast cancer cells and tumors resulted in a dramatic augmentation of tumor size and aggressiveness [Fig. 4 (17)]. Darash-Yahana et al. have also found that cancer cells with enhanced expression of NAF-1 were more tolerant to oxidative stress and undergo less apoptosis and autophagy (17). Moreover, and for the first time, Darash-Yahana et al. reported that the degree of lability of the NAF-1 2Fe-2S cluster is highly important for NAF-1 function in cancer cells [Figs. 4 and 5 (17)]. Thus, in contrast to the overexpression of a wild-type NAF-1 protein, overexpression (to the same level) of a mutated form of NAF-1 with a single point mutation (H114C), which stabilizes the NAF-1 cluster >25-fold (63), resulted in a dramatic decrease in tumor size that was accompanied by enhanced mitochondrial iron and ROS accumulation and reduced cellular tolerance to oxidative stress. Furthermore, Darash-Yahana et al. demonstrated that treating breast cancer cells with pioglitazone [a TZD drug that binds and stabilizes the Fe-S cluster of NEET proteins; (54, 66)] resulted in a similar effect on mitochondrial iron and ROS accumulation [Fig. 5 (17, 66)]. This finding demonstrated for the first time that the cluster of NAF-1 is critical for its function in cancer cells and that drugs that target the lability/stability of NAF-1 and mitoNEET clusters could be used as a viable treatment strategy, at least for patients with tumors that display high expression level of NAF-1 (17). It should also be noted that drugs such as the mitocan cluvanone and its derivatives also alter the stability of NAF-1 and mitoNEET clusters, via either stabilization or destabilization, and affect the growth of breast cancer cells in culture (3).
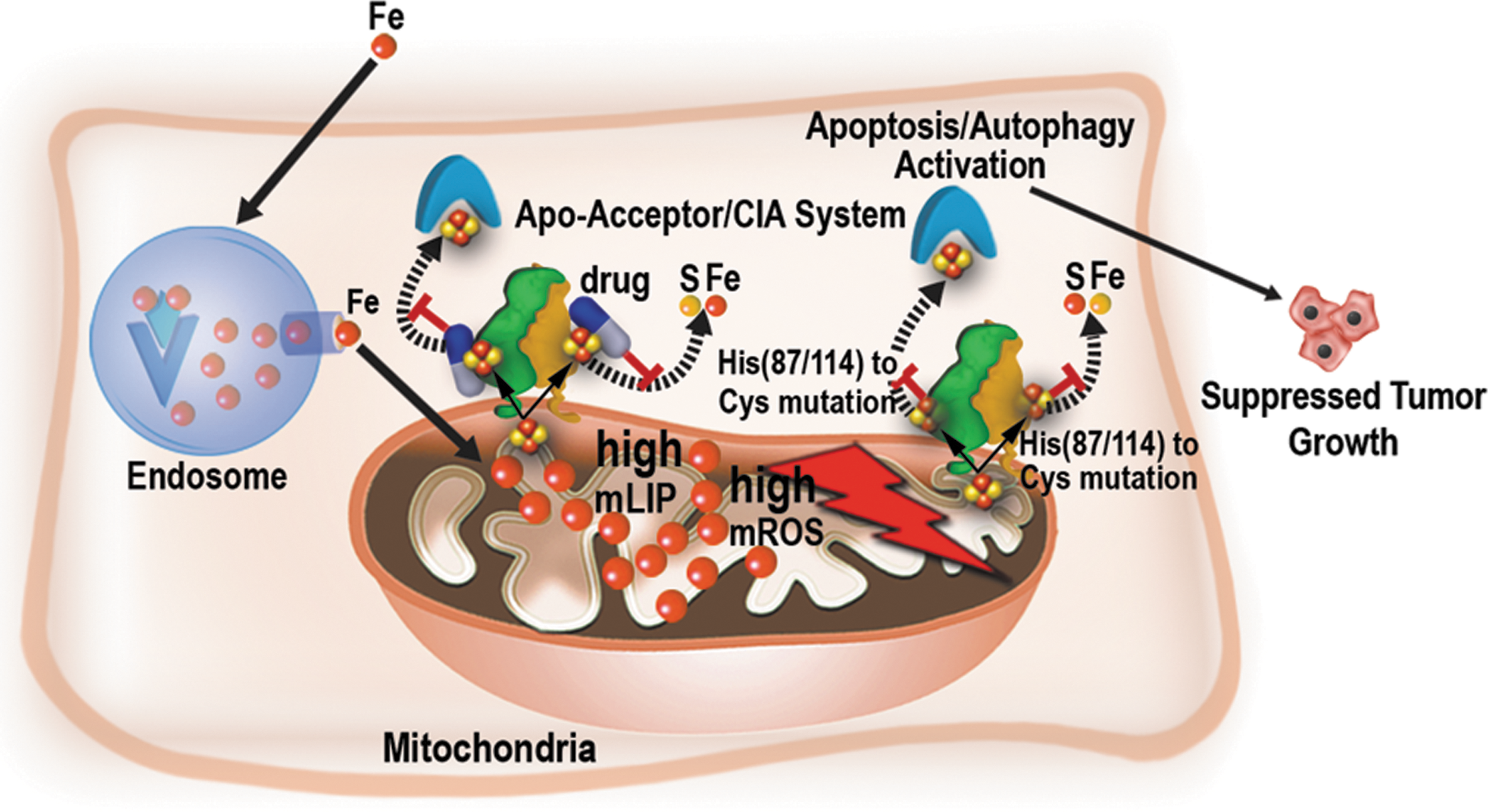
The studies described above support the “iron addiction” phenotype of breast cancer cells and suggest that NEET proteins could promote breast cancer survival and proliferation by protecting the mitochondria of cancer cells from iron overaccumulation, enhancing the tolerance of cancer cells to oxidative stress, and suppressing autophagy and apoptosis (Figs. 4 –6). These effects could be mediated by the stabilization of HIF1α, by controlling iron or Fe-S clusters movement into and out of the mitochondria, or by enhanced interaction with BCL-2 (33, 61, 65): all mediated by the high levels of NEET protein overexpression that accompanies the “iron addiction” phenotype in breast cancer cells (Figs. 2 and 4 –6). In addition to the studies already described on breast cancer cells and tumors, NEET proteins, and, in particular, NAF-1, were shown to play a key role in other cancers and to interact with other cellular pathways as described hereunder.
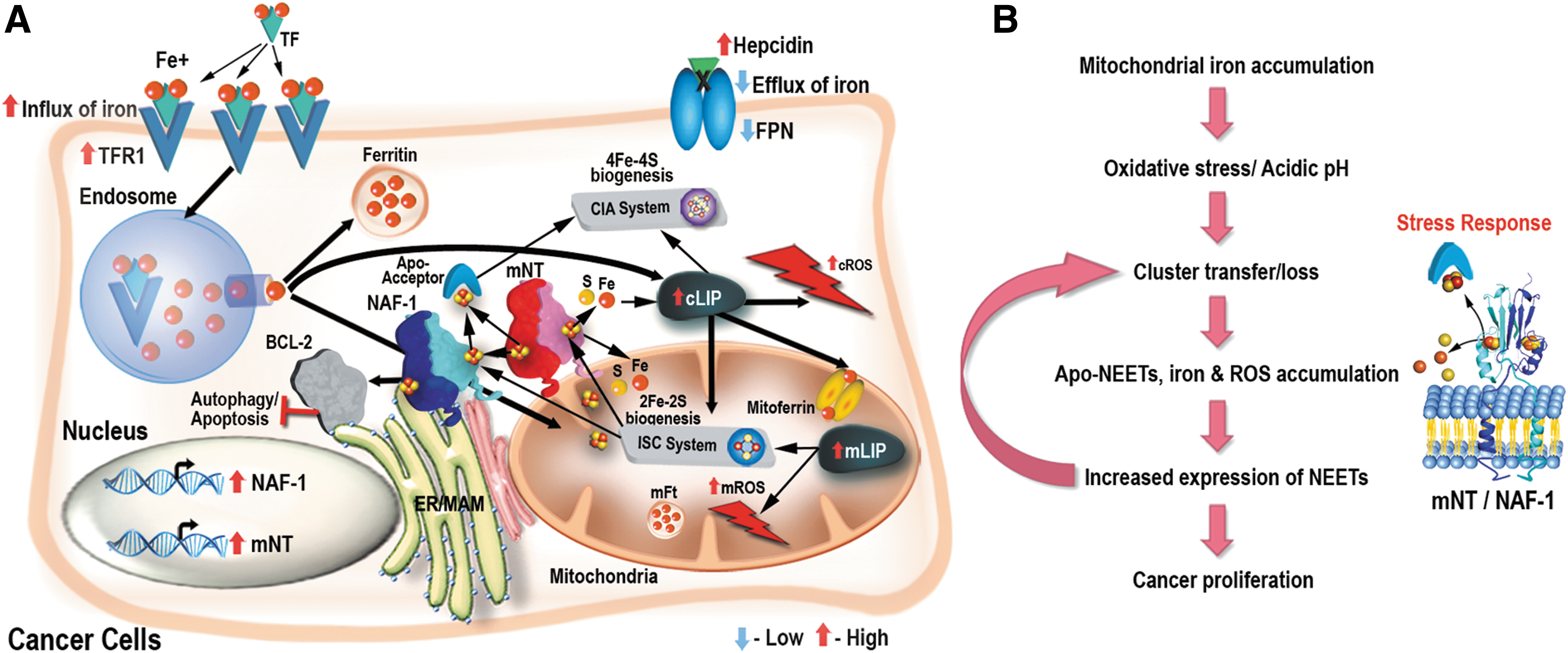
Enhanced expression of NAF-1 (CISD2) was identified in different liver cancer cell lines and tumors (hepatocellular carcinoma [HCC]) by Chen et al. (12). Much like the results with breast cancer (61), suppression of CISD2 expression by shRNA in liver cancer cells and tumors resulted in suppressed cellular proliferation and tumor growth. Moreover, a clinicopathological analysis of CISD2 expression and liver cancer progression, performed with 196 HCC patients, revealed that CISD2 expression could be used as an independent prognostic marker for survival (12). Thus, patients with high NAF-1 expression displayed a shorter overall survival and a higher recurrence rate than those with low CISD2 expression (12). Overexpression of CISD2 was also shown to have an important prognostic value in human gastric cancer (72). In this study, immunohistochemical analysis of 261 gastric tissues and tumors from patients revealed that high CISD2 expression was significantly associated with cancer clinical stage, classification, and venous and lymphatic invasion (72). Overexpression and suppression studies of NAF-1 in gastric cancer cell lines further demonstrated that the level of NAF-1 positively correlates with cell proliferation. In addition, suppression of NAF-1 expression was also shown to result in suppressed tumor growth in xenograft mice (72). Further analysis of signaling pathways activated in response to alterations in NAF-1 expression suggested that the AKT kinase/FOXO signaling pathway is regulated by CISD2. Thus, the phosphorylation of AKT, FOXO4, FOXO3a, and FOXO1 increased in CISD2-overexpressed cells but decreased in CISD2-silenced cells (72). The authors conclude that CISD2 could affect cell proliferation and tumorigenicity of gastric cancer cells by altering the G1-to-S phase transition, and that the pro-proliferation activity of CISD2 could be associated with activation of AKT kinase signaling and the downregulation of cyclin-dependent kinase inhibitors p21CIP1 and p27KIP1 (72). In accordance with the proposed role of NAF-1 in promoting gastric cancer, Mao et al. (48) reported that treatment of gastric cancer cells with ginsenoside F2 (a protopanaxdiol type of saponin that was reported to inhibit human gastric cancer cells) suppressed the expression of NAF-1 and other proteins involved in the NAF-1/BCL-XL/BECLIN-1 pathway.
In a recent study, CISD2 was yet again found to be an independent prognostic factor in pancreatic cancer. Thus, Yang et al. (79) found that NAF-1 overexpression was correlated with advanced clinical- and T-stage vascular invasion, distant metastasis, and tumor size. As with breast, gastric, and liver cancers (12, 61, 72), downregulation of NAF-1 inhibited the survival and growth of pancreatic cancer cells and suppressed the growth of tumors in mice. Of particular interest was the finding that NAF-1 silencing significantly inhibited the epithelial–mesenchymal transition (EMT) via the Wnt/β-catenin pathway (79). This finding revealed for the first time a direct role for NEET proteins in the metastasis process of cancer cells. The expression of NAF-1 was also found to be upregulated in laryngeal squamous cell carcinoma and to significantly correlate with T-stage, lymph node metastasis, clinical stage, and disease progression by Yang et al. (77). The clinicopathological significance of NAF-1 overexpression was additionally demonstrated in patients with early stage cervical cancer by Liu et al. (43). In an attempt to identify single nucleotide polymorphisms (SNPs) that associate with deregulation of prostate-specific antigen, Ge et al. (25) identified SNPs in DNA regulatory regions of mitoNEET (CISD1). These SNPs were correlated with altered mRNA expression of CISD1 and could indicate the involvement of mitoNEET in prostate cancer. Further studies are, however, required to determine this association analysis.
The studies described above point to a key role for NEET proteins in promoting cancer cell proliferation, tumor growth, and metastasis. In addition, they reveal that CISD2 expression levels could be used as an independent prognostic tool in several different cancers. NEET proteins and the iron, Fe-S, and ROS pathways they modulate could, therefore, be used as promising targets for drug development and therapy (26, 27).
Concluding Remarks and Future Perspectives
The iron and ROS addiction phenotype of cancer cells (8, 9, 18, 29, 47, 49, 58, 60, 68, 69), coupled with the mitochondrial iron and ROS regulatory role of NEET proteins (17, 22, 39, 42, 53, 61, 64), and the high expression level of NEET proteins in different cancers (12, 17, 25, 33, 43, 48, 59, 61, 72, 77, 79), suggest that NEET proteins could play a key role in supporting cancer cell proliferation. Indeed, as already described, a number of studies have now reported a direct link between NEET protein overexpression and cancer cell proliferation, tumor growth, and metastasis [(12, 17, 72) Fig. 3). Although a role for NEET proteins in promoting cancer is evident from these studies, an established mechanism for NEET protein function in cancer proliferation is still absent. The different mechanisms proposed for the role of NEET protein in cancer cells thus far include the enhanced regulation of iron/Fe-S homeostasis (17, 33, 61), the protection of mitochondrial OXPHOS proteins from oxidative stress (59), the protection of cancer cells from apoptosis and enhanced autophagy (33, 61), the stabilization of HIF1α (33), the activation of the AKT kinase signaling pathway (72), and the promotion of EMT via the Wnt/β-catenin pathway (79). In principle, these mechanisms could be explained by the metabolic effects of NEET proteins on cells, for example, via the regulation of iron/Fe-S that affects ROS and activity of different metabolic enzymes (i.e., a metabolic link to cancer; Figs. 4 –6). Alternatively, NEET proteins could exert their pro-proliferative function in cancer cells via direct protein–protein interactions with different regulatory proteins, for example, interactions with BCL-2 that control apoptosis and autophagy (i.e., a regulatory link to cancer; Fig. 6). Another possibility is that NEET proteins actually link metabolic effects in the cell with regulatory functions (i.e., a metabolic-regulatory link to cancer).
Of particular interest is the recent discovery by Darash-Yahana et al. that the degree of lability of the NAF-1 2Fe-2S cluster is highly important for NAF-1 function in cancer cells (17). This finding clearly demonstrates that reactions that involve cluster or electron transfer from NEET proteins to other acceptor proteins in the cell are required for NEET protein function in cancer cells. Based on this finding (6, 17), and the report that apo-NAF-1 cannot bind to BCL-2 (11), a possible metabolic-regulatory link model could be proposed for the function of NEET proteins in cancer cells (Figs. 4 –6). In this model, an elevated expression level of NEET proteins in cancer cells is required for alleviating the iron and ROS stress from the mitochondria and ER (a stress that accompanies the iron and ROS addiction state of cancer cells), and this function of NEET proteins is mediated via iron, Fe-S, and/or electron transfer reactions. Thus, as long as NEET proteins are successful in protecting the mitochondria and ER of cancer cells, the level of ROS remains within the tumorigenic range and proliferation proceeds. The elevated levels of NEET proteins, therefore, allow cancer cells to accumulate high levels of ROS without cellular damage and to promote cellular proliferation. The higher NEET-supported ROS accumulation could also stabilize HIF1α and activate AKT signaling, further supporting survival and proliferation (58, 60). The high level of NEET proteins in cancer cells is also needed to suppress the activation of high iron-induced apoptosis and high ROS-induced apoptosis, ferroptosis, and autophagy via interactions with BCL-2, CAPN2, and other yet unknown proteins, thereby allowing cellular proliferation under oxidative stress conditions. If for some reason, the availability of iron, Fe-S clusters, and/or electrons needed for NEET protein function decreases, the cluster-dependent interaction of NEET proteins with proteins such as BCL-2 could be altered or decreased, promoting the activation of apoptosis/autophagy/ferroptosis. Because the lability of the NEET cluster is thought to increase with the increased oxidation of the cellular environment (summarized in Ref. 64), the state of the mitoNEET and/or NAF-1 clusters and its effect on the interaction of NEET proteins with proteins such as BCL-2 could mediate a link between the metabolic state of the cell and regulatory functions that determine life-or-death decisions (Fig. 6). Thus, NEET proteins could function in cells as important sensors for the iron/Fe-S/redox status of the cell and the activation of apoptosis/autophagy/ferroptosis. In cancer cells, due to the high iron and ROS levels, they are needed at much higher levels to prevent the inhibition of cellular proliferation and the activation of cell death pathways that are supposed to occur due to the iron and ROS addiction phenotypes. Because NEET proteins could undergo oxidation and cluster loss under high oxidative stress conditions or low pH (summarized in Ref. 64), the requirement for high levels of NEET proteins in cancer cells could also drive a vicious cycle in which NEET proteins accumulate, undergo oxidation that results in cluster loss, and degradation leading to increased labile iron/Fe-S levels and more oxidative stress that requires even more NEET proteins (Fig. 6). This possibility could also explain why cells with high NEET protein level have high levels of ROS detoxification proteins (17). In summary, NEET proteins could be required in cancer cells to protect the mitochondria and ER from iron-derived stress and ROS-derived stress and to prevent the activation of apoptosis/autophagy/apoptosis. However, due to their potential high turnover, they could also promote oxidative stress by releasing iron/Fe-S into the cytosol under oxidative and low pH conditions (Fig. 6B). Thus, in addition to protecting cancer cells from the activation of apoptosis/autophagy/ferroptosis, the higher ROS levels generated in the presence of high levels of NEET proteins could additionally drive cellular proliferation and mutations (Fig. 6B).
Perhaps the most important question regarding NEET proteins and cancer is whether or not they could be used for the development of anticancer drugs that exploit the iron–ROS balance of cancer cells (Figs. 1 and 2). In this respect, two different classes of drugs could be used: (i) drugs that stabilize the clusters of NEET proteins and prevent them from mediating iron/Fe-S mobilization in or out of the mitochondria, therefore, creating a cellular iron/ROS overload and inducing cell death of cancer cells [e.g., pioglitazone; Fig. 5; (17, 66)] and (ii) drugs that destabilize the clusters of NEET proteins and cause the Fe-S cluster to fall off into the cytosol, thereby increasing ROS production and cell death [e.g., MAD-28 (3]). Both of these drug classes would be highly effective against cancer cells that display high expression levels of NAF-1 and could be used in conjugation with other anticancer drugs, or drugs that enhance the rate of ROS and/or iron accumulation in cancer cells to prevent primary tumor development (17) or metastasis (79). Although further studies are needed to develop additional and/or more efficient version of the drugs already described, the availability of crystal structures for NAF-1 and mitoNEET (15, 54, 64) and the wealth of data concerning binding of small molecules to these proteins (summarized in Ref. 64) should aid in the development of such therapies/drugs. In addition, more studies are needed to determine the function of NEET proteins in the development and progression of other cancers.
Footnotes
Acknowledgments
This work was supported by funding from the Israel Science Foundation (ISF-865/13) awarded to R.N. and funding from the National Science Foundation (IOS- 1353886, IOS-0639964, IOS-0743954, and IOS-1557787), the University of North Texas, College of Arts and Sciences, and the Lady Davis and Jacob and Lena Joels Memorial Foundation fund awarded to R.M. R.N. and R.M. acknowledge the support of the NSF-BSF funding NSF-MCB-1613462 (R.M.) and BSF (Binational Science Foundation (BSF) Grant No. 2015831 (R.N.); work at the laboratory of P.A.J. is supported by the National Institutes of Health Grant DK4441. Work at the Center for Theoretical Biological Physics was sponsored by the National Science Foundation (Grant no. PHY-1427654), by NSF- CHE 1614101, and by NF-1241332, awarded to J.N.O. F.B. was also supported by Welch Foundation Grant C-1792. The funders had no role in the design, data collection, analysis, decision to publish, or preparation of the article.