
Abstract
Aims:
Drug-induced liver injury, especially acetaminophen (APAP)-induced liver injury, is a leading cause of liver failure worldwide. Mouse models were used to evaluate the effect of microelement selenium levels on the cellular redox environment and consequent hepatotoxicity of APAP.
Results:
APAP treatment affected mouse liver selenoprotein thioredoxin reductase (TrxR) activity and glutathione (GSH) level in a dose- and time-dependent manner. Decrease of mouse liver TrxR activity and glutathione level was an early event, and occurred concurrently with liver damage. The decreases in the GSH/glutathione disulfide form (GSSG) ratio and TrxR activity, and the increase of protein S-glutathionylation were correlated with the APAP-induced hepatotoxicity. Moreover, in APAP-treated mice both mild deprivation and excess supplementation with selenium increased the severity of liver injury compared with those observed in mice with normal dietary selenium levels. An increase in the oxidation state of the TrxR-mediated system, including cytosolic thioredoxin1 (Trx1) and peroxiredoxin1/2 (Prx1/2), and mitochondrial Trx2 and Prx3, was found in the livers from mice reared on selenium-deficient and excess selenium-supplemented diets upon APAP treatment.
Innovation:
This work demonstrates that both Trx and GSH systems are susceptible to APAP toxicity in vivo, and that the thiol-dependent redox environment is a key factor in determining the extent of APAP-induced hepatotoxicity. Dietary selenium and selenoproteins play critical roles in protecting mice against APAP overdose.
Conclusion:
APAP treatment in mice interrupts the function of the Trx and GSH systems, which are the main enzymatic antioxidant systems, in both the cytosol and mitochondria. Dietary selenium deficiency and excess supplementation both increase the risk of APAP-induced hepatotoxicity.
Introduction
Drug-induced liver injury has proved to be a leading cause of market withdrawal of approved medicines (27). Acetaminophen (APAP) is used worldwide for the treatment of fever and pain, but it is responsible for a large percentage of acute liver damage cases seen in patients (26). The hepatotoxicity of APAP was first described several decades ago and was recognized as a cause of acute liver damage in the 1980s (26). In 1989, liver injury was defined as occurring when serum alanine transaminase (ALT) levels were more than twice the upper limit of normal levels (36).
The mechanism of APAP-induced liver damage is indicated by the depletion of glutathione (GSH) levels and results from modification of mitochondrial proteins by the electrophilic APAP metabolite N-acetyl-p-benzoquinone imine (NAPQI), resulting in dose-dependent hepatotoxicity (21, 28, 52). At therapeutic doses, most APAP is excreted via conjugation with glucuronic acid or sulfuric acid, with the remainder metabolized by cytochrome P450 enzymes to produce the toxic NAPQI, which reacts with reduced GSH by 1,4-conjugate addition to produce the GS-NAPQI adduct (43, 47). Intentional or unintentional APAP overdose results in the formation of a larger amount of NAPQI, with consequent depletion of GSH and reaction with mitochondrial proteins (2, 43).
Mitochondrial dysfunction and liver damage occur as a consequence of these changes. A recent study showed that the methylation-controlled J protein (MCJ), a negative regulator of mitochondria, can be targeted during APAP-induced liver injury based on the observation that MCJ silencing could protect mice from APAP overdose-mediated liver damage (4). It has also been reported that APAP exposure can deplete GSH levels in mitochondria and promote reactive oxygen species (ROS) generation, which activates the translocation of JNK (c-Jun N-terminal kinase) into mitochondria, thereby affecting mitochondrial function (14, 20, 41). In addition, injection of exogenous mitochondria into APAP-treated mice alleviates the associated liver injury (53). The above evidence indicates the pivotal role of mitochondria in APAP-induced liver injury.
Innovation
This study indicates that selenium could be a major factor in affecting the extent of acetaminophen-induced hepatotoxicity and thus provides a new insight into the mechanism by which both selenium deficiency and selenium overdose could enhance serious liver damage. In addition, this study emphasizes the importance of the overall thioredoxin (Trx) system oxidation state, comprising the mitochondrial and cytosolic Trx systems.
In addition to intentional and unintentional APAP overdose, some other factors contribute to APAP toxicity, including fasting, obesity, alcohol use, malnutrition, and even genetic variability. The involvement of alcohol in hepatotoxicity of APAP has been controversial for a long time. A weak association of APAP-induced hepatotoxicity with alcohol intake was found in 1994 (61), while later studies revealed that chronic alcoholics were susceptible to a therapeutic dose of APAP; simultaneous ingestion of APAP and alcohol attenuated hepatotoxicity as alcohol competes as a substrate for the primary metabolic enzyme CYP2E1, thereby reducing conversion of APAP into NAPQI (27, 46). APAP-induced liver damage is routinely treated by administering the FDA-approved N-acetylcysteine, but this treatment is only effective in the early period after APAP poisoning (9, 16, 60).
The essential trace element selenium has been documented to be beneficial for life for a long time. The physiological significance of selenium has been primarily related to its presence in selenoproteins in the form of selenocysteine (SeC, Se-Cys, U) (30, 39). In addition, the potent anticancer activity of selenium has been linked to the interaction of selenium compounds with selenoproteins (11, 12, 18). Twenty-five selenoprotein genes have been identified in human cells, and SeC is present in selenoproteins, including thioredoxin reductases (TrxRs), glutathione peroxidases (GPxs), and iodothyronine deiodinases (24, 30, 39). TrxRs and GPxs are critical members of the thioredoxin (Trx) and GSH systems, respectively. Trx and GSH systems are two major thiol/selenol-dependent antioxidant systems with overlapping function. They are also involved in in vivo redox equilibrium regulation, protecting the cells from oxidative stress (8, 32, 51, 58, 59). Decreased TrxR (17, 55) and GPx (48) activities were found in selenium-deficient (SD) animal model. The replacement of SeC with cysteine (Cys) in the C-terminus of TrxR occurs in SD rats, resulting in reduced catalytic activity (33). Pretreatment with a single dose of selenite via intraperitoneal injection exhibited protective effects against APAP-induced hepatotoxicity in the rat three decades ago (50), which may be due to the pro-oxidant effects of selenite to cause the increase of glutathione level. However, the regulation of TrxR activity by dietary selenium, and consequently the roles of TrxR-mediated pathways in APAP-induced hepatotoxicity have not been well investigated.
Since SeC has higher nucleophilicity than Cys, it is expected to be highly reactive toward electrophilic agents, including APAP metabolite NAPQI (22, 37). Indeed, additional studies provide proof of the association between APAP-induced hepatotoxicity and the inhibition of cytosolic TrxR1 and mitochondrial TrxR2 activities (40). However, upregulated TrxR1 and GSH levels induced by nuclear factor-E2-related factor 2 (Nrf2) activation may protect the liver from the APAP damage (19). Therefore, the changes of the redox environment regulated by Trx systems upon the treatment with APAP are not well understood. In this study, we investigate the role of the thiol-redox pathways regulated by Trx and GSH systems in APAP-induced liver injury. Particularly, we studied the effects of the selenium deficiency or excess on APAP-caused liver damage and the resulting changes in the related cellular redox environment.
Results
Effects of treatment with APAP on GSH level and TrxR activity in mouse liver
APAP overdose can induce acute liver damage, and a dose-dependent hepatotoxicity of APAP has been indicated in previous studies (25, 44). Here, mice were exposed to 100, 300, or 500 mg/kg APAP, and survival was measured for the following 3 days. Death only occurred in the first 24 h after APAP exposure in a dose-dependent manner, with one mouse dying in the 300 mg/kg APAP group and four mice dying in the 500 mg/kg APAP group (Fig. 1A). We subsequently treated mice with various APAP doses (100, 300, 500 mg/kg; 10 mice in each group) and investigated APAP toxicity after 1, 6, and 24 h. The results showed that APAP toxicity occurred during 24 h after APAP treatment, consistent with the above. In the group treated with 500 mg/kg APAP, 6 of 10 mice died, while no mice died in the other groups. We analyzed serum alanine transaminase (ALT) activity, liver TrxR activity and GSH levels in the surviving mice. There was no significant change in ALT activity in surviving mice between groups 1 and 6 h after APAP treatment. In contrast, a dramatic elevation in ALT activity was observed after 24 h in the groups treated with 300 and 500 mg/kg APAP (Fig. 1B).
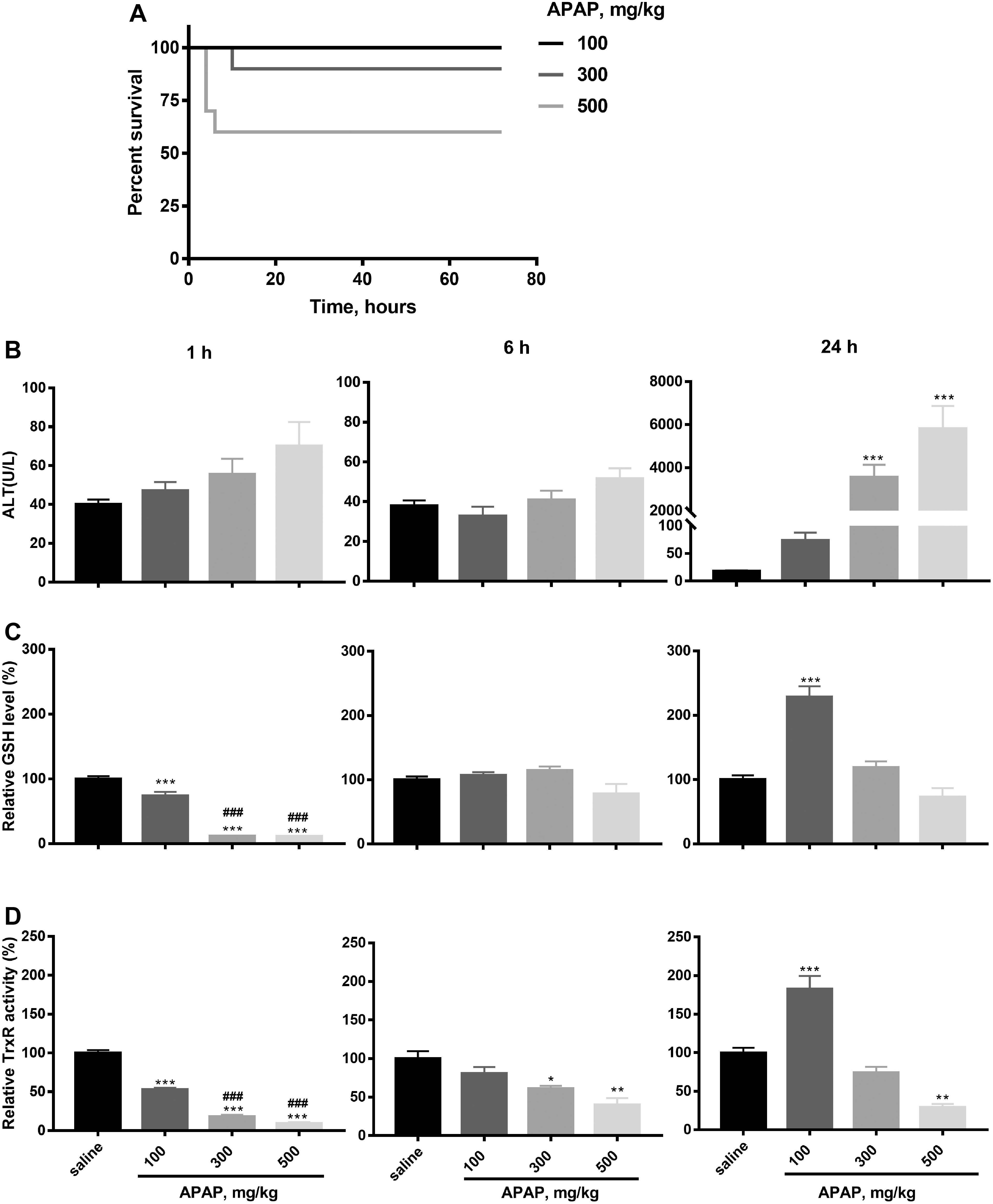
APAP treatment affected GSH levels and TrxR activities in mouse liver extracts in a dose- and time-dependent manner. Treatment with all APAP doses caused a rapid decrease in GSH levels and TrxR activity at 1 h post-treatment (Fig. 1C, D), with GSH levels showing significant recovery at 6 and 24 h post-treatment. It is notable that both TrxR activity and GSH levels were significantly enhanced compared with controls at 24 h post-treatment in the 100 mg/kg group, but not in the 300 and 500 mg/kg groups (Fig. 1C, D). Liver GSH levels had returned to normal levels after 6 h at all doses and were also at normal levels in the 300 and 500 mg/kg groups at 24 h, with the enhanced levels noted above in the 100 mg/kg group (Fig. 1C). Relative TrxR activity was significantly reduced in the 300 and 500 mg/kg groups after 6 h, and TrxR activity was still significantly reduced in the 500 mg/kg group compared with saline controls after 24 h, showing long-lived effects at higher APAP doses (Fig. 1D). We therefore speculated that treatment of mice with low APAP doses activates a compensatory mechanism in the liver resulting in increased TrxR activity and GSH levels at 24 h post-treatment. In dead mice, treatment with 500 mg/kg APAP caused a dramatic decrease in both TrxR activity and GSH levels after 24 h.
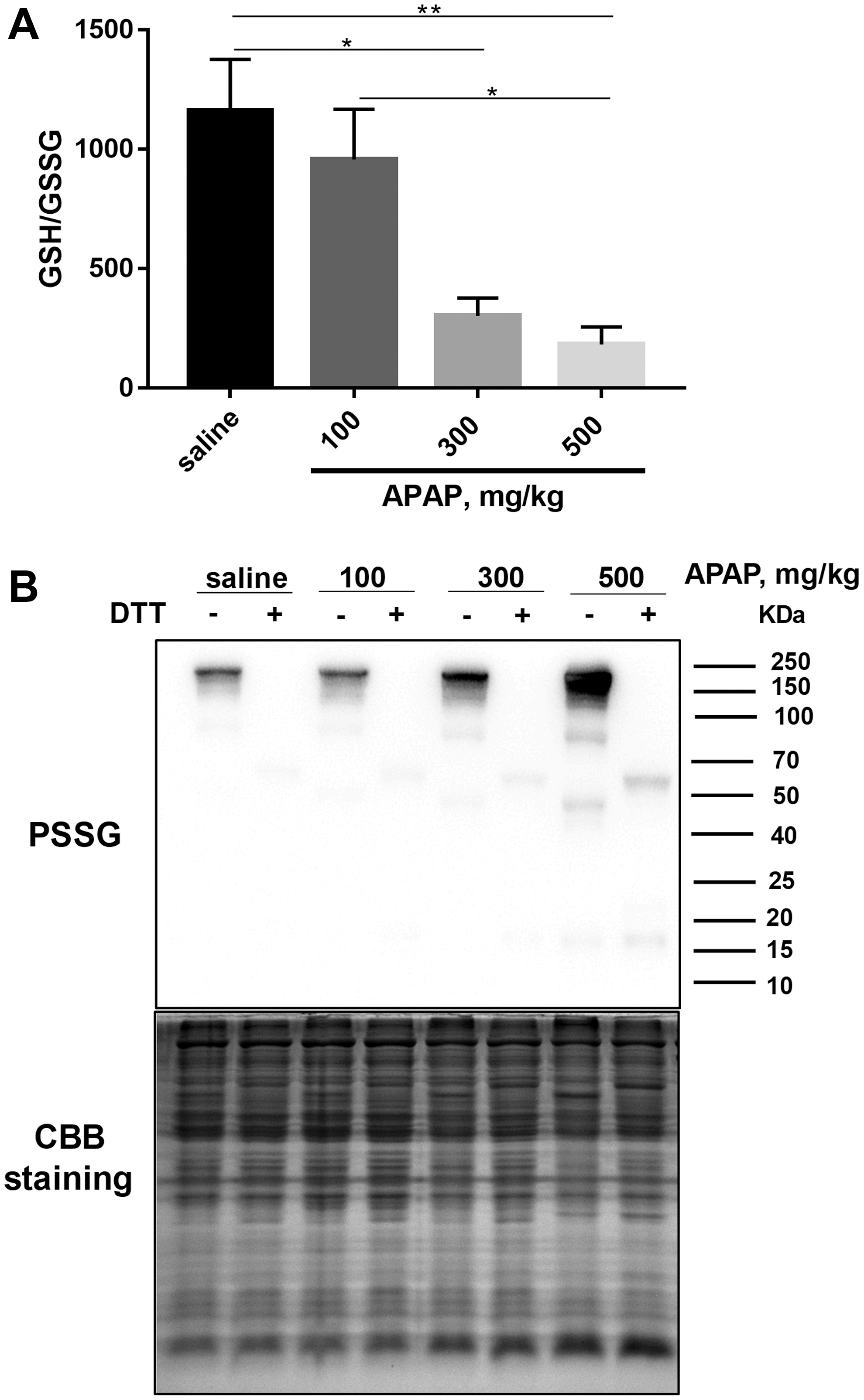
Although total GSH levels in all groups recovered after 24 h, the ratio of GSH/glutathione disulfide form (GSSG) was lower in the livers of mice treated with 300 and 500 mg/kg APAP compared with the control group treated with saline (Fig. 2A), indicating higher oxidative stress levels in these two groups. Under oxidative stress conditions, glutathionylation/deglutathionylation of proteins catalyzed by glutaredoxin is a marker of cellular redox environment (57). Therefore, we investigated the protein glutathionylation level in liver samples and found a dose-dependent increase 24 h after APAP exposure (Fig. 2B), indicating an increase of cellular oxidative stress. We also analyzed cytosolic and mitochondrial Trx protein expression in the livers of mice treated with APAP after 24 h. There was no significant difference in the levels of Trx2, TrxR2, peroxiredoxin (Prx)1, and Prx2 between these groups, but a significant upregulation of TrxR1 expression was observed in the 300 mg/kg (p = <0.01) and 500 mg/kg (p = <0.05) groups and significant (p = <0.05) decrease in Trx1 and Prx3 expression was observed in the 500 mg/kg APAP group (Supplementary Fig. S1).
Effects of selenium status on APAP-induced liver injury
The above results showed that APAP treatment caused a dramatic change in TrxR activity and GSH levels. Since selenoproteins TrxRs and GPxs are the major components in the Trx and GSH antioxidant systems, an understanding is required of the roles of the Trx and GSH systems in mediating the redox environment in APAP-induced liver injury. Consequently, the effects of selenium status on APAP-induced liver injury were investigated. Mild SD and selenium-enriched (SE) mouse models were established by feeding the weanling mice with mild SD or SE diets for 12 weeks.
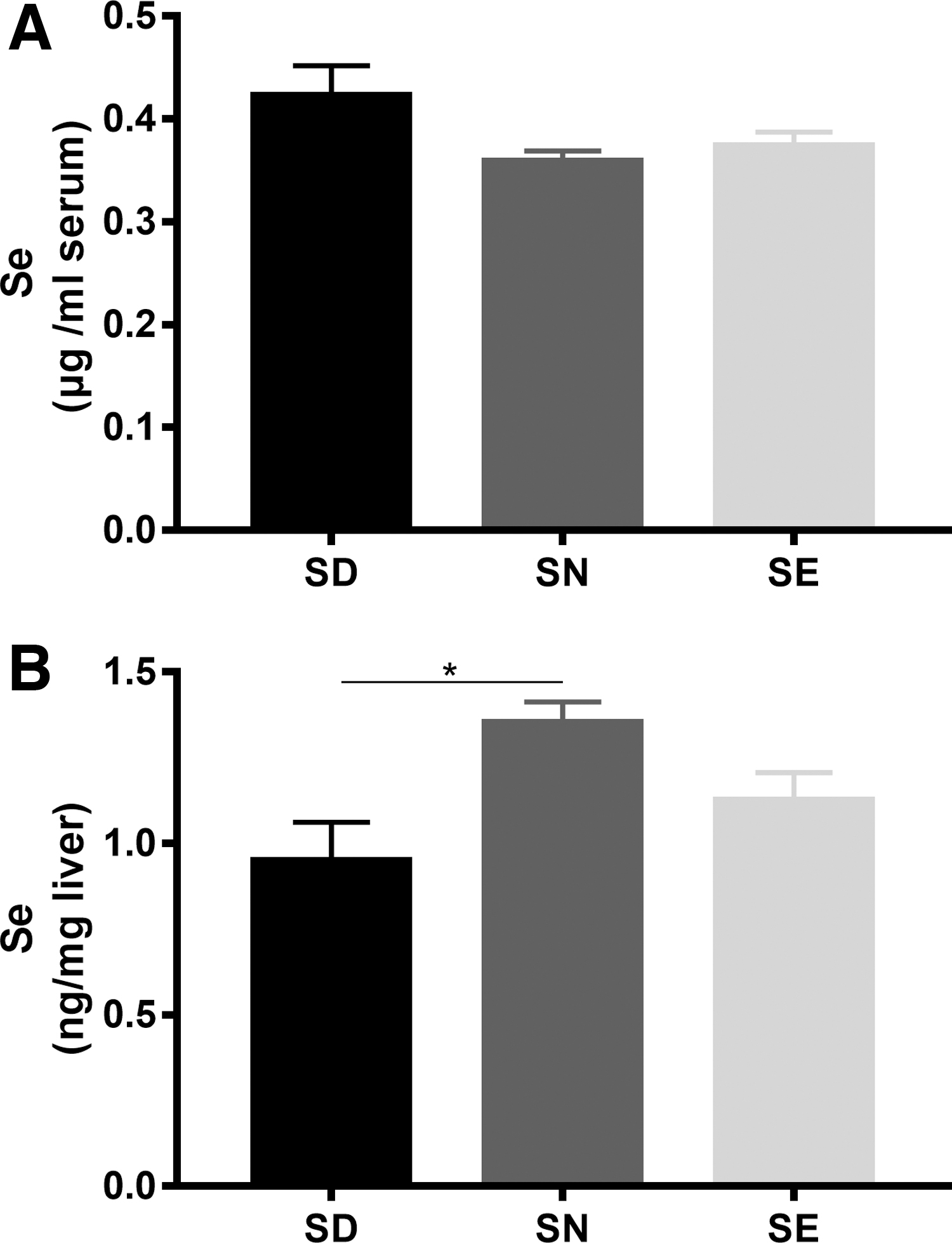
There was no significant difference in the selenium levels in serum between groups (Fig. 3A), but liver selenium level in mice fed with a mild SD diet was significantly (p = <0.05) lower than that in mice fed with selenium-normal (SN) diets (Fig. 3B). Subsequently, mice fed with these diets were treated with 300 mg/kg APAP. This resulted in a significant increase in aspartate transaminase (AST) and ALT levels in all the groups (Supplementary Fig. S2). Compared with the group fed with a diet containing normal selenium levels, the two groups of mice fed with SD and SE diets showed higher sensitivity to APAP at 24 h after treatment, as revealed by histological analysis and survival rates (Fig. 4). Treatment with 300 mg/kg APAP did not cause death in mice fed with a SN diet (Fig. 4A).
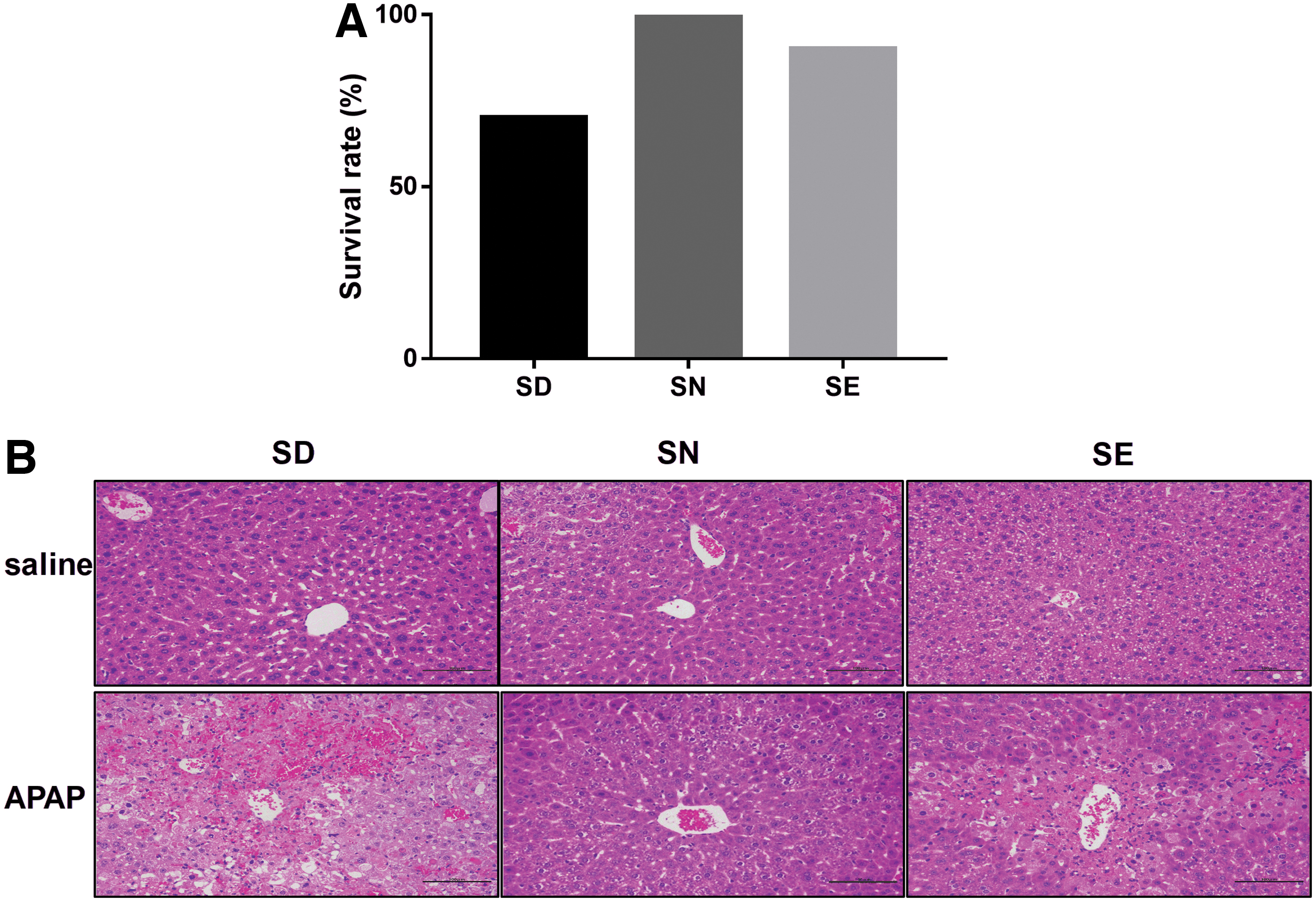
In contrast, feeding mice with a SD or SE diet predisposed them to increased liver damage. Three and one mice died upon the treatment with 300 mg/kg APAP in the SD and SE groups, respectively. Liver damage levels were assessed by hematoxylin and eosin (HE) staining and semiquantified using scores on pathological sections. The scores range from 0 to 4 with increasing values indicating higher levels of liver damage. Results revealed that mildly SD mice were the most sensitive to APAP-induced liver injury (Supplementary Table S1), and displayed disappearing hepatic sinusoids and disorganized hepatocytes upon histological analysis. Excess selenium in the diet did not result in protection of the liver from damage caused by APAP treatment, but instead enhanced liver damage (Fig. 4A, B).
Correlation of TrxR activity with mice susceptibility to APAP
To further explore the relationship between dietary selenium, TrxR activity, and APAP-induced liver damage, we measured TrxR activity and GSH levels in the livers of surviving mice. Changes in liver TrxR activity appeared to be dependent on the selenium dose when exposed to APAP. Selenium deficiency decreased liver TrxR activity (Fig. 5A) in proportion with the decrease in the amount of selenium within the liver (Fig. 3), but had little effect on GSH levels in whole cells (Fig. 5B). Interestingly, mitochondrial TrxR activity did not differ among the three groups (Fig. 5C). At 24 h post-treatment with APAP, total TrxR activity had decreased significantly in both SD and SE groups, which was significantly different from the result obtained for the SN group. Furthermore, compared with the SD group, TrxR activity and GSH levels in SN and selenium-rich mice were higher (Fig. 5A, B), which correlated with the extent of liver damage as detected by HE staining (Fig. 4B).
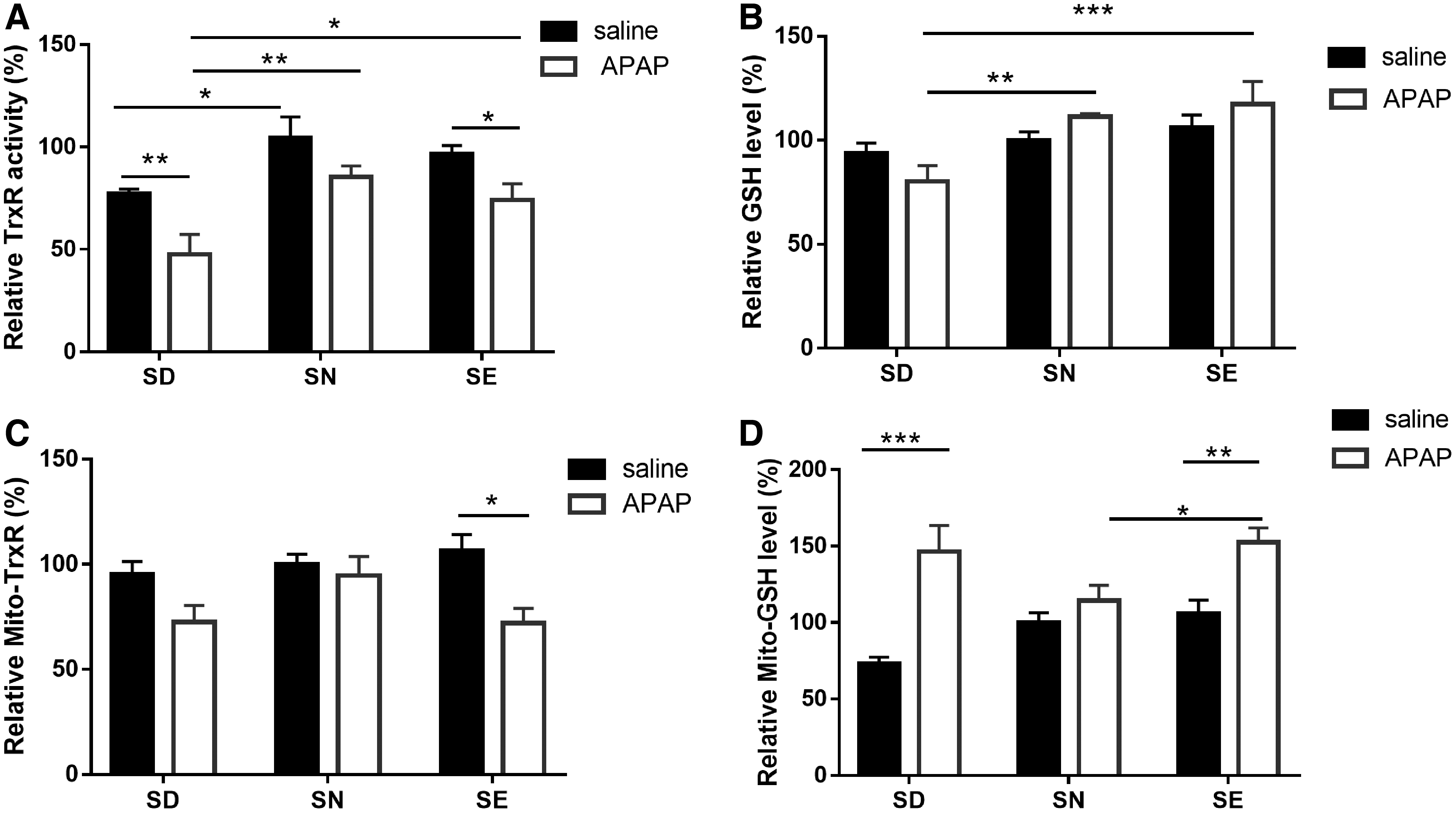
Surprisingly, treatment with APAP increased mitochondrial GSH amount in SD and SE groups, thus making it higher than that in the SN group. However, mitochondrial TrxR activity was only decreased in SE mice after APAP treatment. This result is consistent with a model in which the reduction of overall TrxR activity (Fig. 1D) can be primarily attributed to the decrease in cytosolic TrxR1 activity. TrxR1 and TrxR2 display differences in substrate- and inhibitor specificity, although both selenoenzymes are generally thought to be similar in structure and properties (42). Indeed, cytosolic TrxR1 reacts more rapidly with NAPQI than mitochondrial TrxR2 as judged by the observation that maximum inhibition of TrxR1 activity occurred after 1 h, while maximum inhibition of TrxR2 activity occurred after 6 h following APAP exposure (22).
Elevated oxidation of cytosolic and mitochondrial Trx system in both SD and SE mice
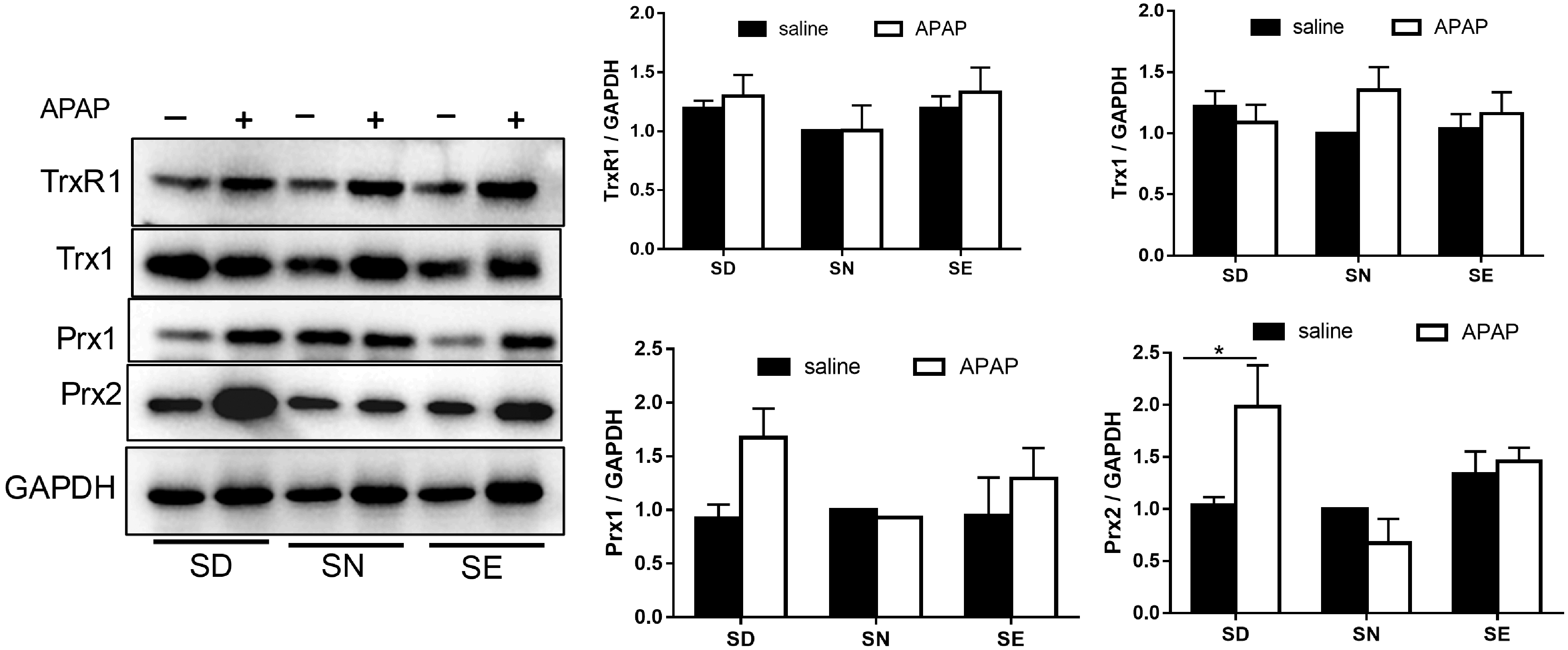
The cytosolic Trx system is involved in ROS scavenging by transferring electrons to Prx1/2. Therefore, decreased TrxR activity could cause ROS accumulation in SD mice, consequently increasing susceptibility to oxidative stress. To test this hypothesis, we detected the expression levels of various enzymes by Western blotting (Fig. 6). Results showed no significant change in Trx1, TrxR1, or Prx1 levels between the three groups with or without APAP treatment. In contrast, Prx2 protein levels were significantly increased in SD group after APAP challenge (Fig. 6).
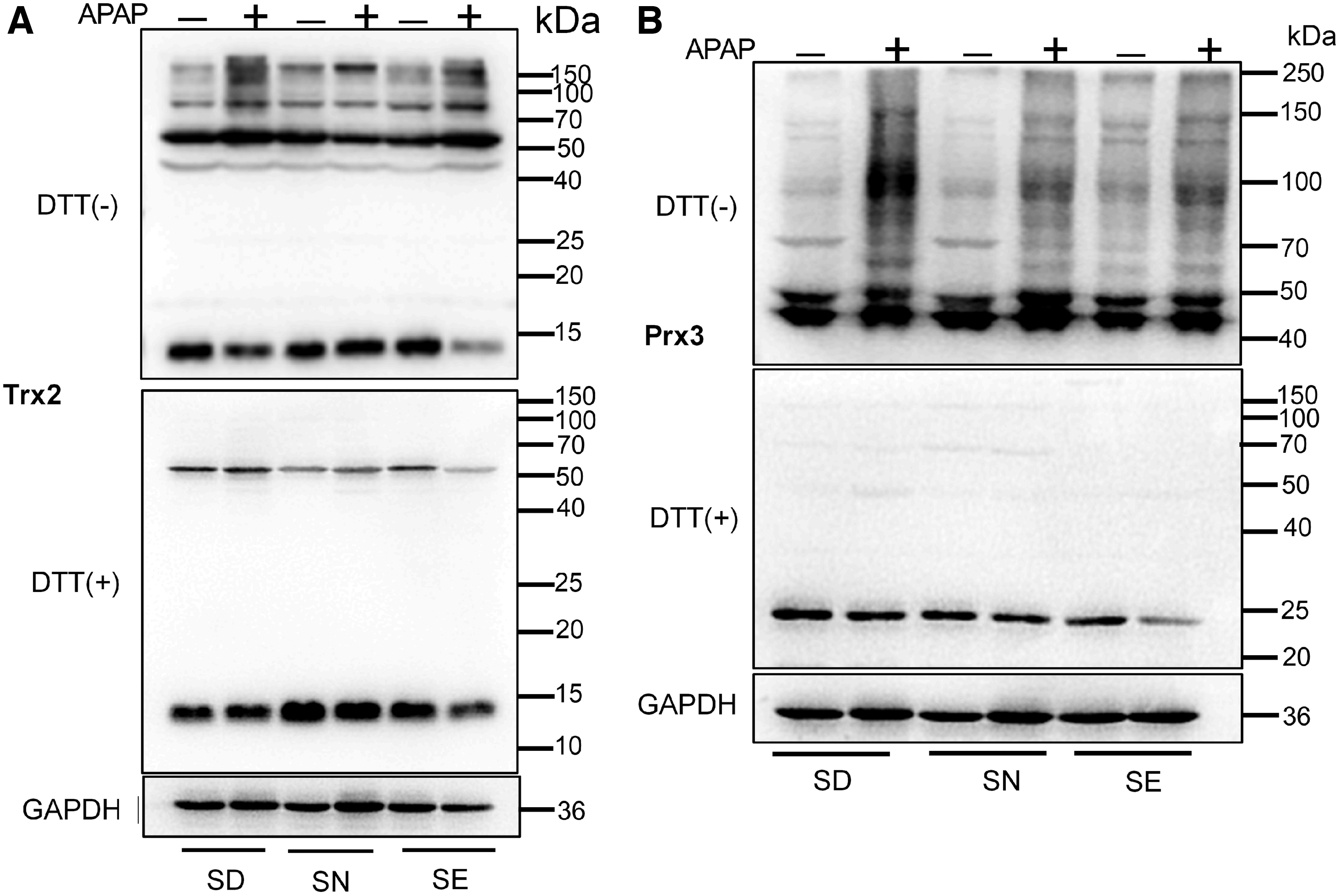
Furthermore, the redox states of Trx1and Prx1/2 were investigated by redox Western blotting after the fixing of free thiols in tissue lysates with iodoacetamide (IAM) (Fig. 7). In nonreducing gels, thiols in the sulfhydral form of these redoxins (Trx1, Prx1/2) are alkylated by IAM, and hence proteins appear as monomers in lower molecular weight positions. When the redoxins are oxidized, they form disulfides with other proteins or themselves, thus appearing at higher molecular weight positions or as fuzzy bands. Addition of dithiothreitol (DTT) to samples before analysis converts the disulfide form of these proteins into the monomeric sulfhydral forms.
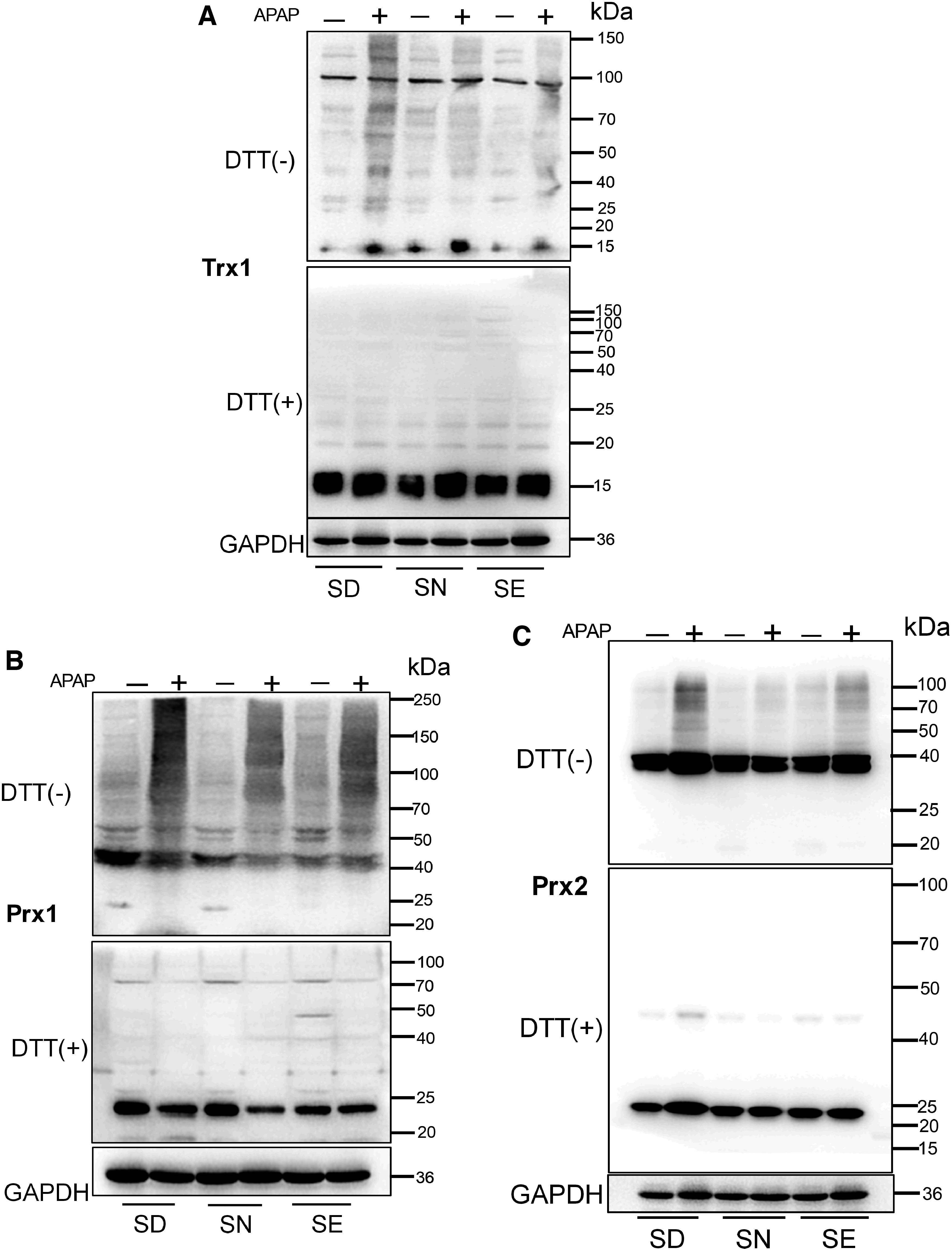
Compared with the group fed with a SN diet, SD and SE groups did not have a dramatic change in the redox status of mice liver redoxins, indicating that the majority of upstream TrxR1 was working correctly and the cellular redox environment was well controlled. Treatment with APAP resulted in oxidation of redoxins in all groups. Compared with the group fed with normal selenium diet, SD and SE groups had more oxidized redoxins, indicating that the Trx-mediated redox environment was more severely disrupted in these groups, particularly in the SD group (Fig. 7). This was consistent with the liver histology results, which showed that APAP induced the severest liver damage in SD mice.
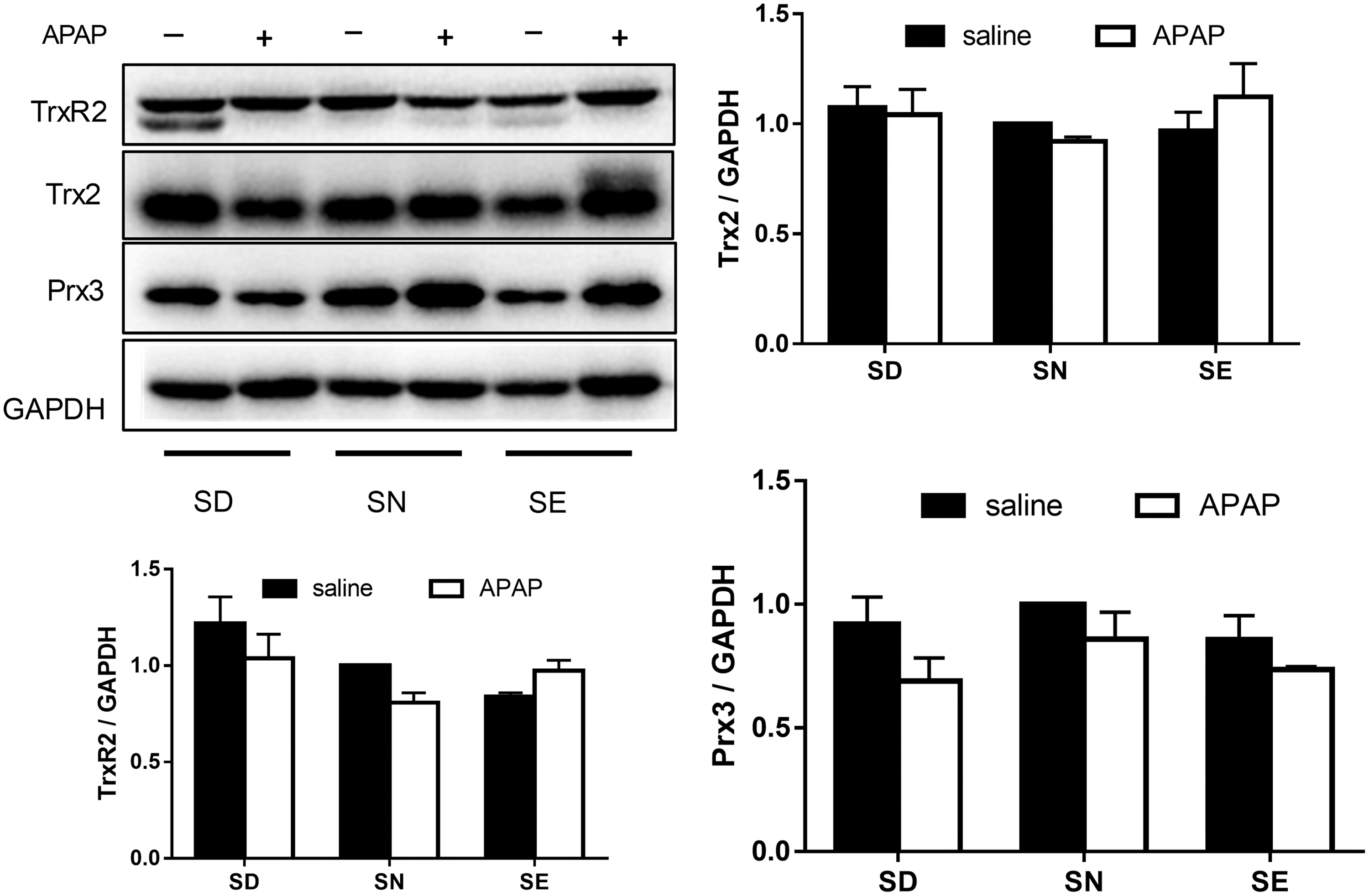
Accumulating evidence demonstrated the correlation between APAP-induced hepatotoxicity and the reaction of toxic metabolite NAPQI with mitochondrial proteins causing mitochondria dysfunction (44, 49). Thus, we investigated the expression of TrxR2, Trx2, and Prx3, which are primarily present in mitochondria. There were no significant changes in the levels of these proteins between these three groups (Fig. 8). Furthermore, we examined their redox states after treatment with APAP. Treatment with APAP caused oxidation of Trx2 and Prx3 in all groups. Compared with the SN group, more Trx2 and Prx3 were oxidized in the SD and SE groups (Fig. 9), particularly in the SD group. This result was similar to the observation made on cytosolic proteins (Figs. 6 and 7). The above results demonstrated that APAP treatment affects redox status of Trx system in both the cytosol and mitochondria. Selenium deficiency or excess supplementation did not induce a dramatic change in the redox environment but disrupted the redox environment upon APAP treatment.
Discussion
Selenium functioning as a component of selenoproteins plays a critical role in human health and disease (6, 38, 39). Thus, its deficiency can contribute to the progression of various diseases, notably Keshan disease, which is prevalent in SD regions in China. In addition, selenium deficiency can sensitize cells to oxidative stress (6, 38), an effect that is also induced by APAP toxicity. It is reasonable to postulate decreased antioxidant capacity in SD mice based on the fact that selenium deficiency could lead to decreased activity of some selenoproteins, such as the important antioxidant enzymes TrxRs and GPxs. Interestingly, selenium deficiency caused decreased TrxR activity, while selenium excess did not result in TrxR activity elevation. Selenium supplementation increased TrxR activity compared with low amounts but plateaued at normal levels. Thus, it is reasonable that we did not detect increased TrxR activity in the livers of mice fed on a SE diet.
Conversely, a more severe liver injury occurred in SE mice compared SN mice. Selenium compounds, typically sodium selenite (5, 13), at a high concentration have been used to kill tumor cells. Selenite is reduced by nicotinamide adenine dinucleotide phosphate (NADPH) and TrxR in mammals to produce hydrogen selenide, which has high reactivity with oxygen resulting in the generation of ROS. Selenomethionine is the major selenium component of the diet in this study. It is metabolized in a different way but ultimately produces the metabolite hydrogen selenide, with consequent increases in oxidative stress (29). Increased ROS levels in SE mice without APAP treatment were indicated by the higher basal oxidation of Prx1 and Prx3 compared with SN mice. Consequently, SE mice showed an increased sensitivity to APAP-induced liver injury.
Many factors including fasting, alcohol, and malnutrition may affect APAP-induced liver injury, but the effects of dietary selenium status on APAP toxicity have been rarely studied (54). This study demonstrates that both selenium deficiency and excess aggravate APAP-induced hepatotoxicity via amplification of the oxidative stress induced by APAP, as evidenced by the higher oxidation status of redoxins in SD and SE mice.
The redox state of proteins in the cultured cells has been well studied, and has been shown to be an important factor in determining enzyme activity and the biological response. For example, the redox state of Trx1 is a marker of the cellular redox environment and determines cell death or survival, with increased Trx1 oxidation resulting in more cell death (31). However, the redox state of proteins in the tissues is rarely investigated. In this study, we examined the redox status of redoxins in liver tissue using redox Western blotting by detecting the intermolecular disulfides by fixing protein-free thiols with IAM. The possibility that selenium deficiency induced loss of antioxidant enzymes (GPx and TrxR) activity, contributing to a decrease of Trx system-mediated antioxidant pathways activity, was investigated.
Thus, the higher sensitivity of SD mice to APAP exposure explains the different responses of mice to APAP challenge. Although nonselenoprotein antioxidant enzymes such as heme oxygenase-1 and NAD(P)H quinone oxidoreductase are upregulated in SD mice by activation of Nrf2, this oxidation state is maintained at a high level, which makes the liver of mice raised on SE diets more likely to reach the toxic threshold when exposed to the same dose of APAP (7).
In this study, we found that selenium status affected hepatotoxicity induced by APAP and SD mice showed the most severe liver damage, as judged by histological analysis. However, serum transaminases activity in SD and SE groups did not increase significantly compared with the SN group. This inconsistency may be explained by the results of Jan et al. (22). In the early stages of APAP challenge, TrxR1 and TrxR2 activities are efficiently inhibited with return-to-normal levels or above by 24 h post-treatment. Serum transaminase activity kept increasing up to 18 h while at 24 h post-APAP treatment both AST and ALT activities had decreased, but histological analysis indicated increased liver injury at 24 h.
Based on these results, we speculate that Nrf2 was activated to enhance antioxidant systems activity and decrease necrotic cell death in the latter stages of APAP-induced hepatotoxicity, but these compensating effects did not halt the progress of liver damage. In response to oxidative stress induced by exogenous stimulus, Nrf2 translocates from the cytosol to the nucleus to upregulate the transcription of redox-related genes such as Txnrd1, Hmox1, and Nqo1. This adaptive activation of Nrf2 may protect cells from death and maintain cellular functions (15). Prx2 was previously shown to have antioxidative activity and scavenge ROS (62). A second study showed that Prx2 cooperated with Nrf2 to defend against oxidative stress, which is of importance for cell survival (34). Therefore, it is not surprising that Prx2 expression largely upregulated in SD mice when exposed to APAP.
Conclusion
Our results demonstrate that mild SD mice are vulnerable to APAP-induced hepatotoxicity. Furthermore, oxidation of both the mitochondrial and cytosolic Trx systems is involved in this process. This indicates an overall increase in oxidative stress in the liver of mice fed on both SD and SE diets after exposure to APAP. Taken together, we conclude that selenium deficiency lowered the activity of selenoenzyme-containing antioxidant systems, thus disrupting the intracellular redox balance. Decreased TrxR activity resulting from selenium deficiency was further exacerbated by APAP metabolite NAPQI, which contributed to a more extreme disruption of the redox environment.
Selenium exerts its beneficial or toxic effects largely depending on its dose (23). High or low selenium levels may decrease the capability of liver cells to defend against oxidative stress induced by exogenous stimuli. However, there is a rather limited dose range for the beneficial biological functions of selenium because of the narrow therapeutic window between selenium deficiency and selenium overdose (10, 56). This emphasizes the importance of safe administration of selenium.
Materials and Methods
Animals were obtained from Ensiwerer Company (Chongqing, China), and RIPA lysis buffer was obtained from Beyotime Biotechnology (P0013B, Shanghai, China). SD (0.09 mg/kg), SN (0.2 mg/kg), and SE (1 mg/kg) diets were manufactured by Medicience Company (Jiangsu, China). APAP (11969B) and IAM (24054A) were purchased from Adamas-Beta (Shanghai, China). Omni-PAGE (polyacrylamide gel electrophoresis) gels were purchased from EpiZyme (Shanghai, China). All the antibodies used in this study were from Proteintech (Hubei, China) except anti-Trx1, anti-TrxR1, anti-Trx2, and anti-TrxR2 antibodies, which were from IMCO (Stockholm, Sweden). 5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB, BS038) and NADPH (MG025) were from Biosharp (Anhui, China). Glutathione reductase (GR, G8810) was purchased from Solarbio (Beijing, China). Aurothioglucose (ATG, 32672321) was purchased from Wako (Osaka, Japan). Sep-Pak C18 cartridges (WAT023590) were purchased from Waters (Massachusetts). All other reagents were obtained from the Sigma-Aldrich Chemical Co. or Fisher Scientific Ltd. and were used without purification.
Animal treatments
The mice experiments carried out in this study were approved by the local Animal Ethics Committee. To investigate the susceptibility of KM mice to APAP, mice were exposed to 100, 300, or 500 mg/kg APAP and survival recorded for 3 days. To explore the effects of dose and time on APAP-induced hepatotoxicity, mice fed with a normal selenium level diet were administered 100, 300, or 500 mg/kg APAP by intragastric injection, and sacrificed at either 1, 6, or 24 h postadministration for further analysis.
In the experiment to investigate the effects of dietary selenium status on APAP hepatotoxicity, 3-week-old weanling KM mice were divided into three groups including mild SD, SN, and SE group (20 mice per group) and fed with enough water and diets with the different levels of selenium (SD: 0.09 mg/kg, SN: 0.2 mg/kg, and SE: 1 mg/kg), with light for 12 h and dark for 12 h. In this study, mild selenium deficiency was used to mimic a more realistic situation. The food and water consumed were recorded per 2 days. After 3 months, 20 mice in each group fed with different selenium level diets were divided into 2 groups (10 mice per group) and treated with 300 mg/kg APAP or saline, respectively, and sacrificed after 24 h. The serum was collected for AST and ALT activity detection, and liver tissue for enzyme activity measurements and Western blot analysis.
Detection of serum ALT and AST activity
ALT and AST activities in serum were detected using automatic biochemistry analyzer (Biobase, BK400) following the standard procedure set by manufacturer after 10-fold dilution with saline. The activity values were recorded and calculated to get the activity before dilution.
Liver histology
Liver tissues were cut into several pieces; one was kept in 4% (w/v) paraformaldehyde for HE staining. In brief, after dehydration, embedding, slicing, and dewaxing, liver tissues were processed to nuclear staining with hematoxylin and cytosolic staining with eosin to observe liver cellular morphology.
GSH/GSSG detection
The ratio of GSH/GSSG in liver was detected using the method described by Calabrese and colleagues (1, 35) with minor modifications. About 10 mg fresh liver tissue was lysed using an autohomogenizer in 1 mL of solution containing 10 mM DTNB (dissolved in buffer 1 containing 100 mM phosphate-buffered saline (PBS) and 5 mM EDTA, pH 7.5) or 10 mM NEM (dissolved in buffer 2 containing 100 mM PBS and 5 mM EDTA, pH 6.8). After centrifugation at 12,000 g, 10 min at 4°C, the protein concentration of supernatant was measured by Bradford assay using bovine serum albumin (BSA) standard protein solution (2 mg/mL) as reference. For GSSG detection, the sample containing NEM (N-ethylmaleimide) was passed through the Sep-Pak C18 cartridge to remove NEM and eluted with buffer 1. The 50 μL collected sample was added to a cuvette containing 50 μL mixture of 50 nmol DTNB and 0.1 U GR in buffer 1. After 1 min incubation, 44 nmol NADPH in 100 μL buffer 1 was added to initiate the reaction. The change of absorbance at 412 nm was recorded using a microplate reader (Bio Tek) for 5 min at 1 min intervals. After 50-fold dilution, the sample with DTNB was processed using the same procedure as that for GSSG detection to measure the total GSH level. The GSSG and total GSH amounts were calculated using a standard curve, which was prepared according to the same procedure for sample detection.
Protein extraction for redox state detection
The method of redox state analysis of target protein was modified from the method to detect the protein redox state in the cultured cells (45). In brief, 100 mg fresh liver tissue was rinsed with cold saline twice, then cut into small pieces and moved into 1 mL radio immunoprecipitation assay (RIPA) lysis buffer containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and 10 mM IAM dissolved in 1 M Tris-HCl buffer (pH 8.3). Homogenization was processed using electric homogenizer (Servicebio, China) with the setting of 120 Hz for 2 min in precold sample mould. Cell extract was centrifuged at 4°C, 13,000 g for 10 min, and supernatant was collected. Protein concentration was determined by bicinchoninic acid (BCA) assay using BSA as reference, and lysis was diluted to certain concentration with saline for redox state analysis. The samples for protein expression analysis were prepared using a similar method but without IAM addition.
Mitochondria isolation
Mitochondria were isolated with classical gradient centrifugation method with small modification (63). In brief, 100 mg liver tissue rinsed was cut into small pieces in plate and washed with cold saline twice, then removed to glass homogenizer. One milliliter mitochondria isolation buffer (containing 210 mM mannitol, 70 mM sucrose, 1 mM EDTA, 10 mM Hepes–NaOH, pH 7.5, and 1 mM PMSF) was added immediately before use, followed by homogenizing with 10 strokes to disrupt cells. The lysis was centrifuged at 4°C, 1000 g for 10 min to remove cell debris, and supernatant was collected for further mitochondria isolation. Mitochondria were obtained by centrifuging at 13,000 g, 4°C for 10 min, and were subsequently resuspended in 200 μL lysis buffer. BCA assay was used to measure mitochondrial protein concentration, and mitochondrial GSH content and TrxR activity were detected.
TrxR activity detection
TrxR activities in liver whole cell and mitochondria extracts were detected using a modified DTNB assay (3). In brief, 25 μg protein was incubated with reaction mixture containing 100 μM NADPH for 2 min at room temperature, then 300 nM ATG was added in reference wells to inhibit TrxR activity. After incubation for another 5 min at room temperature, the mixture of 100 μM NADPH and 2 mM DTNB was added to start the DTNB reduction reaction. The increase of absorbance at 412 nm was recorded by microplate reader (Bio Tek) for 5 min at 51 s intervals. Relative TrxR activity was represented by the increase of absorbance at 412 nm in 1 min, and TrxR activity of sample was calculated by subtracting the value of reference wells from sample wells. TrxR activity of each sample was obtained from the average of triplicate.
GSH amount detection
GSH contents in liver whole cell and mitochondrial extracts were measured with DTNB assay described above. 5 μg liver protein was incubated with reaction mixture containing 100 μM NADPH and 50 nM GR for 5 min at room temperature in sample wells, while the GR was absent in reference wells. After addition of mixture containing 100 μM NADPH and 2 mM DTNB, the increase of absorbance at 412 nm was immediately recorded by microplate reader for 5 min at 51 s intervals. Relative GSH content was showed with slope, which represented the increased value of absorbance at 412 nm in 1 min. Similarly, the value of reference wells was subtracted from sample wells. All samples were measured in triplicate.
Western blot analysis
Lysis buffer was supplemented with 10 mM IAM before addition to cells and homogenization for analysis of the redox state of target proteins. Lysate was diluted with loading buffer without reductant and loaded onto 15% Omni-PAGE gel purchased from EpiZyme, then proteins were separated at 120 V for 60 min. After that, proteins were transferred to polyvinylidene fluoride membrane in Tris-glycine buffer in 20% (v/v) methanol. For protein expression levels analysis, samples without IAM were processed using a similar procedure but 50 mM DTT was added to the loading buffer. After membrane blocking with 10% skim milk solution for 2 h at room temperature, anti-Prx1 (1:1000 dilution), anti-Prx2 (1:1000 dilution), anti-Prx3 (1:5000 dilution), anti-Trx1 (1:1000 dilution), anti-TrxR1 (1:1000 dilution), anti-Trx2 (1:1000 dilution), anti-TrxR2 (1:1000 dilution), and anti-GAPDH (1:10,000 dilution) antibodies were incubated with membrane for 1 h at room temperature. Subsequent incubation with secondary antibody (horseradish peroxidase-conjugated affinipure goat antirabbit IgG[H+L], 1:5000 dilution) for 2 h at room temperature was performed. Finally, chemiluminescence was measured using the appropriate apparatus (Clinx, China).
Data analysis
Data were shown as mean ± standard error of the mean. Data were first processed to outlier analysis using SPSS, then to column statistics and test for homogeneity of variance by GraphPad Prism 7.0 software. The difference among groups was analyzed by one-way analysis of variance (ANOVA) or two-way ANOVA, and the difference between two groups was analyzed by subsequent post hoc test. p < 0.05 indicated the significant difference statistically.
Footnotes
Acknowledgments
We would like to notice the passing away of our coauthor, colleague, and collaborator Arne Holmgren, a redox pioneer in research on thioredoxin and glutaredoxin. Arne will be greatly missed by all who had the privilege of working with him.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
We are grateful for the support of the Hundred Talents Plan of Chongqing and Southwest University “Gathering Talent Project” (SWU116068), Natural Science Foundation of Chongqing (cstc2018jcyjAX0401), and Chongqing Innovation & Entrepreneurship Program for Overseas Returnee (cx2018083).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.