
Abstract
Aims:
Normal mitochondrial function and integrity are crucial for cellular physiology. Given the paramount role of mitochondrial quality control proteases in these processes, our study focused on investigating mechanisms by which the activity of a key quality control protease Oma1 is regulated under normal conditions and in response to homeostatic insults.
Results:
Oma1 was found to be a redox-dependent protein that exists in a semi-oxidized state in yeast and mammalian mitochondria. Biochemical and genetic analyses provide evidence that activity and stability of the Oma1 oligomeric complex can be dynamically tuned in a reduction/oxidation-sensitive manner. Mechanistically, these features appear to be mediated by two intermembrane space (IMS)-exposed highly conserved cysteine residues, Cys272 and Cys332. These residues form a disulfide bond, which likely plays a structural role and influences conformational stability and activity of the Oma1 high-mass complex. Finally, in line with these findings, engineered Oma1 substrate is shown to engage with the protease in a redox-sensitive manner.
Innovation:
This study provides new insights into the function of the Oma1 protease, a central controller of mitochondrial membrane homeostasis and dynamics, and reveals the novel conserved mechanism of the redox-dependent regulation of Oma1.
Conclusion:
Disulfide bonds formed by IMS-exposed residues Cys272 and Cys332 play an important evolutionarily conserved role in the regulation of Oma1 function. We propose that the redox status of these cysteines may act as a redox-tunable switch to optimize Oma1 proteolytic function for specific cellular conditions or homeostatic challenges.
Introduction
Normal mitochondrial functioning and adequate responses to homeostatic challenges are essential for cellular physiology and are regulated by multiple processes. One mechanism includes mitochondrial membrane proteases—a group of highly conserved enzymes that regulate a variety of vital mitochondrial functions, including energy conversion, mitochondrial dynamics, and inner mitochondrial membrane (IM) homeostasis [reviewed in Refs. (6, 11, 28, 33)]. The IM protease Oma1 is a intermembrane space (IMS)-facing, ATP-independent metallopeptidase that has recently emerged as one of the central regulators of the processes just described (2, 21, 34). For example, in higher eukaryotes, Oma1 mediates proteolytic conversion of the guanosine triphosphatase, Opa1, on cellular insults, thereby propelling fragmentation of the mitochondrial network (11, 16, 17, 20)—an event crucial for cellular physiology and processes such as mitophagy and apoptosis (21, 36). Likewise—even though its substrate repertoire remains to be determined—stress-promoted action of yeast Oma1 is required to cope with homeostatic challenges such as mitochondrial uncoupling or oxidative stress (7). Oma1 is also known to perform protein quality control functions (7, 22, 24, 32, 37) and mediate the stability of respiratory supercomplexes, thereby influencing mitochondrial energy metabolism (8, 14).
Many cellular processes are critically dependent on normal mitochondrial function and multiple quality control mechanisms are in place to ensure the organelle's fidelity. Here, we demonstrate that a key mitochondrial quality control protease Oma1 is a redox-dependent protein, the reduction-oxidation modifications of which may influence its biochemical properties and function. Our study establishes a mechanistic framework for the identification of molecular determinants of substrate recognition and redox-tuning of Oma1. We propose that the redox status of these cysteines may act as a conserved redox-regulated switch to optimize Oma1 proteolytic function for specific cellular conditions.
The protease is largely dormant under normal physiological conditions, but it becomes rapidly activated by various stress stimuli such as loss of mitochondrial membrane potential, metabolic crisis, heat, and oxidative insults (11, 13, 16, 17, 20). Although its stress-activation mechanism remains obscure, several reports suggested that Oma1 activation is a post-translational process that involves conformational changes within the homo-oligomeric Oma1 complex (7) and have identified several molecular determinants that are important for the protease's activation (13, 17, 35, 43), including the enzyme's conserved C-terminal region (7). By analogy with the other known proteolytic enzymes, such behavior suggests that the protease's activity could be a subject of precise regulatory control via a conserved mechanism. Indeed, several studies in mammalian cell culture proposed several regulatory mechanisms, including an autocatalytic cleavage model (3, 43) and stress-induced processing by the ATP-dependent protease Yme1 (35) or the m-AAA [matrix-oriented ATPase associated with various cellular activities (AAA)] protease (13). However, these processes appear to be more evolutionarily recent and specific to higher eukaryotes (3, 7). Moreover, being an evolutionary descendant of the ATP-poor bacterial periplasmic space (30), the IMS is likely to employ additional strategies to regulate its proteome. As such, a deeper understanding of mechanisms that control Oma1 activity under physiological and stress conditions is needed.
In this study, we identified two evolutionarily conserved cysteine residues that influence Oma1 activation and stability. We show that Cys332 is crucial for stability and stress-activation of the Oma1 complex. We further demonstrate that mutation of the conserved Cys272 stabilizes the C332A mutant form of Oma1, thereby yielding a functional protease complex, which, however, has an altered conformational state. Further analyses in the yeast model indicate that these two residues exist in the oxidized state and suggest that disulfide bonds involving residues Cys272 and Cys332 are likely to play an important evolutionarily conserved role in the regulation of Oma1 function. The human Oma1 appears to follow the same trend. These data support a model wherein the redox status of these cysteines may act as a redox-tunable switch to optimize Oma1 proteolytic function for specific cellular challenges.
Results
Oma1 exists in a semi-oxidized state and is activated by prolonged hypoxia
To further understand molecular determinants of Oma1 stress-activation, we surveyed for additional stress conditions that may trigger Oma1 activation by using a previously developed proxy assay, wherein changes in the stability of the ∼250 kDa Oma1 high-mass complex stability by blue native gel electrophoresis (BN-PAGE) reflect activation of the enzyme (7, 24). In line with previous reports, we observed that the Oma1 oligomer was more labile in mitochondrial lysates from cells stressed with 1 mM hydrogen peroxide (H2O2) for 1 h—a condition that models an extreme acute oxidative insult—as compared with an untreated control (Fig. 1A, lanes 1 and 2). In contrast, acute osmotic challenge with 1 M sodium chloride (NaCl) did not result in Oma1 activation, as reflected by unaltered stability of the Oma1 complex (Fig. 1A, lanes 5 and 6). Intriguingly, we observed that the Oma1 high-mass complex from cells cultured at 1% oxygen was markedly destabilized by BN-PAGE as compared with the complex from control cells grown under normoxic conditions (Fig. 1A, lanes 3 and 4). The steady-state levels of the protease were comparable in each case. Interestingly, we repeatedly noted the appearance of an additional, slightly heavier Oma1-Myc-specific band in the hypoxia-cultured samples (Fig. 1A).
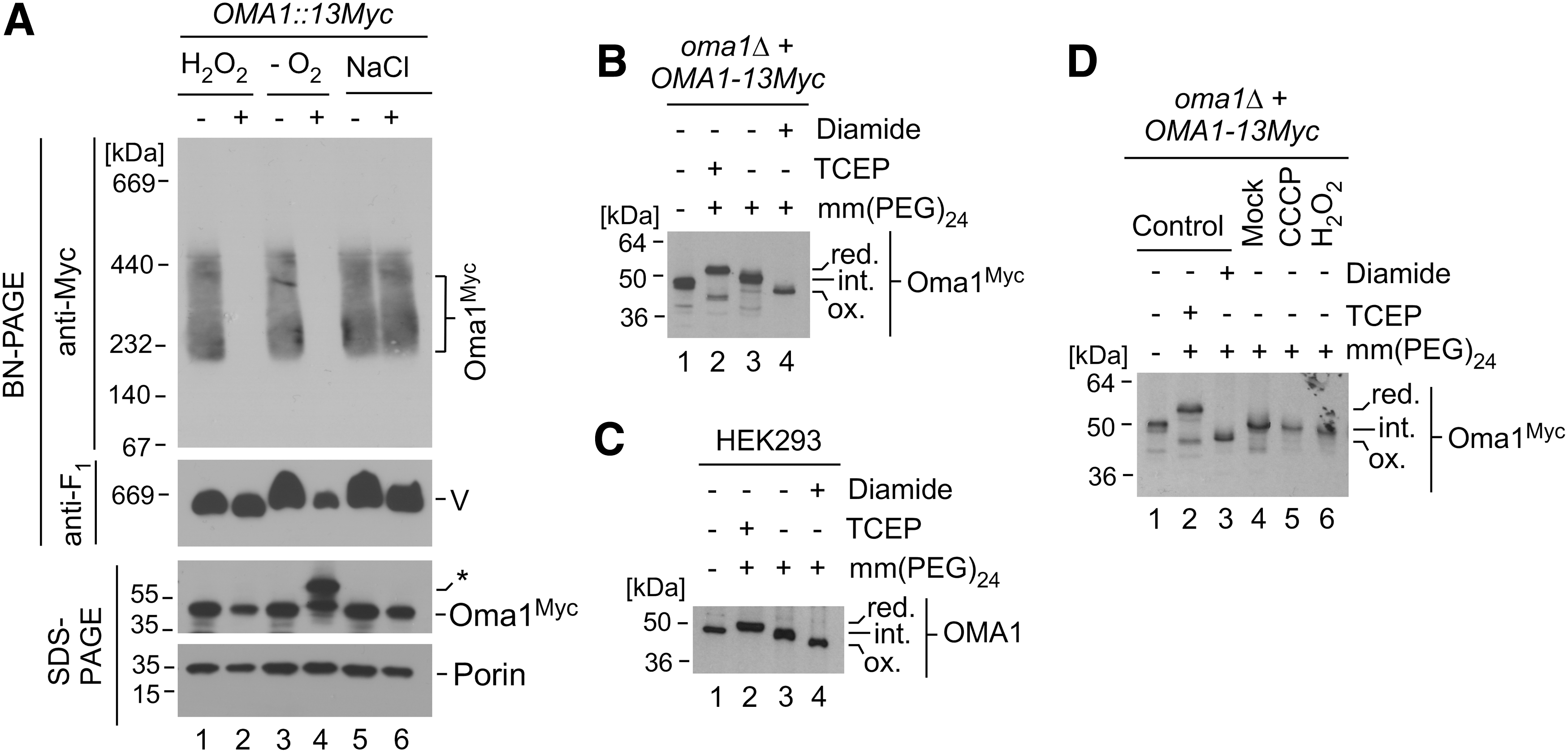
These observations, and the fact that prolonged hypoxia is not associated with enhanced generation of reactive oxygen species (18), prompted us to hypothesize that Oma1 could be responding to changes in the oxygen levels and/or a mitochondrial redox environment. To test this hypothesis, we first sought to determine the redox state of Oma1 under normal physiological conditions. To this end, whole-cell lysates of cells expressing Myc-tagged Oma1 were incubated with the thiol-modifying chemical methyl-polyethylene glycol maleimide 24 [mm(PEG)24], which on its binding to free thiol groups increases the molecular weight of the protein in question, thereby retarding the protein's electrophoretic mobility on a nonreducing polyacrylamide gel. We found that Oma1 was partially modified by mm(PEG)24 when compared with tris(2-carboxyethyl)phosphine (TCEP)-pretreated (fully reduced) or diamide-pretreated (fully oxidized) variants (Fig. 1B), which indicates that Oma1 contains both reduced and oxidized cysteines under basal conditions. Remarkably, mm(PEG)24 modification analysis of human OMA1 in HEK293 cells yielded similar results (Fig. 1C), indicating that the presence of disulfide bond(s) is likely an evolutionarily conserved feature of Oma1. Together, these data suggest that Oma1 might be a subject to thiol-oxidative modifications, which could potentially influence its biochemical properties and function. Interestingly, however, we found that the oxidation state of the protease remained largely unchanged in cells acutely challenged with known stress-activators of Oma1, H2O2 or an ionophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Fig. 1D and Supplementary Fig. S1), suggesting that its redox status is probably not significantly altered on the enzyme's activation by these stimuli.
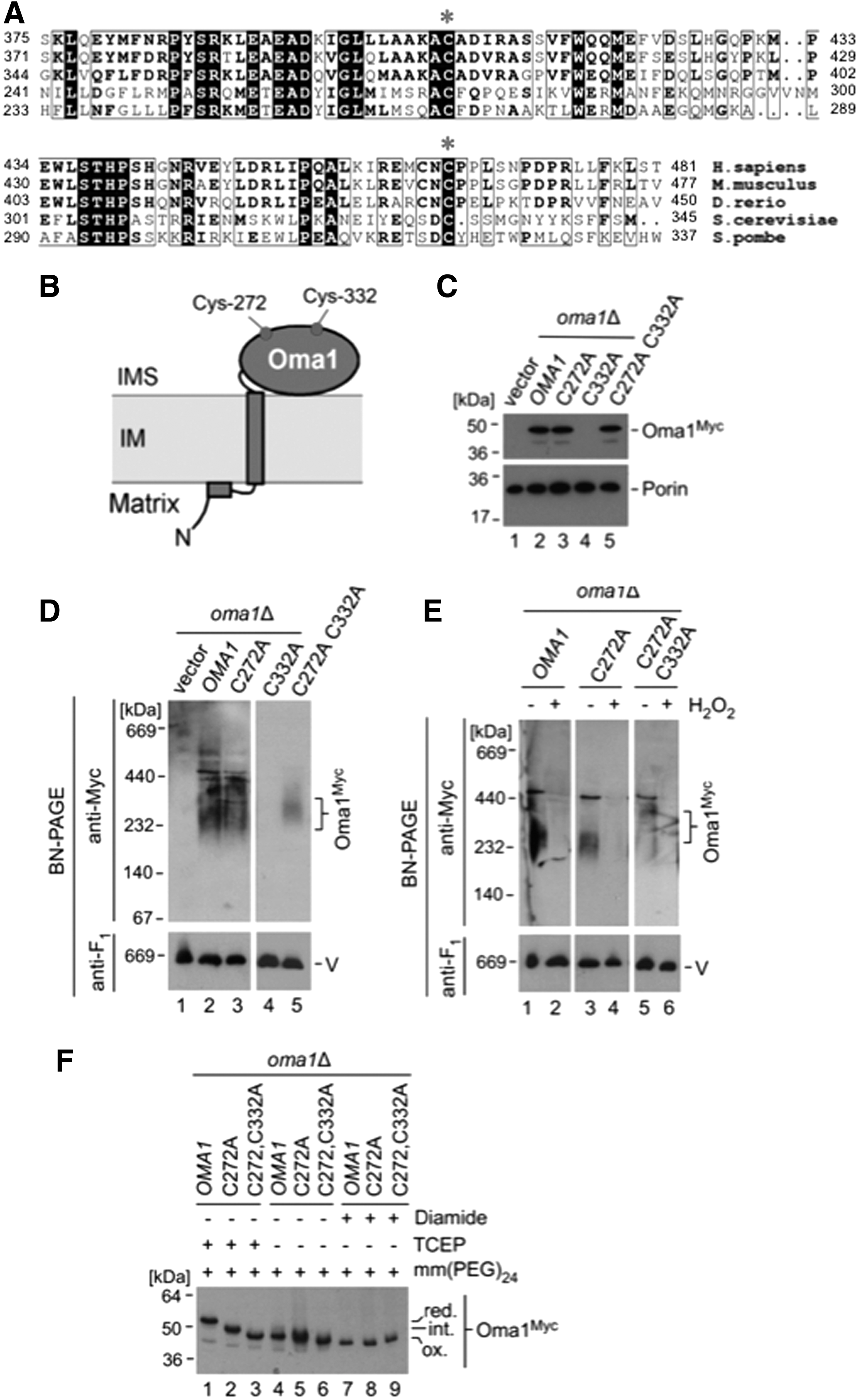
Conserved Cys272 and Cys332 residues mediate the semi-oxidized state of Oma1 and are important for its stability and function
Oma1 harbors several conserved cysteine residues that may be contributing to its semi-oxidized state. Because the IMS is a well-recognized redox-active mitochondrial sub-compartment, we focused our attention on two cysteine residues facing the IMS, Cys272 and Cys332 (Fig. 2A, B). These cysteine residues are highly conserved among eukaryotes, and mutations in Cys332 have been previously shown to affect Oma1 function (7). To better understand the role of these residues in Oma1 function, we generated 13xMyc-tagged variants of Oma1 wherein Cys272, Cys332, or both were mutated to an alanine. Although C272A substitution had no significant effect on Oma1 stability and oligomerization (Fig. 2C, D, lane 3), steady-state levels of the Oma1 C332A mutant were essentially nondetectable relative to controls (Fig. 2C, lane 4). Strikingly, the double C332A C272A mutant was expressed at normal levels (Fig. 2C, lane 5); however, the high-mass Oma1 C332A C272A complex was more labile under BN-PAGE conditions (Fig. 2D, compare lanes 2, 3, and 5). Further, both C272A and C332A C272A Oma1 oligomers were destabilized by BN-PAGE, reflecting a comparable degree of stress activation in H2O2-treated cells (Fig. 2E). These mutations did not affect topological properties of Oma1 (Supplementary Fig. S2). We next assessed the redox state of the mutants in question. We found that the C332A C272A Oma1 mutant was not modified by mm(PEG)24 on pretreatment with TCEP, and the C272A Oma1 mutant displayed only partial modification after such treatment (Fig. 2F). These results demonstrate that the IMS-facing residues Cys272 and Cys332 are key contributors to the oxidized state of Oma1 and form disulfide bonds that are critical for stability of the protein. These data are also consistent with the idea that the redox status of these residues is dispensable for stress-activation of Oma1 (Figs. 1D and 2E).
Being intrigued with the drastic instability of the C332A Oma1 mutant, we wanted to investigate its fate. We first conducted quantitative real-time polymerase chain reaction (qRT-PCR) analysis of OMA1 mRNA levels in wild type (WT) cells or oma1Δ cells expressing WT, C272A, or C332A variants of the protease. The levels of the oma1 C332A transcript were virtually indistinguishable from other OMA1 variants (Fig. 3A), reflecting normal expression of this mutant. This result indicates that stability of the mutant is affected at a post-translational level and suggests two likely scenarios of how the C332A Oma1 mutant could be degraded: (i) The mutant is eliminated by the ubiquitin-proteasome system (UPS) while en route to mitochondria, or (ii) the mutant is transported into mitochondria, but is rapidly degraded by IM protease(s). To test these scenarios, we monitored degradation of WT and C332A Oma1 variants after the inhibition of new protein synthesis by the cytosolic translation-blocking drug cycloheximide, with or without adding the UPS-specific inhibitor MG132. Although MG132 treatment slightly increased steady-state levels of the WT Oma1, we found no appreciable stabilization of the C332A mutant on UPS inhibition (Fig. 1B). This result indicates that UPS is unlikely to be involved in degradation of the C332A Oma1 mutant. Next, we assessed stability of the C332A Oma1 protein expressed in cells lacking Oma1 in combination with the loss of other IM-associated proteases: rhomboid protease Pcp1, the i-AAA protease (Yme1), the m-AAA protease (inactivated via deletion of its Yta10 subunit), and Atp23 metallopeptidase. Again, lack of these proteases did not result in stabilization of the C332A Oma1 mutant (Fig. 3C). Similarly, we were unable to stabilize this mutant by pretreating the cells with known nonselective inhibitors of mitochondrial proteases such as ortho-phenanthroline, ethylenediaminetetraacetic acid (EDTA; to inhibit metallopeptidases), apyrase (to inhibit ATP-dependent proteases), and iodoacetamide (to inhibit cysteine proteases) (Fig. 3D and Supplementary Fig. S3A, B). We conclude that the C332A mutant form of Oma1 may be degraded either by an unidentified mitochondrial protease or through a co-operative action of several mitochondrial quality control proteases. Future studies will attempt to address this intriguing issue.
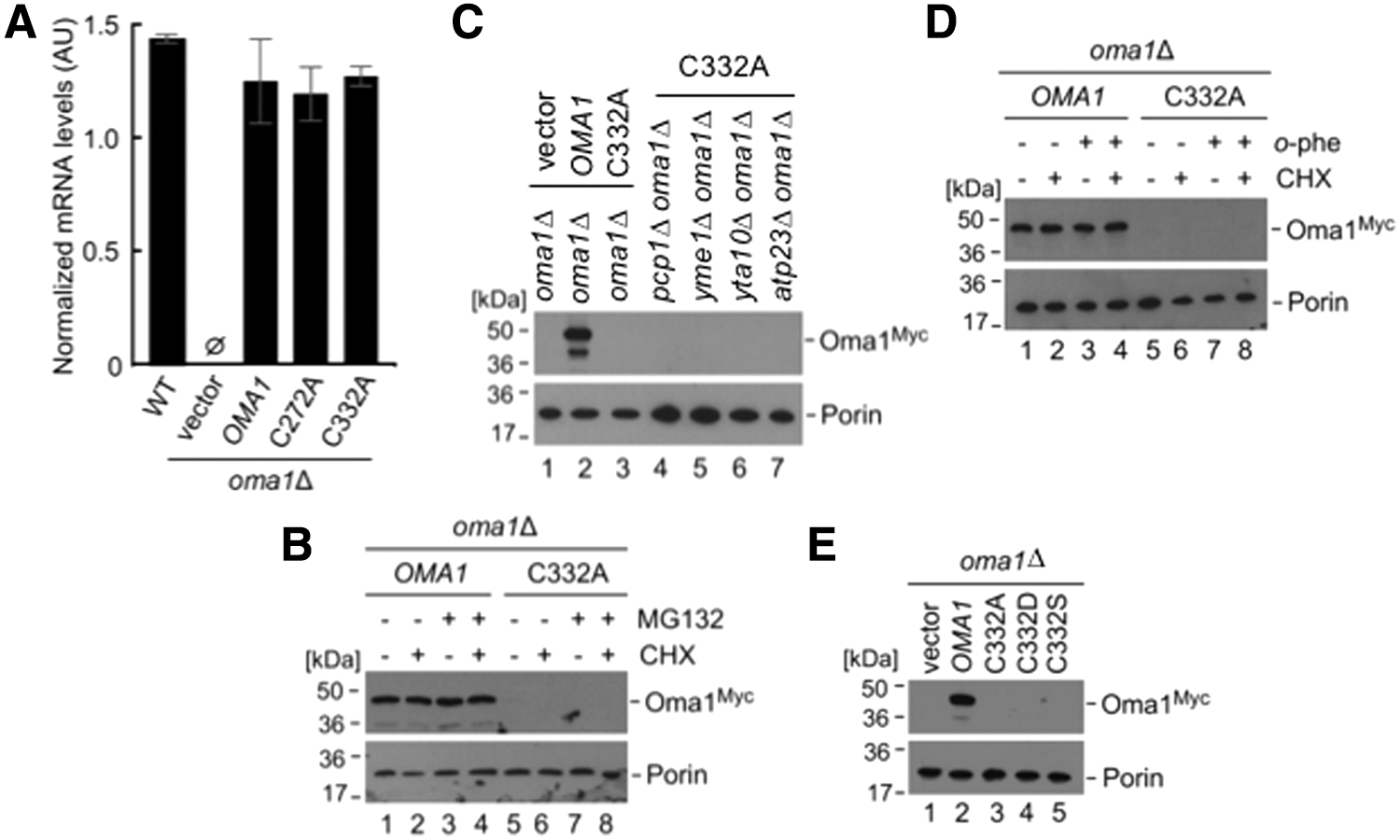
Finally, we probed the specificity of the Cys332 substitution for the stability of Oma1. To this end, two variant mutations were introduced into Oma1 by site-directed mutagenesis: (i) the sulfenylation-mimicking C332D mutation and (ii) the C332S mutation to test the role of the sulfhydryl side chain of Cys332. None of these substitutions at Cys332 resulted in a stable form of Oma1 (Fig. 3E). These findings suggest a crucial role for the cysteine residue at position 332, most likely through its ability to form a disulfide bond with the Cys272. Although the lack of structural data did not allow us to unequivocally determine the nature of this bond (intra- vs. intermolecular), the inability to stabilize the Cys332 mutant variants of Oma1 in trans (Supplementary Fig. S3C) supports the former scenario.
Disulfide bond-breaking mutations in Cys272 and Cys332 alter conformational state of Oma1
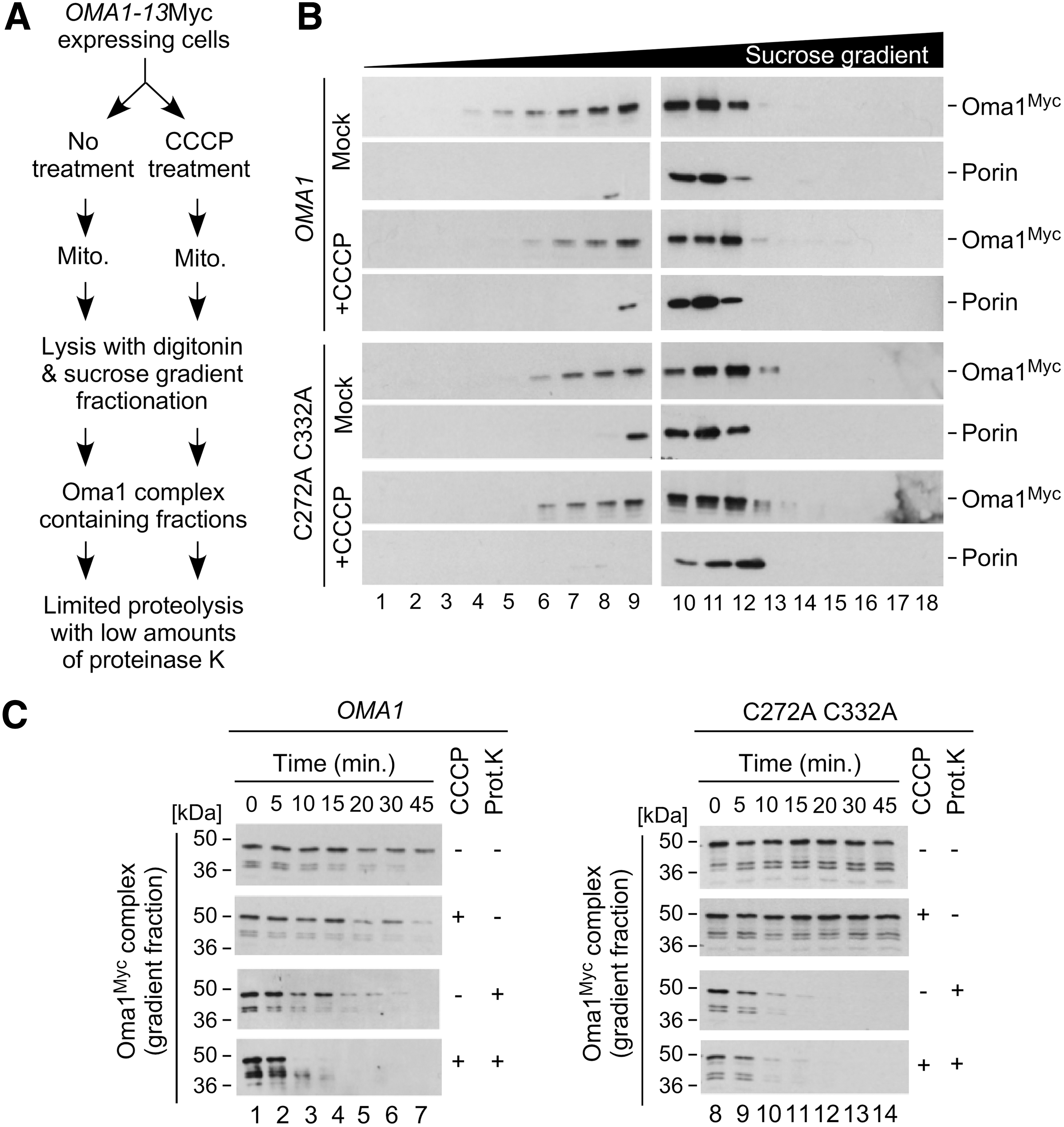
Having determined that disulfide bond-forming Cys272 and Cys332 residues are necessary for Oma1 oligomerization and stability, we next wondered whether loss of a disulfide bond in the C332A C272A Oma1 mutant could influence the conformational state of the protease. This idea is in line with our observation that in unstressed cells the double cysteine mutant Oma1 complex appears to be less stable than the WT Oma1 oligomer under native gel electrophoresis conditions (Fig. 2C). To test this hypothesis, we conducted a previously developed two-step experiment (7) outlined in Figure 4A. First, we isolated mitochondria from either unperturbed or CCCP-treated cells expressing 13xMyc-tagged WT or C332A C272A Oma1 variants. The respective mitochondrial lysates were then subjected to high-velocity sucrose gradient fractionation of the complex in question. Consistent with our previous finding that conformation-altered Oma1 oligomers are not destabilized by the mild (relative to BN-PAGE) conditions of sucrose gradient fractionation (7), we observed no significant differences in the migration pattern of the variant Oma1 high-mass complexes (Fig. 4B). Next, we isolated Oma1 complex-containing fractions and evaluated their resistance toward low amounts of externally added proteinase K. As predicted from the model wherein conformationally altered Oma1 oligomers exhibit increased susceptibility to proteinase K, we found that the C332A C272A Oma1 oligomers from unstressed cells exhibited markedly increased sensitivity to limited proteolysis by this enzyme compared with WT Oma1 complexes (Fig. 4C and Supplementary Fig. S4). Consistently with unaltered steady-state levels of the C332A C272A Oma1 mutant, the stability of the respective gradient fractions that did not receive proteinase K treatment was comparable to that of WT Oma1. Finally, in line with our finding that both the WT and double cysteine mutant Oma1 complexes are destabilized on stress-activation (Fig. 2E), we observed no significant differences in their susceptibility to proteinase K between the oligomeric Oma1 species derived from CCCP-treated cells. Together with the observed destabilizing effect of the C332A mutation, these results support the model that the disulfide bond formed by Cys272 and Cys332 likely plays a structural role and mediates conformational stability of the Oma1 high-mass complex.
Oma1 complex is dynamically tuned in a redox-sensitive manner
To test the hypothesis that the Oma1 complex can be dynamically influenced by changes in its oxidation state, we employed an in vitro assay wherein we used BN-PAGE to monitor stability of the activated WT and C332A C272A Oma1 oligomers subjected to a postisolation reduction. In this assay, mitochondria isolated from CCCP-treated oma1Δ cells expressing relevant Oma1 variants were treated with thiol (DTT) or nonthiol (sodium ascorbate) reducing agents and analyzed by BN-PAGE. In these assays, pretreatment with dithiothreitol (DTT) but not ascorbate led to marked stabilization of the activated WT Oma1 oligomer relative to controls (Fig. 5A, compare lanes 2 and 3). In contrast, pretreatment with DTT had no appreciable stabilizing effect on the complex derived from CCCP-stressed oma1Δ cells expressing the C332A C272A Oma1 mutant. These data validate our prediction that thiol-mediated reduction of Cys272 and Cys332 leads to conformational stabilization of the Oma1 oligomer and further suggest that these residues may also be playing a role in the redox-tuning of Oma1.
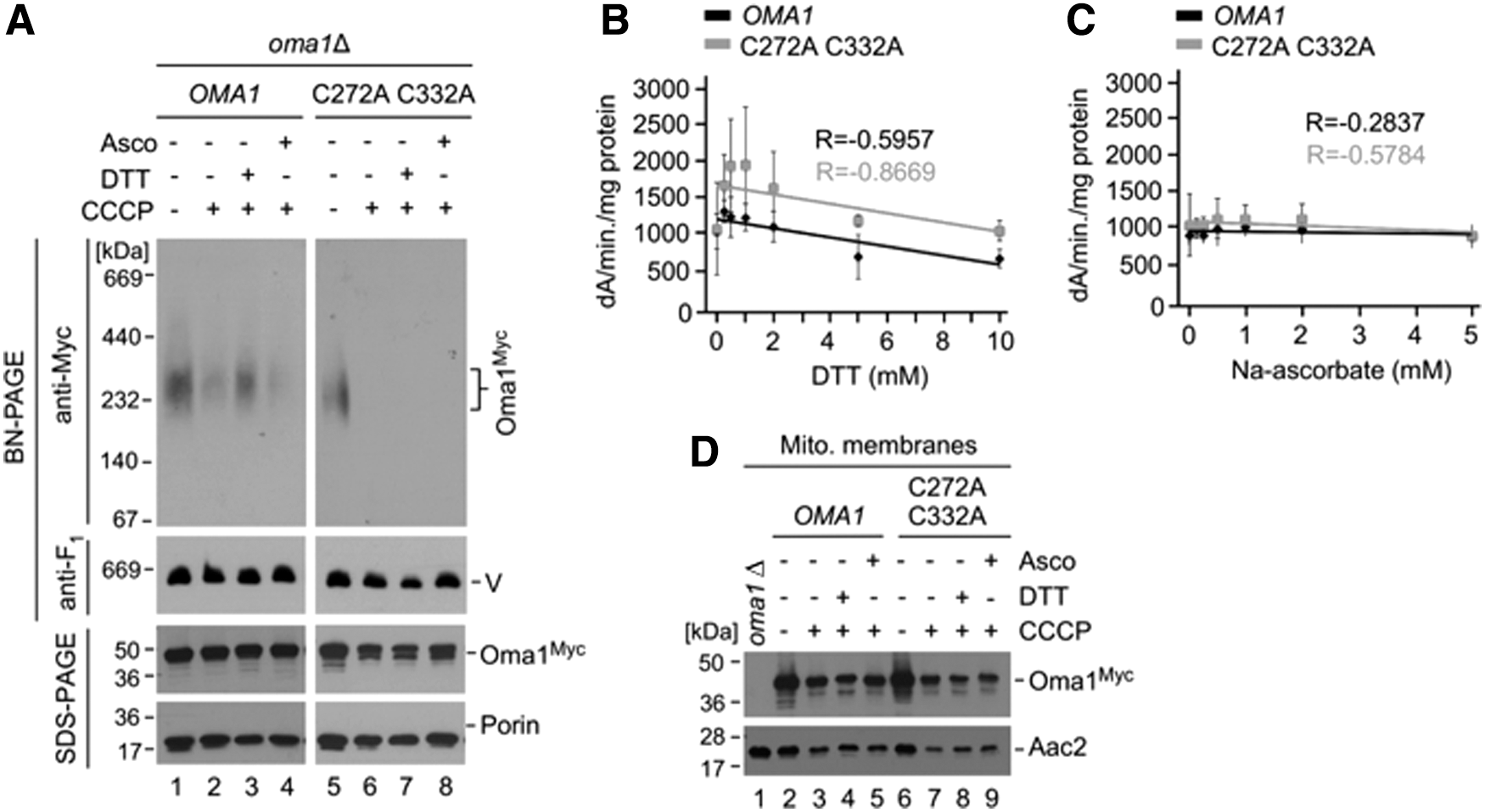
To further test this idea, we examined in vitro proteolytic activity of Oma1 in the presence of increasing amounts of reducing agent, using a previously developed fluorescein isothiocyanate (FITC) casein processing assay (7, 24). To this end, we isolated mitochondrial membranes from Oma1- and C332A C272A Oma1-expressing oma1Δ cells treated with CCCP to activate the protease. We then incubated ATP-deprived membranes with FITC-casein in the presence of 0–10 mM DTT or 0–5 mM sodium ascorbate and monitored changes in FITC fluorescence, which reflects proteolytic conversion of the substrate. The rate of FITC fluorescence generated by either Oma1 variant declined with increasing concentrations of DTT, indicating inhibition of Oma1 proteolytic activity (Fig. 5B). Strikingly, however, sensitivity of Oma1 toward reductant was markedly diminished in the case of the C332A C272A Oma1 mutant, demonstrating that Cys272 and Cys332 are important for redox modulation of the enzyme (Fig. 5B). In contrast, we found no significant changes in the efficiency of FITC-casein proteolysis when the samples were preincubated with sodium ascorbate (Fig. 5C). In all cases, the steady-state levels of each Oma1 variant were not significantly influenced (Fig. 5D). These results indicate that changes in the redox state of mitochondria may regulate Oma1 stability and activity via Cys272 and Cys332.
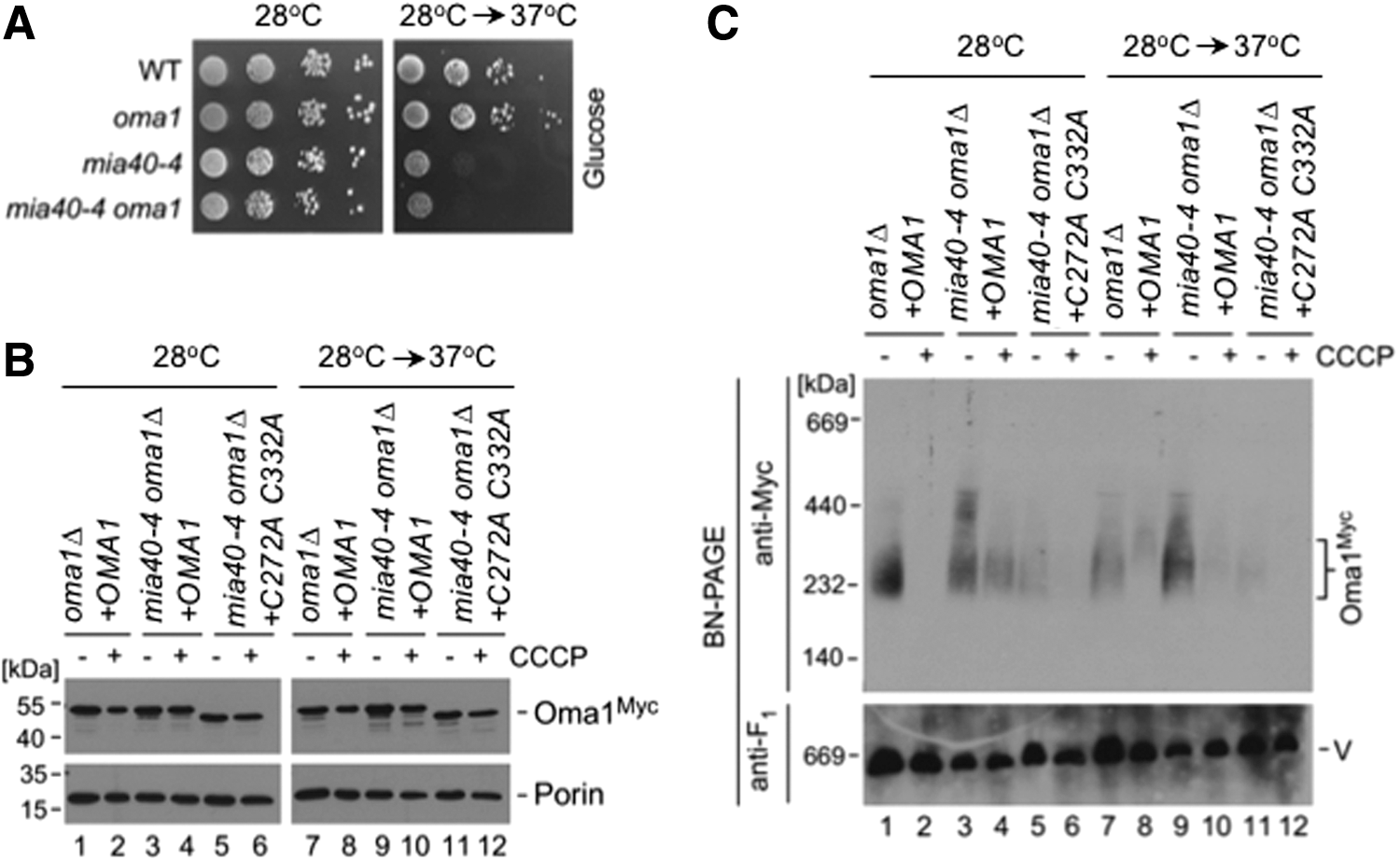
Next, we wanted to corroborate our findings in vivo. Because the redox environment of the IMS is quite unique and cannot be easily genetically modified (9, 26), we decided to focus instead on a downstream condition wherein cells are impaired in their ability to efficiently generate disulfide bonds in the IMS. To this end, we evaluated the biochemical behavior of WT and C332A C272A Oma1 variants in the mia40-4 genetic background. This temperature-sensitive allele of the mitochondrial oxidoreductase Mia40 produces a substrate release-impaired enzyme, thereby affecting cysteine oxidation in the IMS (10). Consistent with the essential role of Mia40 in disulfide formation in the IMS, glucose growth of both mia40-4 and oma1Δ mia40-4 strains was severely impaired when cells were cultured at a nonpermissive temperature (Fig. 6A). We isolated mitochondria from oma1Δ mia40-4 cells expressing WT or C332A C272A Oma1 cultured at normal or restrictive temperatures and with or without addition of CCCP. In all cases tested, steady-state levels of either Oma1 variant were unchanged when compared with relevant controls (Fig. 6B). We next assessed the stability of the Oma1 oligomers by BN-PAGE. Although temperature inactivation of Mia40 had no appreciable stabilization effect on the Oma1 oligomer in CCCP-treated cells—likely due to insufficiently compromised redox equilibrium—we found that stability of the Oma1 complex was strongly increased in mia40-4 cells cultured at 37°C relative to controls (Fig. 6C, compare lanes 3, 7, and 9). In contrast, Mia40 inactivation had no stabilizing effect on the C332A C272A Oma1 complex (Fig. 6C, compare lanes 5 and 11), further supporting the notion that stability and/or activity of Oma1 may be influenced by Mia40 through Cys272 and Cys332 residues of the protease.
Oma1 interacts with its engineered substrate in a redox-dependent manner
We wanted to further refine our understanding of how changes in Oma1 oxidation state might influence the protease's interaction with its substrates. One caveat is that the known repertoire of Oma1 substrates in yeast is limited to proteins that cannot be easily studied due to their large size, hydrophobicity, or mitochondrial genome origin. We, therefore, designed a chimeric protein—designated S1-1–consisting of the 1–104 N-terminal residues of cytochrome c oxidase assembly factor Sco2 (comprising the mitochondrial targeting signal and a transmembrane helix motif) and a 225–244 residue segment derived from the Oma1 substrate protein Oxa1 L240S that harbors an experimentally identified Oma1 cleavage site (22), followed by a 159–250 residue globular domain of cytosolic monothiol glutaredoxin 3, and finally a FLAG epitope tag (Fig. 7A). To minimize any interference from Grx3s cysteine residue on IMS or Oma1 redox balance, its potentially redox-active Cys176 residue was mutated to an alanine. For our subsequent analyses, we co-expressed S1-1 along with the 13xMyc-tagged Oma1 in oma1Δ cells (Supplementary Fig. S5). S1-1 expression yielded a protein of about 36 kDa as well as a ∼17 kDa cleavage product matching the predicted size of the chimera's IMS-exposed moiety, which was not produced in the absence of Oma1 (Fig. 7B). S1-1 was successfully targeted to mitochondria and inserted into the IM, as indicated by its resistance to alkaline carbonate extraction similar to that of IM-anchored protein Cox2 (Fig. 7C). Proteinase K protection experiments further confirmed that S1-1 acquired proper topological orientation in the IM with its C-terminal portion facing the IMS, as reflected by its sensitivity to exogenously added proteinase K in osmotically compromised (swollen) mitochondria (Fig. 7D).
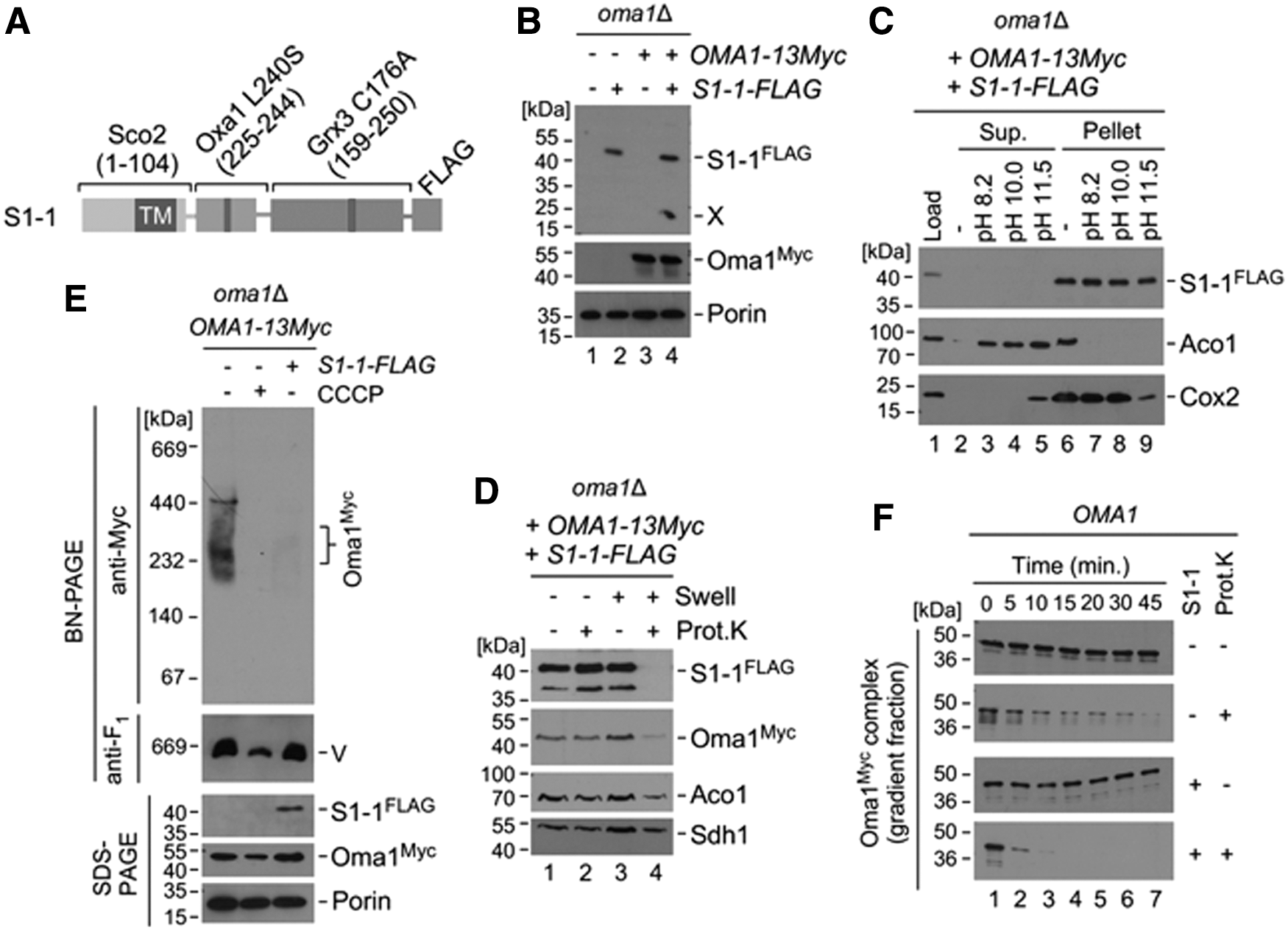
Having confirmed that S1-1 is appropriately targeted and positioned in the IM, we asked whether it could trigger Oma1 activation. To this end, we monitored stability of the Oma1 oligomeric complex in mitochondria from cells expressing S1-1 by BN-PAGE. We found that S1-1 expression resulted in enhanced lability of the Oma1 oligomer relative to untransformed control and was comparable to that observed in CCCP-treated cells (Fig. 7E). As predicted, the steady-state levels of Oma1 remained unchanged at all conditions tested. To further confirm Oma1 activation by S1-1, we examined sensitivity of sucrose gradient-isolated Oma1 oligomers to low amounts of exogenously added proteinase K. Consistent with our earlier observations, expression of S1-1 significantly increased sensitivity of the Oma1 oligomers to limited proteolysis relative to control, thereby reflecting changes in conformation/stability of the complex on S1-1 expression (Fig. 7F). Altogether, these results strongly suggest that S1-1 is a substrate that, indeed, can effectively activate Oma1.
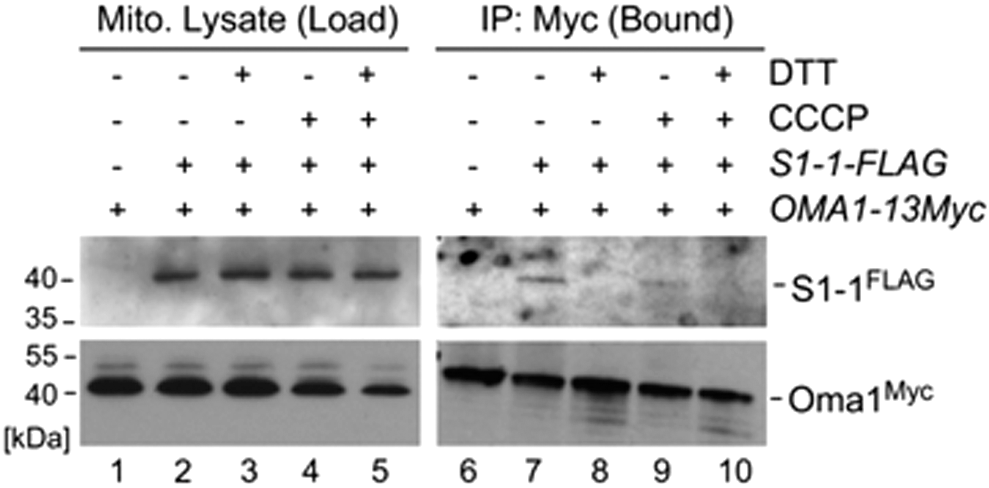
Next, we wanted to determine whether Oma1 can bind to S1-1 and whether this binding could be influenced by changes in redox state of the protease. To address these questions, we conducted co-immunoprecipitations (coIPs) of Oma1 with the S1-1 chimera in mitochondrial lysates from normal and CCCP-stressed cells, in the presence or absence of 10 mM DTT. As predicted, our coIP studies showed a clear interaction of Oma1 with S1-1 in normal and—although slightly less robustly—CCCP-challenged cells (Fig. 8). Strikingly, this interaction was effectively abrogated on addition of DTT (Fig. 8, lanes 7–10). These results demonstrate that Oma1 interacts with S1-1 in a redox-sensitive manner. More generally, these data suggest that redox modification of Oma1 might be influencing its ability to interact with its natural substrates. Future studies are warranted to further investigate this exciting question.
Discussion
The sheer complexity of the IM sub-proteome and inherent risk of generation of byproduct superoxide radicals by mitochondrial respiratory chain enzymes creates a challenging proteostatic environment with a high probability of protein malfunction. This, in turn, can further impair IM permeability, ion homeostasis, and ATP production, ultimately leading to cell demise (29). Unsurprisingly, these conditions are now recognized as a root cause of a wide variety of prevalent human pathologies, including cardiovascular and neurodegenerative diseases (29, 33). A network of mitochondrial quality control mechanisms is in place to avert these scenarios and maintain normal mitochondrial functions. The stress-activated metallopeptidase Oma1 operates on the IMS side of the IM, has emerged as a potential molecular nexus for several quality control activities (3, 28), and is a prospective therapeutic target in mitochondria (1, 27, 39). In spite of its importance, only limited structural and molecular data on Oma1 are available, which offer very little insight into its function. As such, it is important to elucidate molecular details of Oma1 function in the context of the dynamic redox environment of the IMS.
In this work, we sought to better understand how Oma1 activity is regulated and adjusted in response to various physiological demands. Using the yeast model, we discovered that Oma1 shares features of a bona fide redox-dependent protein and determined that the protease exists in a semi-oxidized state under both normal and stressed conditions. Importantly, we also found that the human ortholog of Oma1 shares these biochemical properties, thereby underscoring the evolutionary conservation of this biochemical trait. Interestingly, the oxidation state of the protease was largely unaltered on stress-activation by acute cellular insults with H2O2 or the uncoupler CCCP. These results suggest two possible scenarios: (i) Changes in the redox status of Oma1 may not be sufficient to trigger its activation in response to these stimuli, or (ii) these stressors may be causing drastic changes in the IMS environment wherein a delicate redox-tuning may not be an optimal regulatory response. Consistent with the first idea is our finding that acutely challenging cells with 100 mM DTT had no effect on Oma1 activation (data not shown). On the other hand, our in vitro functional analyses indicated that increased thiol reduction of Oma1 may reduce its proteolytic activity concomitant with stabilization of the Oma1 oligomeric complex. These data are in line with a previously observed inverse correlation between Oma1 complex activity and proteolytic activity of the enzyme (7). Our experiments in mia40-4 background that aimed at examining Oma1 behavior under a condition wherein normal disulfide bond formation in the IMS is impeded indicate that the oxidation state of the protease correlates with its stability. It is noteworthy that in vivo the IMS redox environment appears to be better buffered by reduced glutathione that can freely diffuse from the cytosol (9, 26), which could potentially explain the only modest stabilizing effect observed in the mia40-4 mutant.
These results also raise an interesting possibility that Mia40 may be involved in the redox-tuning of the Oma1 complex, in a manner reminiscent of that reported for the Atp23 protease (40) and the Mic19 subunit of the MICOS (mitochondrial contact site and cristae organizing system) machinery (38). How might Oma1 be activated when redox changes can be excluded? Our data suggest that additional molecular features of the protease could be important for conformational rearrangements required for Oma1 stress-activation. This notion is consistent with previous reports showing Oma1 activation in certain mutants or on conditions that do not appear to elicit any significant changes in mitochondrial redox homeostasis (4, 5, 24). Fundamental understanding of these molecular features and events is limited due to complete lack of structural data on Oma1. It is possible, however, that changes in the redox status of Oma1 might influence the enzyme's activation, as discussed later.
Some mechanistic insights into molecular determinants of Oma1 stabilization also emerged from this study. Our analyses identified the conserved IMS-exposed Cys332 and Cys272 residues of Oma1 as key contributors to the protease's oxidized state. The nearly invariant nature of the C-terminal Cys332 points at its involvement in a structural, most likely intramolecular, disulfide bond. Such molecular architecture is reminiscent of an intramolecular disulfide bond in the enterobacterial Lon protease (31), which has been proposed to function as a switch to optimize Lon's proteolytic activity depending on the redox environment. The stabilizing effect of the C272A mutation in the C332A backbone is remarkable and implicates Cys272 as a second residue involved in the bond. Notably, the C332A C272A Oma1 mutant was shown to be less stable than the WT protease, thereby suggesting a structural role for the Cys272-Cys332 bond in conformational stability of the Oma1 high-mass complex. Of note, in our earlier study (7), we were able to observe a partially stabilized C332A mutant, likely because of the miniature His-Myc epitope tag that was used to visualize steady-state levels of the mutant protein, as opposed to the much larger 13xMyc tag used in this study. It is, thus, possible that the latter tag further affects the already compromised stability of the Cys272-Cys332 bond-deprived protein.
Despite extensive efforts, we were unable to identify factors responsible for rapid post-translational degradation of the C332A Oma1 mutant. Two potential explanations for such a fate are envisioned. First, degradation of the C322A Oma1 mutant may occur through a concerted action of several mitochondrial proteases, and inhibition or removal of one proteolytic component may not significantly impair its proteolysis. Indeed, similar degradation mechanisms have been suggested for mutant forms of the IM insertase, Oxa1 (22), and mitochondrial phosphatidylserine decarboxylase Psd1 (32). A second, less likely, scenario considers the possibility that the C322A Oma1 mutant could be degraded by an unidentified mitochondrial protease that eluded our prediction-based analyses.
Finally, we engineered an S1-1 chimeric protein that can efficiently activate Oma1 when targeted to mitochondria. S1-1 readily binds to Oma1 regardless of its activation state, thus indicating that the 19-residue segment derived from L240S Oxa1 mutant is sufficient to mediate S1-1 recognition and cleavage by the protease. These data also suggest the relative promiscuity of Oma1 toward its substrates. Importantly, we found that Oma1 interacts with S1-1 in a redox-sensitive manner, wherein S1-1 binding to Oma1 is effectively abrogated in the presence of DTT. As S1-1 is designed in such a way that it contains no cysteine residues, the sensitivity of Oma1-S1-1 interaction is unlikely to arise from the disruption of intermolecular disulfide bonding between the two proteins. Taken together, our results suggest that redox modifications of the Cys272-Cys332 bond might be influencing the enzyme's ability to either interact or maintain an association with its natural substrates. It is also tempting to speculate that this disulfide bond might be dynamically influenced by additional cysteine-rich proteins in the IMS such as Atp23 via reversible formation of intermolecular mixed disulfides. Our study establishes the framework for further structural studies directed at resolving molecular determinants for substrate recognition and redox-tuning of Oma1. Given the central role of Oma1 in mitochondrial function and integrity, this study opens an exciting research avenue regarding how changes to the cellular and mitochondrial redox environment might be influencing the proteolytic control of these processes in normal and pathological states.
Materials and Methods
Yeast strains, plasmids, and growth conditions
Yeast strains used in this work are listed in Supplementary Table S1. Yeast cells were cultured in rich YPD (1% yeast extract, 2% peptone, and 2% glucose), or complete synthetic medium supplemented with relevant amino acids and containing 2% glucose or 2% glycerol/2%
The ability of strains to utilize various carbon sources was tested by spotting serial dilutions of the cells onto complete synthetic plates containing 2% glucose or 2% glycerol and 2%
Gene expression analysis
Total mRNA was isolated from 5-mL cultures grown overnight in selective minimal medium. The cells were pelleted by quick centrifugation (5000 g for 1 min), washed twice with water, and immediately used for RNA isolation with yeast RNA purification kit (Epicentre) according to the manufacturer's instructions. cDNA synthesis was performed by using Maxima H Minus cDNA Synthesis Master Mix (Thermo Fisher Scientific). The qRT-PCR was carried out on a Bio-Rad iCycler by using SYBR GreenER™ qPCR SuperMix Universal (Thermo Fisher Scientific) in a 96-well plate using the following conditions: 50°C for 2 min, 95°C for 10 min, 95°C for 10 s, and 60°C for 60 s for 40 cycles. All samples were analyzed in triplicate. The mRNA levels were normalized to ACT1 transcript levels. The parametric Student's t-test was used to determine significance. p-Values of <0.05 were considered statistically significant.
Alkaline lysis of yeast cells
Whole-cell extracts were prepared according to a published protocol (24). Briefly, pelleted cells (A600 = 1.0) were resuspended in 250 μL of 50 mM Tris-HCl, pH 8.0, followed by addition of 50 μL of the lysis buffer containing 10 M NaOH, 7.5% β-mercaptoethanol, and 2 mM phenylmethylsulfonyl fluoride (PMSF). The samples were mixed vigorously and incubated on ice for 15 min, and after addition of 220 μL of cold 100% trichloroacetic acid (TCA), they were incubated for another 15 min on ice. The samples were then precipitated by centrifugation at 20,000 g for 10 min at 4°C. The resulting pellets were washed twice with 1 mL of absolute acetone, air-dried on ice, and resuspended in 133 μL of 1 × Laemmli buffer followed by incubation at 85°C for 10 min. Twenty microliters of obtained samples were then used for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis.
Mitochondria isolation and purification
Yeast mitochondria were isolated from 1-L cultures grown overnight in a relevant selective media essentially as described (15) Cells were collected by centrifugation at 3500 g for 5 min and washed once with water. The pellets were then resuspended in 0.1 M Tris-SO4, pH 9.4 (buffer A) with 10 mM DTT according to the ratio of 1 mL of the buffer per 0.5 g wet weight of pellets. After incubation at 30°C for 10 min, the cells were washed once with 20 mL of 1.2 M sorbitol (buffer C). The spheroplasts were obtained by incubation in 1.2 M sorbitol, 20 mM KH2PO4, pH 7.4 (1 mL of buffer B per 0.15 g wet weight) with lyticase (Sigma-Aldrich) at 4 mg/g wet weight ratio for 1 h at 30°C and gentle shaking. The samples were pelleted, washed once with 30 mL of 1.2 M sorbitol, and resuspended in 10 mL of 0.65 M sorbitol, 10 mM Tris-HCl, pH 7.4, 1 mM PMSF (buffer D). The spheroplasts were homogenized by 15 strokes in a 40-mL dounce homogenizer (Kontes Glass Co.) on ice and centrifuged at 3500 g for 5 min at 4°C. The supernatants were collected in separate tubes, and the pellets were homogenized two more times. Total supernatants were centrifuged at 12,000 g for 10 min at 4°C. The supernatants and the lipid layer on the tube walls were removed; the pellets were gently resuspended in 10 mL of buffer D and centrifuged at 2500 g for 5 min at 4°C. The supernatants were transferred into new tubes and precipitated by centrifugation at 12,000 g for 10 min at 4°C. The obtained mitochondria-enriched pellets were then resuspended in 750 μL of buffer D.
Isolated mitochondria-enriched fractions were further purified by using a discontinuous Histodenz (Sigma-Aldrich) gradient as previously described (42). Briefly, the gradient was formed by layering 10 mL of 14% (w/v) Histodenz solution in 1.2 M sorbitol on top of 5 mL of 22% (w/v) Histodenz soulution in 1.2 M sorbitol. Then, 1.5 mL of crude mitochondria were layered on top of the gradient and centrifuged at 50,000 g for 1.5 h at 4°C. Pure mitochondria were transferred from the interphase fraction between the two Histodenz densities into a new tube, mixed with 20 mL of buffer D, and centrifuged at 12,000 g for 10 min at 4°C. The pellets were gently resuspended in 1.5 mL of the same buffer, transferred into microtubes, and reisolated again. The final pellets were resuspended in 500 μL of buffer D.
Mitochondria from HEK293T human embryonic kidney cells were isolated according to Chen et al. (12). Total mitochondrial protein concentrations were determined by using the Coomassie Plus protein assay kit (Thermo Fisher Scientific).
Mitochondrial assays
High-velocity fractionation of protein complexes was performed in continuous 10%–50% sucrose gradient as previously described (23, 24). In brief, the gradient was formed by a gentle layering of 100 μL 80% sucrose (bottom cushion) and 2.15 mL of 50% sucrose in the stock buffer (0.1% digitonin, 20 mM HEPES, pH 7.4, 150 mM KCl, 1.2 mM MgCl2, 0.5 mM PMSF), followed by 2.25 mL of 10% sucrose in the stock buffer in 5-mL tubes (Beckman-Coulter). The tubes were then set at a 45°C angle for 3 h at 4°C to allow diffusion-based equilibration. Seven hundred micrograms of mitochondria were reisolated at 12,000 g for 10 min at 4°C and lysed in 450 μL of lysis buffer (1% digitonin, 20 mM HEPES, pH 7.4, 150 mM KCl, 1.2 mM MgCl2, 0.5 mM PMSF) for 15 min on ice. The samples were then centrifuged at 20,000 g for 15 min at 4°C, and clarified lysates were loaded onto the gradient and fractionated at 148,000 g for 6 h at 4°C. Fractions were manually collected as 250-μL aliquots, flash-frozen in liquid nitrogen, and stored at −80°C.
Sucrose fractions #10–12 were subjected to limited proteolysis by proteinase K (7). One hundred microliters of each fraction were combined together, mixed well, and split into two separate aliquots. One aliquot was treated with 1.5 μL of Proteinase K solution (40 μg/mL), and the other was used as a untreated control; all samples were kept on ice. The 15-μL aliquots were collected on addition of Proteinase K (0 min), and during the following time periods of 5, 10, 15, 20, 30, and 45 min. Collected samples were immediately mixed with 5 μL of 6 × Laemmli buffer with 5% β-mercaptoethanol and 0.8 μL of 200 mM PMSF, and they were incubated for 10 min at 100°C. The samples were subjected to SDS-PAGE analysis no later than 1 h after the end of the treatment.
Mitochondrial topology of the S1-1 chimeric protein was analyzed according to previously published protocols (25, 42). Six aliquots of gradient-purified mitochondria (20 μg/sample) were pelleted by centrifugation at 12,000 g for 10 min at 4°C and subjected to the following treatments: (i) intact mitochondria in 500 μL of 0.65 M Sorbitol, 10 mM Tris-HCl, pH 7.4; (ii) intact mitochondria in 500 μL of 0.65 M Sorbitol, 10 mM Tris-HCl, pH 7.4 with 5 μL Proteinase K (100 μg/mL); (iii) hypotonic swelling in 500 μL of 20 mM HEPES, pH 7.4; (iv) in 500 μL of 20 mM HEPES, pH 7.4 with 5 μL Proteinase K (100 μg/mL); (v) lysed mitochondria in 450 μL of 20 mM HEPES, pH 7.4 with 50 μL 10% DDM (n-dodecyl-β-
Membrane association of the S1-1 chimera was determined by carbonate extraction, as described earlier (25, 42). Four aliquots of 50 μg of gradient-purified mitochondria were pelleted by centrifugation at 12,000 g for 10 min at 4°C. The pellets were then resuspended in 500 μL of the following buffers containing 1 mM PMSF: (i) 0.65 M sorbitol, 10 mM Tris-HCl, pH 7.4; (ii) 0.1 M NaHCO3, pH 8.3; (iii) 0.1 M NaHCO3, pH 10.0; (iv) 0.1 M NaHCO3, pH 11.5. All samples were incubated on ice for 1 h with periodic mixing and then centrifuged at 60,000 g for 45 min at 4°C. The supernatants were transferred into separate tubes and subjected to TCA precipitation as described earlier. The pellets were dissolved in 50 μL of 1 × Laemmli buffer with 5% β-mercaptoethanol and incubated for 10 min at 85°C. The load control was obtained by pelleting 50 μg of gradient-purified mitochondria, which were then resuspended in 50 μL of 1 × Laemmli buffer with 5% β-mercaptoethanol.
The redox state of mitochondrial proteins was determined as described by Bode et al. (5) with slight modifications. Four aliquots of isolated mitochondria (50 μg/sample) were reisolated in 500 μL of 0.65 M sorbitol, 10 mM Tris-HCl, pH 7.4, 1 mM PMSF. Then, 60 μL of cold 100% TCA was mixed with the protein samples, followed by incubation on ice for 15 min. The samples were centrifuged for 10 min at 20,000 g at 4°C. The obtained pellets were washed twice with acetone and air-dried on ice for 20 min. All pellets were resuspended within 50 μL of modifying buffer (80 mM Tris-HCl, pH 7.0, 10% glycerol, 2% SDS, 0.04% bromocresol green). For complete reduction of all cysteine residues, one sample was additionally treated with 2 μL of 0.5 M TCEP, pH 7.0. Complete oxidation was achieved by treatment with 2.5 μL of freshly prepared 100 mM diamide. All samples were incubated at 96°C for 15 min, followed by 5-min incubation at RT. After incubations, one sample was mock-treated with 3 μL DMSO and was used as an unmodified control. The remaining samples were treated with 3 μL of 250 mM mm(PEG)24 (Thermo Fisher Scientific) and incubated for 1 h in the dark. After the treatment, the samples were resuspended in 1 × Laemmli buffer without any reactants and analyzed by nonreducing SDS-PAGE immunoblotting using 8% polyacrylamide gels.
Blue native gel electrophoretic analysis of digitonin-solubilized mitochondrial lysates produced as described earlier—except that 70 μg of mitochondria was used—was carried out by using home-made 5%–13% gradient gels as previously described (23, 41).
Immunochemical assays
Immunoprecipitation (IP) assays were conducted as previously described (19). Gradient-purified mitochondria (500 μg) were pelleted by centrifugation at 12,000 g for 10 min at 4°C, resuspended in 100-μL lysis buffer [1% (w/v) digitonin, 10 mM HEPES, pH 7.4, 50 mM NaCl, 2 mM EDTA, pH 8.0, 1 mM PMSF], and incubated at 4°C with gentle agitation for 15 min. The lysates were further resuspended in 1 mL of washing buffer [0.1% (w/v) digitonin, 10 mM HEPES, pH 7.4, 50 mM NaCl, 2 mM EDTA, pH 8.0, 1 mM PMSF] and spun down at 16,000 g for 15 min at 4°C. The supernatants were then transferred into new tubes. An aliquot of 40 μL (prepreclearance loading control) was collected. The remaining supernatants were mixed with 40 μL of preclearance IgG beads slurry (MBL International) followed by incubation at 4°C with gentle agitation for 1 h. The beads were then precipitated by a quick centrifugation at 15,000 g for 15 s, and clarified supernatants were transferred into new tubes. Pelleted beads were resuspended in 40 μL of lysis buffer and stored. The samples were pelleted by quick centrifugation, and the resulting supernatants were transferred into new tubes. Forty-microliter aliquots of these supernatants (postpreclearance control) were stored. The remaining supernatants were mixed with 40 μL of anti-Myc-tag magnetic beads (Santa Cruz Biotechnology) and incubated overnight at 4°C with gentle agitation. DTT (final concentration of 10 mM) was added to the relevant samples, whereas the other samples were left untreated. After incubation, the samples were precipitated by quick centrifugation and 40-μL aliquots of supernatants (unbound control) were collected. The beads were washed thrice with the washing buffer. Ten millimolar DTT was included in the buffer used to wash DTT-treated IP samples. After the final wash, the supernatants were transferred into a new tube and precipitated with 100 μL of ice-cold 100% TCA. The resulting protein pellets (wash controls) were resuspended in 50 μL of 1 × Laemmli buffer and incubated for 10 min at 85°C. Previously collected fractions were mixed with 8 μL of 6 × Laemmli buffer and 1 μL of 1 M DTT and incubated as described earlier with vigorous agitation. The final pelleted beads were resuspended in 40 μL of lysis buffer, mixed with 8 μL of DTT-supplemented 6 × Laemmli buffer, and handled as described earlier. The samples were centrifuged for 1 min at 12,000 g, and the resulting supernatants were transferred into new tubes and used for Western blotting by using standard 10% polyacrylamide gels.
For immunoblot analyses, gel-separated proteins were transferred onto nitrocellulose (denaturing gels) or polyvinylidene fluoride (blue native gels) membranes by tank blotting. The membranes were blocked in 5% nonfat milk in phosphate-buffered saline buffer containing 0.1% Tween-20 at room temperature and subsequently incubated with relevant primary antibodies at 4°C. The following antibodies and sera were used in this study: mouse anti-Myc epitope (11667149001, 1:1000; Roche); rabbit anti-Myc epitope (A-14, 1:2500; Santa Cruz Biotechnology); rabbit anti-FLAG epitope (2368, 1:1,000; Cell Signaling); mouse anti-Porin (459500, 1:5000; Invitrogen); mouse anti-Cox2 (ab110271, 1:2000; Abcam); rabbit anti-Aac2 (a gift from Dr. Carla Koehler, 1:5000); rabbit anti-β subunit of F1–F0 ATPase (a gift from Dr. Alexander Tzagoloff, 1:5000); mouse anti-Sdh1 (a gift from Dr. Dennis Winge, 1:2000); and rabbit anti-Aco1 (a gift from Dr. Roland Lill, 1:5000). The secondary horseradish peroxidase-coupled antibodies were as follows: goat anti-mouse IgG (115-035-068, 1:5000; Jackson ImmunoResearch Laboratories) and goat anti-rabbit IgG (7074S, 1:5000; Cell Signaling). The protein-antibody signals were detected by using either homemade (5 mM luminol, 0.4 mM p-coumaric acid, 100 mM Tris-HCl, pH 8.5 mixed with 0.2% H2O2 100 mM Tris-HCl, pH 8.5 in 1:1 proportions and used immediately) or Clarity (BioRad) chemiluminescence reagents and X-ray film (Phenix Research Products).
ATP-independent proteolytic activity
Proteolytic activity was measured by using ATP-depleted mitochondrial membranes according to the previously described procedure (24). Membranes were isolated from 300 μg of mitochondria pelleted by centrifugation at 12,000 g for 10 min at 4°C, resuspended in 700 μL of hypotonic 20 mM HEPES buffer, pH 7.4 with 1 mM PMSF, and subjected to gentle sonication (3 × 30 s, 50% duty cycle). Samples were centrifuged at 65,000 g for 1 h at 4°C. Pellets were then resuspended in 300 μL of buffer D (0.65 M sorbitol, 10 mM Tris-HCl, pH 7.4). Ten micrograms of obtained membranes were then resuspended in 200 μL of the filter-sterilized reaction buffer (10 mM MgCl2, 50 mM Tris-HCl, pH 8.0, 0.05% Triton X-100) containing DTT or ascorbate. Samples were incubated at 25°C for 5 min, followed by transfer of 150 μL of the reaction mixture into a new tube containing 7.3 μL of 102.4 μM FITC-casein (Sigma-Aldrich). The samples were then incubated in the dark for 35 min with periodic mixing; finally, they were centrifuged at 20,000 g for 10 min at 25°C. One hundred microliters of supernatants were transferred into clear 96-well plates and used for fluorescent measurements by a Synergy 2 multi-mode microplate reader (BioTek Instruments) at 485-nm excitation and 528-nm emission wavelengths. The measurements were taken every 45 s for 10 min. The obtained measurements were exported into Microsoft Excel, used to calculate ΔA per minute, and normalized to protein concentration.
Footnotes
Acknowledgments
The authors thank Dr. Jennifer Fox for critical reading of the article. They thank D. Winge (University of Utah), A. Tzagoloff (Columbia University), T. Langer (University of Cologne), C. Koehler (UCLA), and S. Claypool (Johns Hopkins University) for providing reagents. This work was supported by the NIH grant GM108975 to O.K. J.V.D. was supported by the NIH training grant T32 GM107001-01A1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.