
Abstract
Significance:
Most brains affected by neurodegenerative diseases manifest mitochondrial dysfunction as well as elevated production of reactive oxygen species and reactive nitrogen species (RNS), contributing to synapse loss and neuronal injury.
Recent Advances:
Excessive production of RNS triggers nitric oxide (NO)-mediated post-translational modifications of proteins, such as S-nitrosylation of cysteine residues and nitration of tyrosine residues. Proteins thus affected impair mitochondrial metabolism, mitochondrial dynamics, and mitophagy in the nervous system.
Critical Issues:
Identification and better characterization of underlying molecular mechanisms for NO-mediated mitochondrial dysfunction will provide important insights into the pathogenesis of neurodegenerative disorders. In this review, we highlight recent discoveries concerning S-nitrosylation of the tricarboxylic acid cycle enzymes, mitochondrial fission GTPase dynamin-related protein 1, and mitophagy-related proteins Parkin and phosphatase and tensin homolog-induced putative kinase protein 1. We delineate signaling cascades affected by pathologically S-nitrosylated proteins that diminish mitochondrial function in neurodegenerative diseases.
Future Directions:
Further elucidation of the pathological events resulting from aberrant S-nitrosothiol or nitrotyrosine formation may lead to new therapeutic approaches to ameliorate neurodegenerative disorders.
Introduction
The incidence of neurodegenerative disorders, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia, is rising with the increasing longevity of the global population. The majority of patients suffering from neurodegenerative disorders manifest progressive impairment of cognitive, behavioral, and motor function; a hallmark of these disorders is synaptic dysfunction and loss, with eventual irreversible neuronal dropout. In AD, synaptic dysfunction may start early in presymptomatic stages and correlates with cognitive decline as the disease progresses (74, 111). Growing evidence in a variety of neurodegenerative disorders has suggested that a number of factors may contribute to synaptic loss and neuronal damage, including misfolded proteins or lack of adequate degradation of these proteins (e.g., amyloid-β [Aβ], α-synuclein, tau, and TDP-43), mitochondrial dysfunction, endoplasmic reticulum (ER) stress, excitotoxicity, and neuroinflammation. Moreover, recent studies have presented evidence that, as the disease progresses, pathologically misfolded proteins may spread throughout the nervous system via cell-to-cell transmission, propagating the toxic effects of the abnormal proteins to recipient cells, including neurons and glia (39). Although the mechanisms for these key neurodegenerative features remain unclear, increased generation of reactive oxygen species (ROS)/reactive nitrogen species (RNS) is known to precede or accompany these pathological processes, consistent with the notion that oxidative and nitrosative stress represents an important contributor to pathogenesis (4, 43).
Along these lines, we and others have mounted evidence that elevated production of RNS, including nitric oxide (NO)-related species, engenders aberrant post-translational modifications on specific cysteine residues (i.e., protein S-nitrosylation), contributing to pathological disturbances in neural networks, synaptic function, and neuronal survival (81, 82). Analogous to other post-translational modifications, S-nitrosylation can affect a protein's enzymatic activity, cause conformational changes (including protein aggregation), alter protein–protein interactions, and influence protein localization, thus acting as an important signaling mechanism under both physiological and pathological conditions. In distinction to other post-translational modifications such as phosphorylation, protein S-nitrosylation can occur both intracellularly and extracellularly. Moreover, at low levels of RNS, protein S-nitrosylation represents a normal signaling process. For example, basal levels of NO support normal mitochondrial function in part via S-nitrosylation of transcription factors, such as CREB and p53 (8, 121), driving the expression of proliferator-activated receptor gamma coactivator 1α that promotes mitochondrial biogenesis. In contrast, at high levels of RNS, as occurs in many neurodegenerative disorders, aberrant protein S-nitrosylation involves reaction with cysteine residues that would not normally become nitrosylated and often do not have a full consensus motif of flanking amino acids that facilitate S-nitrosylation by lower levels of RNS (82, 122). Given the fact that cysteines are often located in the active site of enzymes, aberrant S-nitrosylation at these sites can seriously compromise normal function. For instance, aberrant S-nitrosylation of dynamin-related protein 1 (Drp1) and phosphatase and tensin homolog (PTEN)-induced putative kinase protein 1 (PINK1) alters their enzymatic activities, leading to abnormalities in mitochondrial bioenergetics, morphology, and quality control, as discussed in detail in subsequent sections of this review (22, 87). Additionally, another type of RNS, peroxynitrite (ONOO−), can lead to post-translational modification of tyrosine residues, known as protein nitration, and also contributes to mitochondrial dysfunction, among other disturbances (33, 51). In this review article, dedicated to the Special Forum Issue of “Mitochondrial Metabolism and Mitophagy,” we will focus on the pathological effects of NO-dependent post-translational modifications leading to mitochondrial dysfunction in neurodegenerative disorders.
There are at least two classifications of neurodegenerative diseases: rare familial cases (typically <5%–10% of all patients) and the much more common sporadic cases (>90%–95%). The familial cases are generally early onset (before age 65) and due to a mutation in a single disease-causing gene, such as α-synuclein, PINK1, or Parkin in PD, and amyloid precursor protein or presenilin 1 in AD. In contrast, sporadic forms are generally of late onset, have no familial predisposition, and likely arise from a combination of genetic risk factors (such as ApoE polymorphism) and environmental influences (e.g., agricultural chemicals, air pollution, head trauma, and lifestyle [such as disturbance of sleeping]). Importantly, from work on experimental models of neurodegeneration, exposure to many disease-linked environmental factors is known to increase ROS/RNS production and thus overcomes the brain's antioxidant capacity, resulting in oxidative/nitrosative stress (10, 108). Accordingly, in this review, we propose the hypothesis that elevated production of ROS/RNS is a critical pathogenic mediator of common sporadic cases of neurodegenerative disorders via triggering post-translational modifications on specific proteins, thus mimicking pathological manifestations of rare genetic mutations. To support this notion, we highlight classical findings and new studies with particular focus on impairment of mitochondrial metabolism and function. We show that aberrantly S-nitrosylated (or nitrated) proteins can contribute to disease pathogenesis.
RNS Generation in the Central Nervous System
In mammalian cells, three isoforms of nitric oxide synthase (NOS) predominantly generate NO, involving enzymatic conversion of
In the brain, when produced at physiological levels, RNS/ROS generally support normal neuronal signaling, function, and survival, whereas elevated production of RNS/ROS can contribute to protein misfolding, mitochondrial/bioenergetic dysfunction, ER stress, synaptic injury, and other forms of neuronal damage (82). For example, mild activation of N-methyl-
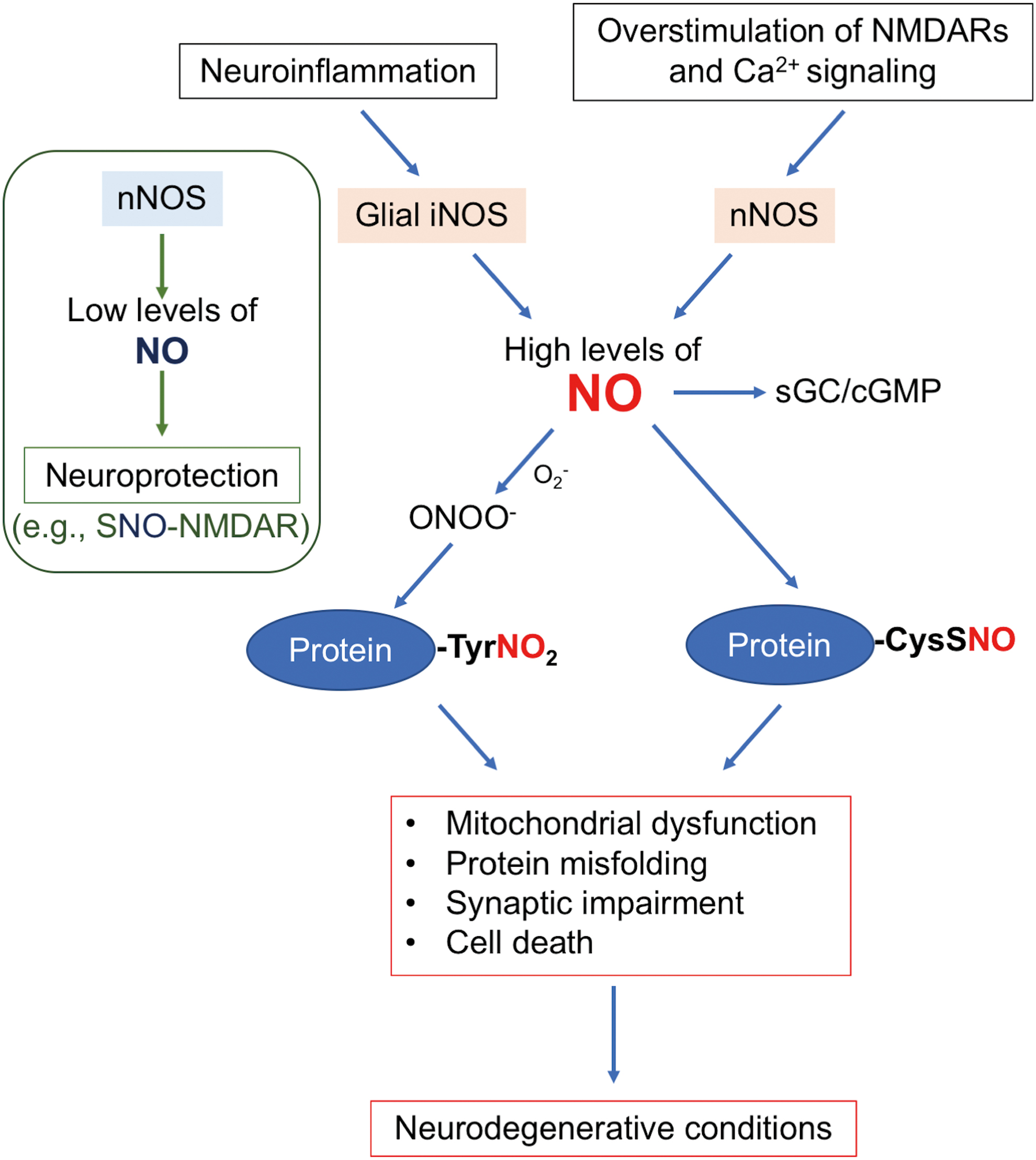
NO-Dependent Post-Translational Modifications
Protein S-nitrosylation
NO was initially demonstrated to exert its biological function via activation of guanylyl cyclase (GC), followed by the production of cyclic guanosine monophosphate (50, 78, 90). However, emerging evidence suggests that, in large part, protein S-nitrosylation mediates NO-dependent signal transduction pathways (even regulating GC activity), affecting both physiological and pathophysiological processes (33, 48, 82). S-nitrosylation is a reversible post-translational modification in which an NO group is covalently attached to a specific cysteine thiol to form an S-nitrosothiol (SNO). Mechanistically, we favor this reaction proceeding through the chemical intermediate of a nitrosonium cation (NO+, which is not present in a free state at physiological pH). This intermediate reacts with a free cysteine thiol (or more properly thiolate anion, –S−) (48, 68, 82). For this reaction to occur, a transition metal is needed for electron transfer, although oxidation may potentially take place very slowly in vitro in the absence of a transition metal. For example, copper ions may act as an electron acceptor to facilitate the transfer of [NO+] equivalents (brackets to indicate this generally represents a chemical intermediate rather than the free cation) (36). It should be noted, however, that some authorities have proposed direct thiyl radical/•NO radical reactions to mediate protein S-nitros(yl)ation (128). Moreover, S-nitrosylating enzymes (nitrosylases) and denitrosylating enzymes (denitrosylases) generally control the reactions in vivo when physiological amounts of NO are involved. Enzymatically mediated S-nitrosylation may not be required when excessive levels of NO are present that can aberrantly S-nitrosylate a protein. NO-related species engender S-nitrosylation on specific/critical thiol groups of proteins (forming SNO-proteins). Additionally, low molecular weight (LMW) compounds, such as
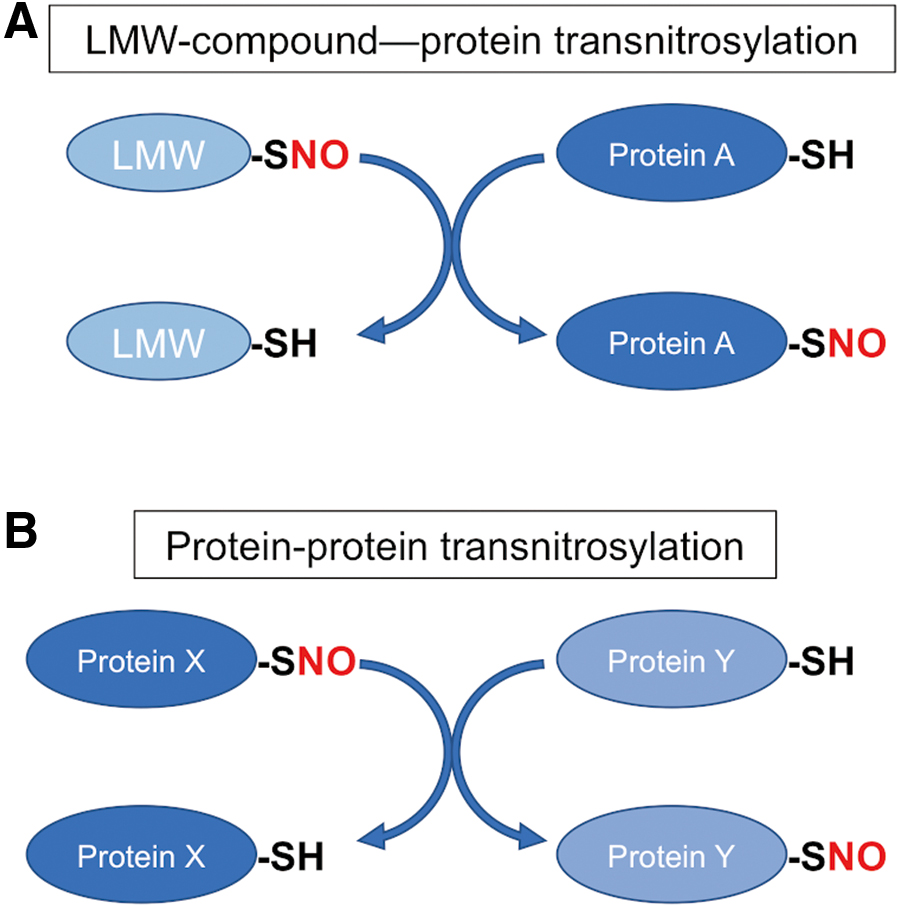
SNO (S-nitrosylation reaction) specificity
Although most cellular proteins contain multiple cysteine residues, NO groups only target a specific population of thiols to form SNO-proteins (48, 65, 67, 82, 124). Several contributing mechanisms have been implicated in governing SNO specificity: acid–base and hydrophobic motifs, proximity to the NOS complex, and protein–protein transnitrosylation. Concerning the acid–base motif, our group originally proposed that the presence of the charged acidic and basic amino acid sidechains that are located in close proximity to the target cysteine lowers its pKa to facilitate SNO formation (48, 122). Notably, later proteomics and structural studies confirmed that the acid–base SNO motif can support protein S-nitrosylation when it exists within a 6–8 Å distance from the target site in a tertiary protein structure (29, 73). In addition, a hydrophobic environment, as occurs in lipid membranes or within a particular protein structure, aids in SNO formation because hydrophobicity promotes the formation of the nitrosylating species (124).
Importantly, with excessive generation of RNS, as occurs during normal aging or in many neurodegenerative disorders, a partial SNO motif will support S-nitrosylation of a cysteine residue, which is not targeted for S-nitrosylation by physiological levels of NO-related species. Thus, elevated production of RNS often leads to aberrant formation of SNO-proteins, appearing only under disease conditions. Additionally, localization of a SNO target cysteine in a protein complex containing NOS can facilitate S-nitrosylation. In this case, high local concentrations of NO may (i) result in auto-S-nitrosylation of NOS, possibly via the transition metal pathway; or (ii) contribute to the generation of nitrosylating species that S-nitrosylate nearby proteins (82, 120, 124). For example, tethered via a PSD-domain, nNOS is located in a complex with the NMDA receptor. Calcium influx through the NMDA receptor-associated ion channel stimulates nNOS activity, resulting in S-nitrosylation of both the NMDA receptor and PSD-95 (65, 67, 82).
Also, as introduced above, accumulating evidence implies that protein–protein transnitrosylation plays a major role in cellular SNO formation (Fig. 2). In this case, the protein–protein interaction underlies precisely regulated transfer of the NO group from the donor (transnitrosylating) protein to an acceptor (denitrosylating) protein. As an example, we demonstrated that SNO-caspase-3 acts as a transnitrosylase toward XIAP, resulting in the formation of SNO-XIAP and thus inhibition of the protein's antiapoptotic activity; concurrently, denitrosylation of the active/cleaved form of caspase-3 results in its reactivation (83). Interestingly, interfering with this caspase-3–XIAP interaction, for example, by using a pro (uncleaved)-form of SNO-caspase-3 or D148A mutant XIAP, inhibited the transnitrosylation reaction, consistent with the notion that specific protein–protein interaction is required for the transnitrosylation reaction to occur.
Additionally, transnitrosylation reactions occur not only from one SNO-protein to another protein but also from LMW-SNOs to proteins (Fig. 2) (48, 82, 124). Enzymes that can mediate denitrosylation include S-nitroso-glutathione reductase (GSNOR), SNO-CoA reductase (SCoR), thioredoxin (Trx), and Trx-related protein 14 kDa (3, 13, 124). As an example, the active site cysteine of Trx can facilitate denitrosylation of target SNO-proteins, including SNO-caspases and SNO-GAPDH, leading to the liberation of an NO group and the formation of an intermolecular disulfide bond between Trx and the target protein (12, 18).
In addition, GSNOR- and SCoR-dependent metabolism of LMW-SNOs can prevent the formation of SNO-proteins due to decreased transnitrosylation from these LMW-SNOs. In contrast, GSNOR-deficient mice show greatly increased protein S-nitrosylation, producing phenotypes in part resembling advanced aging and presymptomatic processes of neurodegenerative disorders, such as protein misfolding and mitochondrial dysfunction (104). Moreover, in the kidney, decreased SCoR activity results in increased S-nitrosylation of pyruvate kinase M2, which inhibits the final steps of glycolysis. This inhibition results in a metabolic switch from glycolysis to the pentose phosphate pathway, reportedly increasing antioxidant capacity to enhance cell survival (150). Despite being expressed in the brain, the possible role of SCoR in protein S-nitrosylation in the nervous system is as yet unknown (57, 150).
Tyrosine nitration
After production from NOS, •NO can very rapidly react with another free radical, superoxide, to produce ONOO− (100). Apparent decomposition of peroxynitrite generates •NO2, which can promote tyrosine nitration with the formation of 3-nitrotyrosine, often associated with aberrant and pathological NO signaling (11, 34, 51) (Fig. 1). Chemically, tyrosine nitration is thought to be generated by a two-step, free radical-mediated process: a one-electron oxidation of tyrosine, producing tyrosyl radical, is followed by a radical−radical coupling reaction of the tyrosyl radical and •NO2, thus attaching a nitro group (–NO2) to the three-position of the phenolic ring of the tyrosine residue. Although nitration appears to favor certain tyrosine residues, the mechanism for selective tyrosine nitration remains largely unknown (34). Notably, peroxynitrite can also facilitate disulfide bond formation between vicinal cysteines, as can S-nitrosylation of one of the cysteines (68, 101). In addition, similar to other post-translational modifications such as protein S-nitrosylation, tyrosine nitration is thought to affect the conformation and activity of target proteins. For example, nitration of α-synuclein, Aβ, and Tau accelerates their aggregation and occurs in human brains with neurodegenerative disorders (38, 59, 103), consistent with the notion that the augmented NO/nitration pathways contribute to disease pathogenesis. Additionally, tyrosine nitration of α-synuclein is associated with an autophagic response in peripheral blood cells of individuals with idiopathic PD (96). Interestingly, peroxynitrite can also react with free thiol to yield the sulfenic acid (–SOH) derivative, at least in vitro (101), suggesting that peroxynitrite-mediated oxidative modifications of cysteine residues (i.e., forming –SOH, –SO2H, or –SO3H) can also occur, and this may potentially contribute to the disease process. In recent years, mitochondrial function has been reported to be influenced by increasing numbers of S-nitrosylated or nitrotyrosinated proteins [e.g., MEF2C (88, 108), Bcl-2 (6), PPARγ (119), and mitochondria-associated caspases (12, 117)]. Therefore, in the following sections, we focus on protein S-nitrosylation and tyrosine nitration that directly affect mitochondrial metabolism, morphology, and mitophagy (Fig. 3).
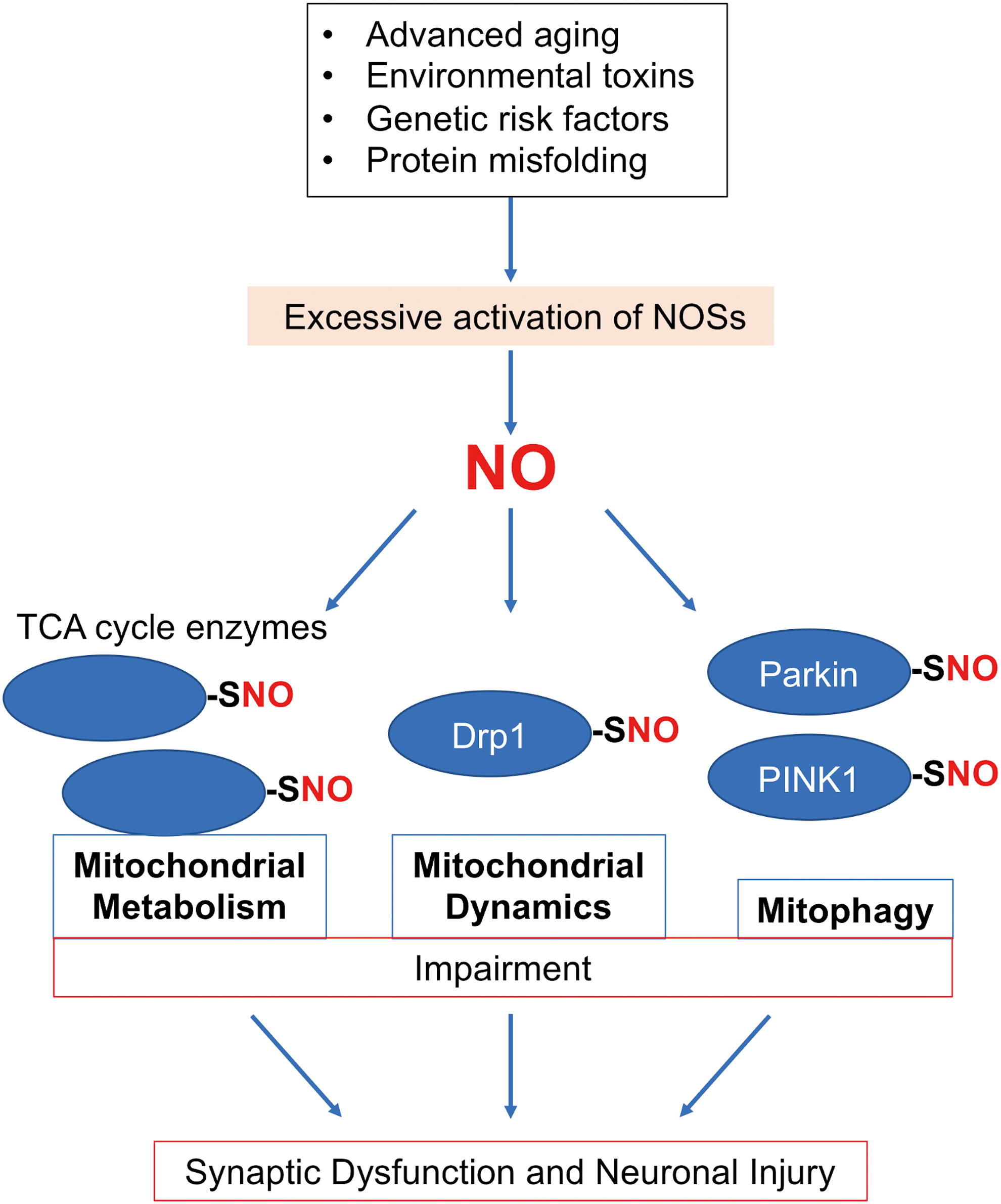
Impairment of Mitochondrial Metabolism in Neurodegenerative Diseases
Mitochondrial metabolism in the brain
Neurons demand substantial energy for the maintenance of membrane potential and synaptic function. Hence, efficient energy production from functional mitochondria represents a vital cellular event for neuronal/synaptic activity, plasticity, and survival. Additionally, mitochondria are known to regulate apoptosis and cellular calcium storage, making them an even more important organelle for neurons. Consistent with these notions, dysregulation of mitochondrial metabolism/function is linked to the pathogenesis of neurodegenerative diseases. The first evidence to support such a concept came from a human study whereby the inadvertent use (via illicit drug contamination) of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a prodrug to MPP+, interfered with mitochondrial respiration and increased ROS/RNS production in dopaminergic neurons, thereby causing parkinsonian-like symptoms in the patients (62). Subsequently, over the next several decades, numerous studies demonstrated that mitochondrial dysfunction, including not only impairment in respiration/energy (i.e., ATP) production but also mitochondrial DNA defects, abnormal Ca2+ handling, imbalance in redox homeostasis, alterations in mitochondrial morphology, and aberrant mitochondrial quality control, may contribute to and even underlie the pathogenesis of neurodegenerative conditions. For instance, our group demonstrated that PD-related environmental toxins decrease mitochondrial respiration in dopaminergic neurons derived from human induced pluripotent stem cells (hiPSCs), and the presence of genetic factors aggravate such impairment (108). Interestingly, this type of mitochondrial dysfunction resulted, at least in part, from the formation of SNO-MEF2C.
Interestingly, while adult neurons mainly rely on mitochondrial oxidative phosphorylation (OXPHOS) for ATP production, glial cells predominantly leverage glycolysis for this purpose (72). In the adult brain, glucose is the major source of energy/ATP production, although ketone bodies may be utilized during development or glucose starvation. Additionally, neuronal stimulation in adult brains can temporarily trigger neuronal glycolysis (28). Concerning glucose metabolism in the brain, the Magistretti group proposed an interesting model termed the astrocyte–neuron lactate shuttle (72, 92), whereby astrocytes generate lactate via glycolysis and export lactate to neurons for conversion into pyruvate, feeding into the tricarboxylic acid (TCA) cycle, which appears to be critical, at least under basal conditions (Fig. 4). These findings suggest that disturbances in either astrocytic glycolysis or neuronal oxidative metabolism can lead to impairment in energy metabolism in the brain.
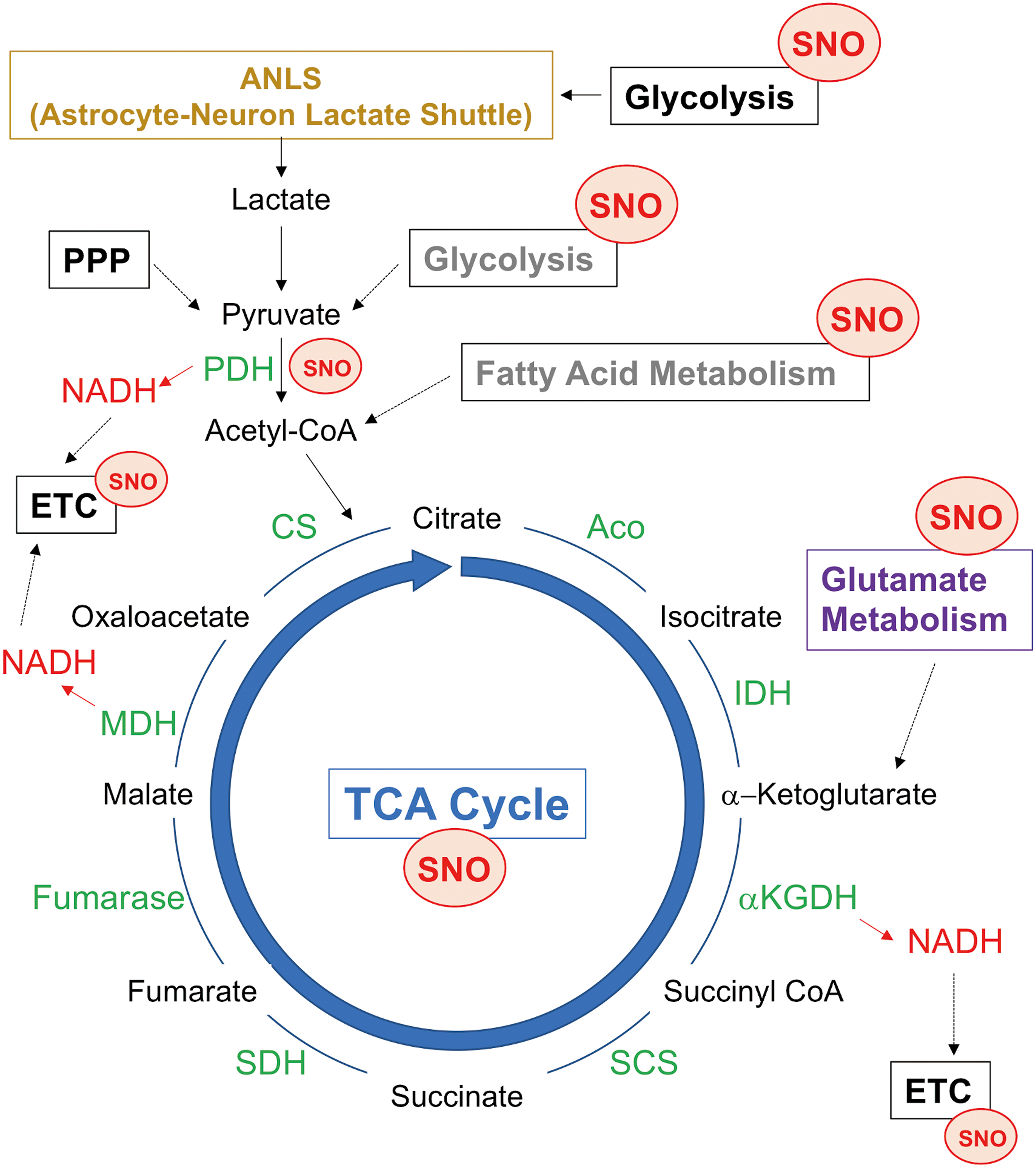
Emerging evidence suggests that pathological interactions between aberrant NO signaling and mitochondrial metabolism contribute to synaptic abnormalities and eventual cell death during neurodegenerative diseases. Along these lines, elevated levels of NO can lead to aberrant S-nitrosylation of enzymes in the TCA cycle as well as proteins in the electron transport chain (ETC), compromising their enzymatic activity (81). For instance, direct S-nitrosylation and/or nitration of key subunits in complexes I, II, IV, and V in the ETC inhibit their respiration activity and consequently their ability to generate ATP. In this review, we highlight the relationship between aberrant NO signaling and TCA cycle enzymes/metabolites; the effects of NO-dependent post-translational modifications on mitochondrial OXPHOS have been extensively discussed in multiple recent review articles [see reviews (37, 81, 95)].
Effects of NO signaling on the TCA cycle
Enzymatic activities required for the TCA cycle to run in the forward direction include aconitase (ACO), isocitrate dehydrogenase, α-ketoglutarate dehydrogenase (αKGDH), succinyl-CoA synthetase, succinate dehydrogenase (SDH), fumarase, malate dehydrogenase, and citrate synthase. Recent S-nitrosoproteome analyses identified all the TCA cycle proteins as potential targets of S-nitrosylation (7, 25, 30, 64, 93, 115, 127) (Fig. 4). Generally, S-nitrosylation of these TCA proteins is inhibitory, thereby decreasing reduced nicotinamide adenine dinucleotide (NADH) production needed for activity of complex I and the ETC to generate ATP. For instance, S-nitrosylation of ACO occurs at cysteine residues responsible for the formation of the Fe–S cluster in the enzyme, possibly disrupting iron–sulfur binding (112). As a second example, the αKGDH complex, comprising three enzymatic components, αKGDH (E1), dihydrolipoamide succinyltransferase (E2), and dihydrolipoamide dehydrogenase (E3), produces succinyl-CoA, NADH, and FADH2, using FAD, NAD+, lipoic acid, Co-A, and thiamine pyrophosphate as cofactors. S-nitrosylation of the αKGDH complex reportedly can occur on the E1 component leading to reversible inhibition (25), although another study showed that S-nitrosylation of the E1 component augments the αKGDH activity (127). Moreover, S-nitrosylation can also occur on the E3 component of the αKGDH complex (35), possibly inhibiting the enzymatic activity of E3 (143). However, since most of these studies used exogenous NO donors for monitoring the αKGDH activity, further studies using models of neurodegenerative diseases are needed to determine how endogenously produced NO might affect the αKGDH activity. In addition, peroxynitrite induces nitration of all three subunits in the αKGDH complex, inactivating the enzyme (118).
In contrast to these inhibitory effects of NO, at least in cancer cells, S-nitrosylation at Cys501 of the mitochondrial chaperone TRAP1, which acts as an SDH inhibitor, facilitates TRAP1 degradation, thereby increasing SDH levels and activity (105). Direct effects of S-nitrosylation of SDH remain unclear (127). Interestingly, because SDH also functions as complex II in the ETC, SNO-TRAP1-dependent regulation of SDH may simultaneously affect the activity of both the TCA cycle and ETC. Notwithstanding these prior protein S-nitrosylation/nitration findings, human brains with neurodegenerative diseases, such as AD, have been reported to manifest decreased TCA cycle enzymatic activity, including ACO, isocitrate dehydrogenase, and αKGDH (17). Hence, these findings are consistent with the hypothesis that S-nitrosylation and possibly oxidation of TCA cycle-related proteins compromise mitochondrial TCA cycle activity, contributing to the development of neurodegenerative conditions because of bioenergetic compromise.
Effects of NO signaling on other aspects of mitochondrial metabolism
NO may also affect mitochondrial metabolism through S-nitrosylation of proteins associated with fatty acid β-oxidation, producing acetyl-CoA from fatty acids for the TCA cycle. For instance, basal levels of NO promote S-nitrosylation of very long-chain acyl-CoA dehydrogenase, possibly enhancing fatty acid metabolism, whereas high levels of NO can aberrantly S-nitrosylate mitochondrial carnitine/acylcarnitine transporter, potentially impairing fatty acid β-oxidation (30, 130). While glucose as a fuel plays a major role in homeostatic adult brain metabolism, as discussed above, it has been suggested that neurons may also use fatty acids as a fuel under specific circumstances, for example, during early developmental stages or nutrient depletion (106, 113). The brain may not favor fatty acid metabolism, possibly because neuronal fatty acid β-oxidation is linked to high levels of oxidative stress, and intermediates of β-oxidation are toxic to the brain mitochondria (114). Additionally, analysis of the S-nitrosoproteome has identified mitochondrial enzymes involved in ketone body utilization (ketolysis), including succinyl-CoA:3-ketoacid coenzyme A transferase 1 that catalyzes the rate-determining step in ketolysis, as substrates for S-nitrosylation (30). Future studies will be necessary to examine whether S-nitrosylation of proteins involved in β-oxidation or ketolysis serves as an important regulator of energy metabolism during periods when fatty acid metabolism influences brain function.
Moreover, S-nitrosylation of several key enzymes in glutamate metabolism and clearance, including glutamate transporter 1 or excitatory amino acid transporter 2, glutamate dehydrogenase (GDH), mitochondrial aspartate aminotransferase (mAspAT), and glutamine synthetase (GS), appears to regulate mitochondrial metabolism (102). During glutamate catabolism, GDH and mAspAT catalyze oxidation of glutamate to produce α-ketoglutarate, which enters into the TCA cycle as a substrate for αKGDH; however, S-nitrosylation can inhibit the activity of GDH and mAspAT, thus potentially decreasing α-ketoglutarate availability.
Defects in Mitochondrial Dynamics Contribute to Neurodegenerative Diseases
Mitochondrial dynamics
Mitochondria constantly undergo fusion and fission (collectively termed mitochondrial dynamics) under physiological conditions. These processes result in distinct mitochondrial morphologies. The dynamic nature of mitochondrial fusion and biogenesis (resulting from fission) ensures proper distribution of mitochondria to subcellular locations, including neuronal axons, presynaptic terminals, and postsynaptic dendritic spines. Critically, in these spines, Ca2+ homeostasis and ATP production are required for normal synaptic maintenance and transmission. Additionally, mitochondrial dynamics modulate apoptosis, ROS generation, mitophagy, and bioenergetics (19, 126). Along these lines, an imbalance in mitochondrial fission and fusion has been linked to a variety of human diseases, including neurodegenerative disorders (23, 125).
While many proteins affect mitochondrial morphology, four large GTPases constitute the core machinery for mitochondrial dynamics: mitofusin 1 (Mfn1), Mfn2, optic atrophy 1 (OPA1), and Drp1 (Fig. 5). Mfns and OPA1 mediate mitochondrial fusion; Mfn1 and Mfn2 are responsible for the fusion of outer mitochondrial membranes, and OPA1 supports the fusion of inner mitochondrial membranes. By contrast, Drp1 acts as a mitochondrial fission protein. To initiate mitochondrial fission, Drp1 translocates from the cytosol to mitochondria, binds to mitochondria fission factor, and oligomerizes to form a ring-like structure at the mitochondrial scission site. Constriction of the Drp1 ring structure upon GTP hydrolysis then initiates mitochondrial fission, producing two mitochondria from a single mitochondrion. Drp1 contains several functional domains that execute mitochondrial fission, including a GTPase domain, a dynamin-like middle domain, and a GTPase effector domain (GED). The intermolecular interactions of the GED domain facilitate the assembly of higher order structures, for example, Drp1 dimers and tetramers (147, 154), thus stimulating assembly-driven GTPase activity.
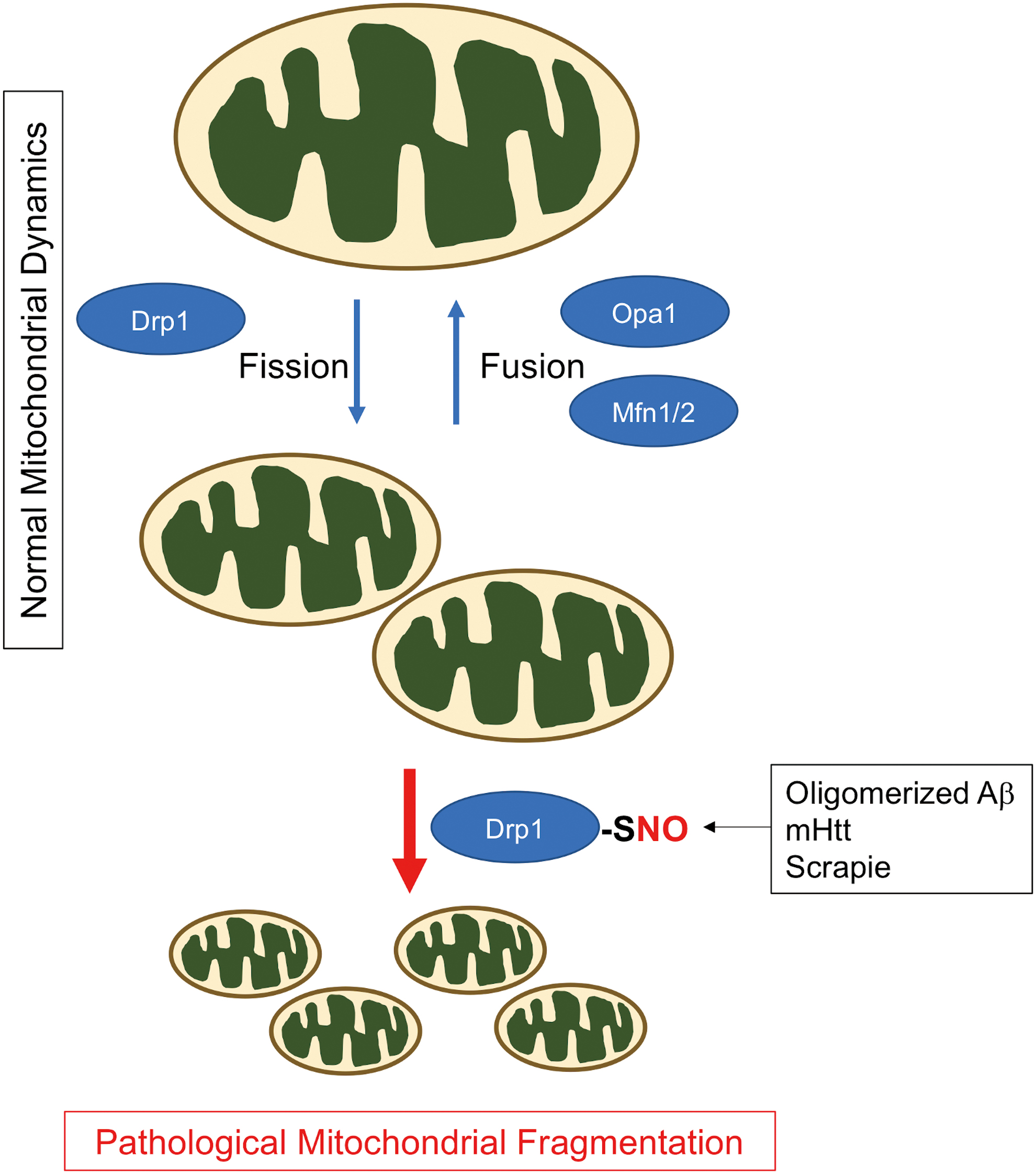
Several lines of evidence indicate that dysregulated Drp1 activity leads to an imbalance in mitochondrial dynamics, contributing to the development of several neurological conditions. In humans, a dominant-negative mutation (A395D) in the Drp1 gene was encountered that causes lethality in the newborn (138). With this mutation, mitochondria display elongated structures due to decreased mitochondrial fission. Consistent with this human case, Drp1-deficient mice demonstrate elongated mitochondria, abnormalities in synapse formation, and embryonic lethality (52, 133). Thus, these results argue that basal Drp1 activity supports physiological mitochondrial fission for maintenance of proper mitochondria distribution and bioenergetics in neurons, as well as in other cell types. Additionally, normal levels of fission can facilitate removal of small mitochondria through mitophagy as discussed in the following sections.
S-nitrosylation of Drp1 in neurodegenerative diseases
Models of neurodegenerative disorders simulated by the application of AD-related Aβ oligomers, exposure to PD-related environmental toxins (e.g., rotenone, 6-hydroxydopamine), or expression of mutant huntingtin (mHtt) to mimic Huntington's disease (HD) all induce Drp-1 dependent, excessive mitochondrial fragmentation, in part via an increase in ROS/RNS (9, 40, 70). Along these lines, our group and subsequently others found that Drp1 activity became overactivated by S-nitrosylation and possibly further oxidation of Drp1 at Cys644 (1, 2, 22, 47, 55, 63, 79, 85, 104, 134, 135). While S-nitrosylated Drp1 (SNO-Drp1) appears in the brains with various neurodegenerative disorders, as discussed below, studies with AD models exemplify the pathophysiological relevance of formation of SNO-Drp1 (2, 22, 99, 134, 135). In response to oligomeric Aβ or nitrosative stress, Drp1 is S-nitrosylated within the GED domain, facilitating dimerization and allosteric upregulation of the GTPase activity (22). Accordingly, SNO-Drp1 formation leads to excessive mitochondrial fragmentation, bioenergetic failure, and consequently synapse loss/neuronal injury (Fig. 5) (22). Recent studies further identified Cdk5 and possibly protein disulfide isomerase as S-nitrosylases toward Drp1, enhancing SNO-Drp1 formation through direct transnitrosylation (63, 99, 132). Notably, expression of a Drp1 mutant lacking the S-nitrosylation site ameliorated synaptic loss induced by Aβ oligomers, consistent with our hypothesis that SNO-Drp1 plays a causal role in the pathogenesis of AD. In agreement with this notion, we and subsequently others found significantly increased levels of SNO-Drp1 in the postmortem human AD brains (22, 135). Intriguingly, we also showed that elevated levels of glucose, as seen in metabolic syndrome (MetS) and type 2 diabetes mellitus (T2DM), increase NO production and subsequent SNO-Drp1 formation coordinately with Aβ oligomers (2). Because T2DM/MetS represents a key risk factor for the development of AD (14), these results provide mechanistic insight into the epidemiological association between these metabolic and cognitive diseases. By contrast, calorie restriction (CR) delays the onset of neurodegenerative phenotypes in some mouse models (41), and it has been suggested that NO-dependent signaling pathways may mediate, at least in part, the anti-aging effects of CR (116). Whether CR decreases S-nitrosylation of Drp1 or other mitochondrial-related proteins remains as an open question.
More recent studies have identified multiple cellular mechanisms acting upstream or downstream of SNO-Drp1 to enhance its neurotoxic effects. First, as an upstream effector of SNO-Drp1 in AD, marked downregulation of microRNA-132 results in elevated NO production due to increased expression of nNOS (137). This leads to aberrant S-nitrosylation of critical neuronal proteins, including not only Drp1 but also Cdk5 and GAPDH. Additionally, as a downstream event of SNO-Drp1-dependent excessive mitochondrial fragmentation, another study found that ROS released from fragmented mitochondria trigger elevated cytokine production in immune cells (107). Since many cytokines stimulate NO production, this finding may represent a positive-feedback loop involving SNO-Drp1 and neuroinflammation during Alzheimer pathogenesis.
Interestingly, several research groups have now reported SNO-Drp1-mediated mitochondrial fragmentation and associated synaptic injury in models of other neurological disease conditions, pointing to the possibility that S-nitrosylation of Drp1 may serve as a common mediator of synaptic damage neurodegenerative processes. For example, in models of HD and transmissible spongiform encephalopathies (TSEs) (47, 144), the formation of SNO-Drp1 was shown to contribute to mHtt or scrapie-induced mitochondrial dysfunction and synaptic injury. In the case of HD, aberrant expansion of a trinucleotide CAG repeat in the htt gene translates into the expanded polyglutamine repeat of mHtt protein, resulting in toxic misfolding of mHtt. The misfolded mHtt initially affects the striatum, triggering an adult onset, neurodegenerative movement disorder. As the disease progresses, the neuropathology also occurs in the cerebral cortex. Consistent with this aspect of disease progression, expression of mHtt in the striatum and cortex was found to correlate with increased SNO-Drp1 formation in HD transgenic mouse and human postmortem HD brains. Using cell-based models, we further demonstrated that expression of non-nitrosylatable mutant Drp1 mitigates mHtt-induced mitochondrial fragmentation and synaptic damage. Hence, these findings suggest that SNO-Drp1 acts downstream of mHtt, mediating signaling cascades contributing to degeneration of synapses and neurons in HD. Since S-nitrosylation of Drp1 appears to be associated with AD, HD, TSE, and possibly PD, characterization of SNO-Drp1-dependent pathways in other disease states could further strengthen the unifying role of SNO-Drp1 as a critical mediator of mitochondrial pathology in neurodegenerative disorders.
As another example of SNO-regulated mitochondrial dynamics, a knock-in mouse line, in which endogenous type 1 ryanodine receptor (RyR1) was replaced with its non-nitrosylatable mutant (C3636A), demonstrated a decrease in mitochondrial fragmentation, thereby rescuing neuronal cell death in the hippocampus of a kainic acid-induced model of temporal lobe epilepsy (75). S-nitrosylation of RyR1 is known to increase Ca2+ release through the RyR1 channel in the ER (31). Hence, it will be important for future experiments to study the possible interaction between the SNO-RyR1/ER/Ca2+ pathway and SNO-Drp1-mediated mitochondrial fragmentation.
NO-Induced Dysfunction in Mitophagy/Autophagy Neurodegenerative Disorders
NO/SNO and mitophagy
Mitophagy involves a selective elimination of damaged mitochondria through the autophagy–lysosomal degradation pathway. Impairment of this cellular defense machinery results in the accumulation of dysfunctional mitochondria typically associated with excessive production of ROS, eventually leading to mitochondrial-dependent neuronal cell death. Numerous studies have suggested that PINK1 and Parkin, whose gene mutations are linked to familial forms of PD, regulate the process of mammalian mitophagy (94), although Parkin/PINK1-independent mitophagy also exists (139). PINK1 represents a mitochondrially targeted serine/threonine kinase, whereas the cytosolic protein Parkin possesses ubiquitin E3 ligase activity. Under basal conditions, PINK1 protein levels remain low due to its high turnover rate; however, when the mitochondrial membrane potential drops, a sign of mitochondrial damage, PINK1 accumulates on the outer membrane of the damaged mitochondrion. PINK1 then phosphorylates ubiquitin, facilitating the selective recruitment of Parkin to the mitochondrial membrane, which enhances Parkin's E3 ligase activity (58). Consequently, Parkin ubiquitinates mitochondrial membrane proteins to recruit autophagy-related proteins, such as microtubule-associated protein 1A/1B-light chain 3 and p62, thus initiating mitophagy (Fig. 6). Notably, defects in Parkin/PINK1-dependent mitophagy have been linked not only to PD but also to many other neurodegenerative diseases, including AD, ALS, and HD [reviewed in ref. (136)].
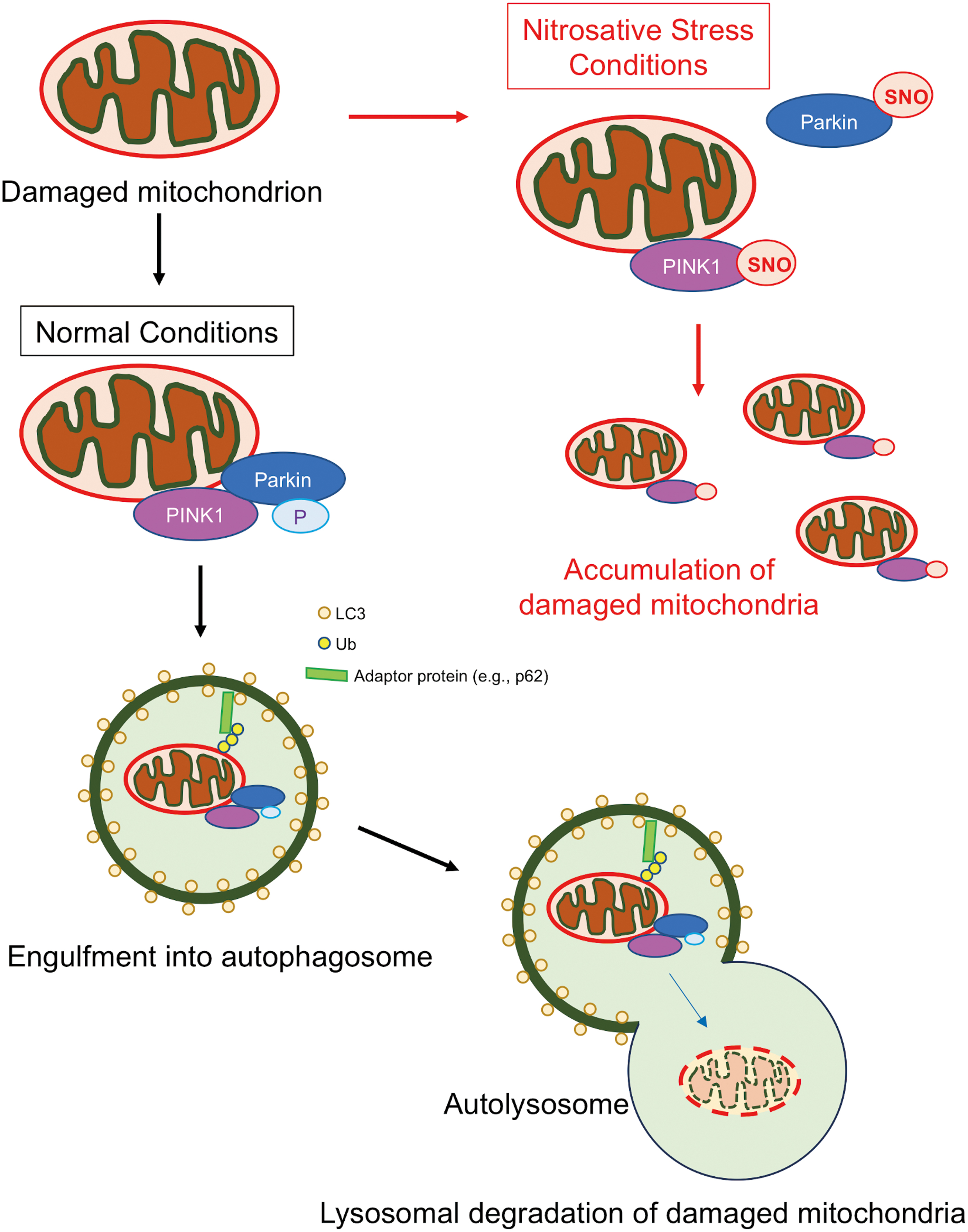
An initial study from our group provided some of the first evidence that NO could affect neuronal mitochondrial dynamics and mitophagy (9). In that study, NO triggered excessive fragmentation of mitochondria, and these injured mitochondria were engulfed in autophagosomes. Additional evidence has revealed that NO signaling plays a critical role in mitophagy at multiple steps. For instance, under normal conditions, PINK1 recruits nNOS to mitochondria, and physiological levels of NO from nNOS enhance Parkin translocation to mitochondria to increase mitophagy (46). However, until recently exactly how pathological levels of NO impair mitophagy remained unclear. Our group recently demonstrated that following mitochondrial insult, aberrant S-nitrosylation of PINK1 at Cys568 inhibits its kinase activity and disrupts Parkin translocation to the mitochondrial membrane, thus impairing Parkin/PINK1-dependent mitophagy in dopaminergic neurons derived from hiPSCs (87) (Fig. 6). Intriguingly, we found that SNO-PINK1 formation occurs at the presymptomatic stage in mouse PD models, consistent with the notion that SNO-PINK1 contributes to mitophagy deficits early in the pathogenesis of PD.
In addition, SNO-Parkin levels have been reported to be greatly elevated in the postmortem human brains of patients diagnosed with PD (26, 145). Moreover, in cell-based and animal models, exposure of neurons to PD-related mitochondrial toxins (e.g., MPP+ or rotenone) or oligomeric α-synuclein triggers aberrant S-nitrosylation of Parkin, leading to abnormal mitochondrial morphology and mitophagy (89, 141, 148) (Fig. 6). Mechanistically, in the later stages of PD, S-nitrosylation of Parkin results in an increase in its ubiquitin-E3 ligase activity; intriguingly, this is followed temporally by a decrease in this activity (26, 145). Accordingly, the SNO-Parkin thus produced contributes not only to deficits in mitophagy but also to the accumulation of misfolded proteins, such as synphilin-1 (26, 89, 145). In addition, S-nitrosylation of Parkin inhibits its binding to Drp1, thus decreasing Parkin ubiquitination of Drp1 and relatively increasing Drp1 levels. The resulting increase in SNO-Parkin-dependent Drp1 activity coincides with the appearance of excessively fragmented mitochondria in neurons exposed to mitochondrial toxins (148), implicating SNO-Parkin in aberrant mitochondrial dynamics.
Further concerning the role of SNO-Parkin in mitophagy, a recent study demonstrated that the initial increase in Parkin activity following its S-nitrosylation promotes ubiquitination of mitochondrial proteins destined for mitophagy, whereas the subsequent decrease in Parkin ubiquitin E3 ligase activity decreases mitophagy (89). Moreover, SNO-Parkin and SNO-Drp1 are present in the GSNOR knockout model of aging/senescence, and these S-nitrosylated proteins contribute to impairment of mitochondrial dynamics and mitophagy (104). Since this experimental model exhibits early signs of neurodegeneration, for example, aggregated α-synuclein, the formation of SNO-Parkin and SNO-Drp1 may represent an early pathogenic event in neurodegenerative diseases.
Prior studies have also found that prolonged nitrosative stress results in tyrosine nitration of Parkin, thus inhibiting mitophagy (89). Nonetheless, in a different in vivo model system, another group demonstrated that peroxynitrite activates mitophagy, possibly through nitration of Drp1 (32). Hence, the precise role of nitration on mitophagy merits further study. In addition to nitration of Drp1 and Parkin (32, 89), recent work has also suggested that nitrotyrosine formation on other proteins may affect mitophagy. For example, nitration of Mn superoxide dismutase (SOD2) inhibits its activity; the resulting increase in ROS may contribute to an imbalance in mitochondrial redox homeostasis and increased mitophagy (27). However, the exact mechanism by which nitrated MnSOD upregulates mitophagy remains unclear, as does the potential pathological role of this event.
SNO-proteins and autophagy
Macroautophagy (referred to here as autophagy) eliminates unwanted cellular components via a lysosomal degradation pathway that helps maintain proper cellular homeostasis and proteostasis. As described above, mitophagy is a special form of autophagy that clears damaged mitochondria. In neurons, autophagic dysregulation (via either increased or decreased activity) disrupts the normal neuroprotective function of autophagy, thus contributing to various disease states including a number of neurodegenerative disorders. In this context, aberrant protein S-nitrosylation of autophagy-related proteins can affect autophagy. Two classical pathways that regulate autophagy involve the c-Jun terminal kinase (JNK)/Bcl-2/Beclin 1 cascade and the IκB kinase (IKK)β/AMPK/mTORC1 cascade. During autophagy, JNK1 activation leads to Bcl-2 phosphorylation, which decreases Bcl-2–Beclin 1 interaction and allows Beclin 1 to participate in autophagosome formation. In addition, activation of IKKβ results in phosphorylation of AMPK, which inactivates mTORC1 (a potent inhibitor of autophagy); this occurs in a tuberous sclerosis complex (TSC) 2-dependent manner. Using cell culture models of HD, Rubinsztein and colleagues presented evidence that inhibition of JNK1 and IKKβ, possibly via protein S-nitrosylation of each, mediates an inhibitory effect on autophagy (109).
Moreover, subsequent studies found that additional SNO-proteins were involved in autophagy. For example, S-nitrosylation of Bcl-2 stabilizes Bcl-2–Beclin 1 interaction (142) (71), and S-nitrosylation of TSC2 increases mTORC1 activity (69); thus, both of these SNO reactions inhibit autophagy. It has also been demonstrated that S-nitrosylation and thus inhibition of PTEN, known to affect the mTORC1 activity, promote cell survival (24, 45, 60, 86, 91). A recent study showed that SNO-PTEN affords prosurvival effects in part via activation of mTORC1, causing inhibition of autophagy (152, 153). However, other studies have demonstrated that NO can elicit cytotoxic activity via excessive augmentation of autophagy. Notwithstanding the SNO-TSC2 study described above (69), NO has also been reported to activate TSC2 and suppress the mTORC1 activity to increase autophagy; whether protein S-nitrosylation plays a role in this setting, however, remains to be established (131). Another effect of protein S-nitrosylation on autophagy has been reported by Snyder and colleagues. They found that cocaine stimulates the formation of SNO-GAPDH, which in turn triggers a Beclin 1-dependent form of autophagy that exerts a cytotoxic effect (44). Obviously, further studies will be essential to elucidate the differential effects of S-nitrosylation of all these proteins and potentially others on autophagy, particularly under conditions relevant to neurodegenerative diseases.
Additionally, S-nitrosylation of lysosomal proteins may contribute to impaired autophagy. Obesity, as observed in high-fat diet mice, leads to downregulation of the GSNOR activity in the liver, increasing S-nitrosylation of two lysosomal enzymes, hexosaminidase subunit β and cathepsin B, thus inhibiting their activities (98). Moreover, S-nitrosylation of lysosomal ATPase (Atp6v1a1 subunit) and cathepsin D has been reported (53, 151). However, whether these SNO-lysosomal proteins play a causal role in inhibition of autophagy remains uncertain. Importantly, further work is needed to elucidate the effects of these autophagy/lysosome-related SNO-proteins on autophagy in general and mitophagy in particular using experimental models of neurodegenerative disorders.
Finally, another study demonstrated that nitration of transient receptor potential melastatin-related 2 (TRPM2) protein at tyrosine 1485 inhibits its ion channel activity, stimulating autophagy during injury to brain pericytes; this form of autophagy activation may possibly involve the HIF (hypoxia-inducible factor)-1/2 pathways (21, 54). Notably, mutation of the nitration site in TRPM2 attenuates autophagy, consistent with the notion that TRPM2 nitration contributes to autophagy-dependent pericyte injury. Intriguingly, pericyte injury is linked to dysfunction of the blood–brain barrier, as occurs in stroke, AD, and other neurodegenerative disorders (146). Thus, in future studies, it will be interesting to determine if nitrated TRPM2 affects autophagy in other cell types, and consequent cell loss in neurodegenerative diseases.
Concluding Remarks
In the present review article, we have highlighted emerging roles for S-nitrosylated and tyrosine nitrated proteins in mitochondrial metabolism, mitochondrial dynamics, and mitophagy/autophagy with a focus on neurodegenerative diseases. With recent advancement in the analysis of the SNO-proteome (30, 115), we anticipate that increasing numbers of SNO-proteins that directly or indirectly regulate mitochondrial function will be identified. Interestingly, while many proteins are aberrantly S-nitrosylated in and contribute to the pathogenesis of neurodegenerative disorders, intervention in just a few or even a single critical SNO pathway(s), such as SNO-Drp1, SNO-Parkin, or SNO-PINK1, can alleviate mitochondrial dysfunction and protect cells in models of neurodegenerative diseases. Hence, identification of the SNO-proteome in each neurodegenerative disorder is only the beginning, as characterization of critical SNO-dependent pathways leading to mitochondrial impairment and neuronal injury will be critical to our understanding of the pathophysiological role of protein S-nitrosylation in these conditions. Moreover, future studies aimed at abating aberrant protein S-nitrosylation may lead to new therapies for dysregulated mitochondrial dynamics, metabolism, and mitophagy in neurodegenerative disorders.
Footnotes
Acknowledgment
We thank Scott R. McKercher (The Scripps Research Institute) for critical reading of the article.
Funding Information
This work was supported in part by the National Institutes of Health (NIH) grants RF1 AG057409, R01 AG056259, R01 NS086890 and DP1 DA041722 (to S.A.L.), R01 AG061845 (to T.N.), an award from the California Tobacco-Related Disease Research Program (TDRP 27IR-0010), a Distinguished Investigator Award from the Brain and Behavior Research Foundation (to S.A.L.), and the Michael J. Fox Foundation (to S.A.L. and T.N.).