
Abstract
Significance:
Heme oxygenase-1 (HO-1) is a ubiquitous 32-kDa protein expressed in many tissues and highly inducible. They catalyze the degradation of the heme group and the release of free iron, carbon monoxide, and biliverdin; the latter converted to bilirubin by biliverdin reductase. Its role in the regulation of cellular homeostasis is widely documented. Studying regulation of HO-1 expression is important not only to understand the life of healthy cells but also the unbalances in cell metabolism that lead to disease.
Recent Advances:
The regulation of its enzymatic activity depends heavily upon changes in expression studied mainly at the transcriptional level. Current knowledge regarding HO-1 gene expression focuses primarily on transcription factors such as Nrf2 (nuclear factor erythroid 2-related factor 2), AP-1 (activator protein-1), and hypoxia-inducible factor, which collect signal transduction pathway information at the HO-1 gene promoter. Understanding of gene expression regulation is not limited to transcription factor activity but also involves an extended range of post- or cotranscriptional regulated events.
Critical Issues:
In addition to the regulation of gene promoter activity, alternative splicing, alternative polyadenylation, and regulation of messenger RNA stability play critical roles in changes in HO-1 gene expression levels, involving specific factors, proteins, and microRNAs. All potential targets for diagnosis or treatment of diseases are related to HO-1 dysregulation.
Future Directions:
Unbalances in the tightly regulated gene expression mechanisms lead to cell transformation and cancer development. Knowledge of these events and signal transduction cascades triggered by oncogenes in which HO-1 plays a critical role is of upmost importance for research in this field.
Introduction
Heme oxygenase-1 (HO-1) catalyzes the degradation of heme with consequent production of free iron, carbon monoxide, and the linear tetrapyrrole biliverdin. The latter is converted to bilirubin by the enzyme biliverdin reductase (1, 40). Different stimuli trigger HO-1 gene expression. This isozyme is a phase II enzyme that is transcriptionally regulated by a large variety of stimuli: tumor necrosis factor-alpha (TNF-α), nerve growth factor, interleukin (IL)-1, and interferon-gamma, its own substrate, heme oxidative stress, and phenolic compounds (curcumin, caffeic acid, etc.) as well as the expression of several oncogenes (7, 37, 58). The products of this reaction display potent antioxidant activity (7, 78). The cytoprotective actions of carbon monoxide on vascular endothelium and nerve cells have also been well documented. Current knowledge indicates that the induction of HO-1 expression and/or activity is a cellular adaptive response that provides cell resistance to oxidative injury (1, 16, 75).
To date, three mammalian HO isozymes have been reported: HO-1, HO-2, and HO-3. The only inducible form is HO-1, while HO-2 is constitutively expressed (16). HO-3 finally proved to be an artifact (19). HO-1 is present in cells as a 32-kDa isoform highly expressed in spleen and liver, and it is also found in a variety of other tissues although at a lower level (1). In vascular tissue, HO-1 protects endothelial cells from a wide variety of apoptotic stimuli, regulating endothelial cell-cycle control, proliferation, vascular endothelial growth factor (VEGF) secretion, and also angiogenesis (38, 54, 78). Its role as an angiogenic mediator in particular context is supported by the fact that a variety of angiogenic factors such as IL-1 and IL-6, prolactin, 15-deoxy-12,14-prostaglandin J are able to trigger HO-1 expression (8, 30). In this regard, we have shown that HO-1 expression and activity are induced by an angiogenic oncovirus, the Kaposi sarcoma-associated herpesvirus (KSHV), in endothelial cells, and elevated levels of the enzyme are detectable in biopsy tissue from oral acquired immune deficiency syndrome (AIDS)-Kaposi sarcoma (KS) lesions (6).
HO-1 Promoter
The structure of the HO-1 promoter has been characterized quite some time ago (65). The human heme oxygenase gene (HO-1 or Hmox-1 gene) is ∼14 kb long and organized into five exons. Induction of HO-1 promoter expression is mediated through cis-regulatory DNA sequences located in the promoter 5′ flanking region of the gene. A wide variety of putative regulatory sites can be found in human, rat, mouse, and chicken HO-1 genes, although, due to the physiological involvement of the activity of HOs, most investigations focused on those implicated in the inducible responses to oxidative signals (65). Structure of the promoter and sequence analysis reveal the existence of enhancer sequences responsible for the binding of well-known transcription factors, and these are aligned in three main clusters located approximately half a kilobase, 4 and 10 kb upstream from the transcription start site (2, 4). The data regarding promoter nucleotide sequence are aligned with multiple lines of evidence coming from signal transduction approaches that support a role in HO-1 promoter activation for a battery of redox-sensitive transcription factors (2), such as activator protein-1 (AP-1) (52), nuclear factor-kappa B (NF-κB) (2), hypoxia-inducible factor (HIF) (36), and, especially, nuclear factor erythroid 2-related factor 2 (Nrf2) (4, 5, 7) (Fig. 1).
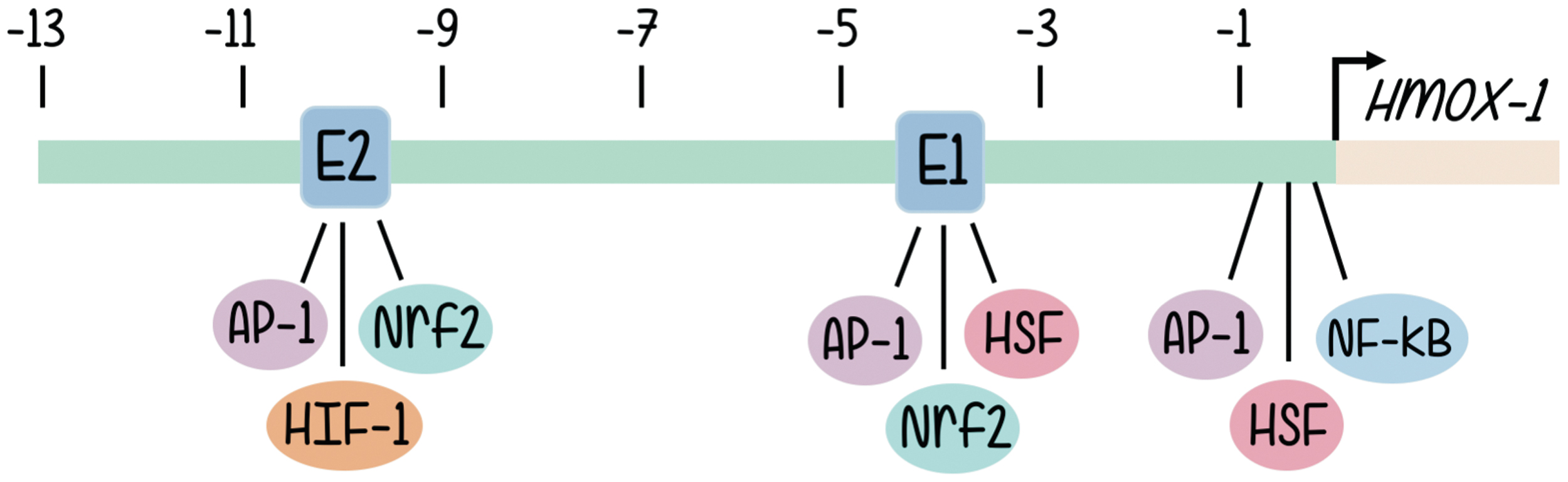
The involvement of AP-1 proteins in HO-1 gene regulation has been supported by studies demonstrating that pharmacological inhibition of AP-1 activity attenuates IL-1a- or TNF-α-mediated induction of HO-1 messenger RNA (mRNA) levels in human endothelial cells (4). An alternative line of evidence showed that AP-1 silencing using a dominant-negative mutant of c-Jun inhibits arsenite-mediated activation of the chicken HO-1 promoter in hepatoma cells (4). The fact that expression and activities of some members of the Jun and Fos family of protein are stimulated by many of the same agents that induce HO-1 expression is also suggestive of a common involvement in signal transduction ensembles. The 5′ flanking region of the HO-1 gene contains sequences termed the stress response element (StRE). The consensus StRE (T/C)GCTGAGTCA resembles the consensus binding site, TGA(C/G)TCA, for the AP-1 class of transcription factors (5).
The AP-1 transcription factor family (2, 44, 77) plays a key role in many instances of cell development that involve changes in cell fate, including the commitment to cell growth and differentiation. This transcription factor is formed by dimers of proteins encoded by the Fos family (c-Fos, FosB, Fra-1, and Fra-2) and the Jun family (c-Jun, JunD, and JunB). Homodimerization of Jun family proteins or heterodimerization between proteins of the two subfamilies results in the formation of AP-1 protein complexes, which recognize specific DNA sequences known as tetradecanoylphorbol acetate-responsive elements (TREs) or AP-1-binding sites, in a wide variety of gene promoters. Examples of those gene promoters include cell-cycle-related genes, AP-1 genes themselves, and the HO-1 gene.
AP-1 proteins (either Fos or Jun monomers) are final targets of signal-transducing kinase cascades. Its phosphorylation confers transcriptional activation, triggers the activity of AP-1-driven promoters and expression of their regulated genes. mitogen-activated protein kinases (MAPKs) are the best known examples of AP-1-related signaling pathways and the phosphorylation of c-Jun by activated JNK (c-Jun N-terminal kinase), the most widely reported (13, 20, 23, 29, 79). Regarding Fos, the activation of c-Fos proteins by MAPKs in response to extracellular activated signaling pathways has not been investigated that extensively. However, we have shown that phosphorylation by p38 or Erk1/2 phosphorylates the transcriptional activation domain of c-Fos in a parallel manner to c-Jun/JNK when cells are stimulated by ultraviolet (UV) or platelet-derived growth factor, respectively (Fig. 2a) (2, 11, 44, 77).
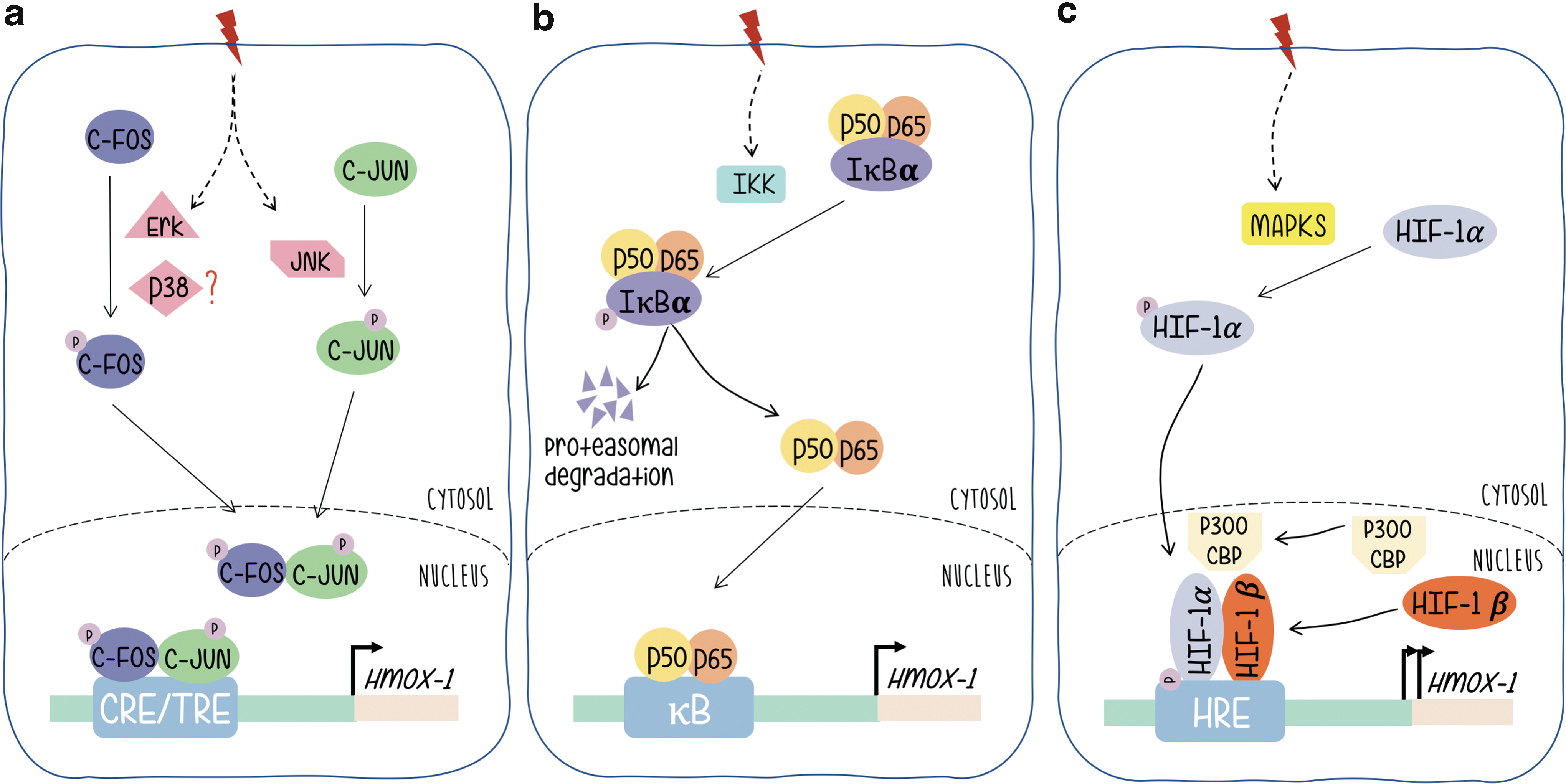
The NF-κB family of transcription factor proteins (NF-κB/Rel) regulates gene expression in a variety of immunological or antioxidant-protective responses. Genes harboring an NF-κB-like enhancer include those of proinflammatory cytokines and antioxidant stress proteins such as HO-1. Basically, and without signal stimulation NF-κB rests in a basal condition with NF-κB linked to the inhibitor of NF-κB (I-κB) that retains the heterodimer composed of p50 and p65 subunits in the cytoplasm (Fig. 2b). Upon signal stimulation, the NF-κB subunits p50 and p65 dissociate from the regulatory subunit I-κB (which is phosphorylated by IKK and sent to proteasomal degradation) and subsequently translocate to the nucleus binding to specific enhancer sequences and triggering expression of target genes. Evidence that supports a role for NF-κB in HO-1 gene expression regulation includes the following: NF-κB induced by stimuli that also upregulate HO-1 gene expression; functional binding sites for NF-κB in the promoters of the HO-1 genes of different mammalian species have been identified. A κB element of the proximal rat HO-1 gene promoter region controls HO-1 upregulation by the phorbol myristate acetate, which is an activator of macrophages, phenomenon that is not granted in cells from NF-κB subunit p65-deficient mice, etc. (52).
Exposure of rats to hypoxia increases HO-1 expression (36). In reporter gene assays, hypoxia-dependent induction of the chloramphenicol acetyltransferase reporter gene was mediated by a sequence found ∼9.5 kb upstream of the transcription start site (Fig. 1). Several lines of evidence support a role for HIF-1 in HO-1 gene regulation: HIF-1 proteins bind specifically to an oligonucleotide that displays these sequences, binding that is not found when the sequences are altered by mutation; the increase in HO-1 mRNA that is hypoxia dependent is not observed in mutant hepatoma cells that lack HIF-1 DNA-binding activity. HIF-1α/HIF-1β heterodimers are essential for hypoxia-induced gene expression regulation. HIF-1α is the target of hypoxia and HIF-1β, although unaffected by hypoxia, is required for DNA binding. As a classical transcription factor, HIF-1α contains a DNA binding domain (amino acids 1–390), heterodimerization domain included (amino acids 1–166), and a transcriptional activation domain (amino acids 531–826). HIF-1α is the target of phosphorylation by MAPKs, p38a, and p38g, thus providing a possible mechanism for the induction of HIF-1-dependent transcription by dual specificity kinases such as MKK6 (71) (Fig. 2c).
The HO-1 promoter presents regulatory sequences for the binding of Nrf2 (3 –5, 25, 31, 35) and associated proteins (33, 36, 51, 57, 61, 64). They constitute the group of transcription factors most widely studied regarding HO-1 expression with a great deal of information present in the currently available literature. Nrf2 is the redox-sensitive transcription factor (also named NF-E2-related factor-2), is a leucine zipper type of protein, member of the Cap'n'Collar family of transcription factors (4). It binds to particular sequence motifs termed StREs or antioxidant response elements (AREs), which are AP-1-related sites. The consensus reported binding sites somehow resemble but differ from the AP-1 (TRE) sequences, and the related CRE, TGCTGAGTCAGCA and (T/C)GCTGA(G/C)TCA(C/T), respectively.
Under basal conditions, Nrf2 is found in the cytoplasm associated with a dimer of other proteins called Keap1, also reported as an actin binding protein (27). Interaction in this heterodimer causes the continuous degradation of Nrf2 that is ubiquitinated and directed toward the proteasome, and consequently inactivated (Fig. 3 left panel).
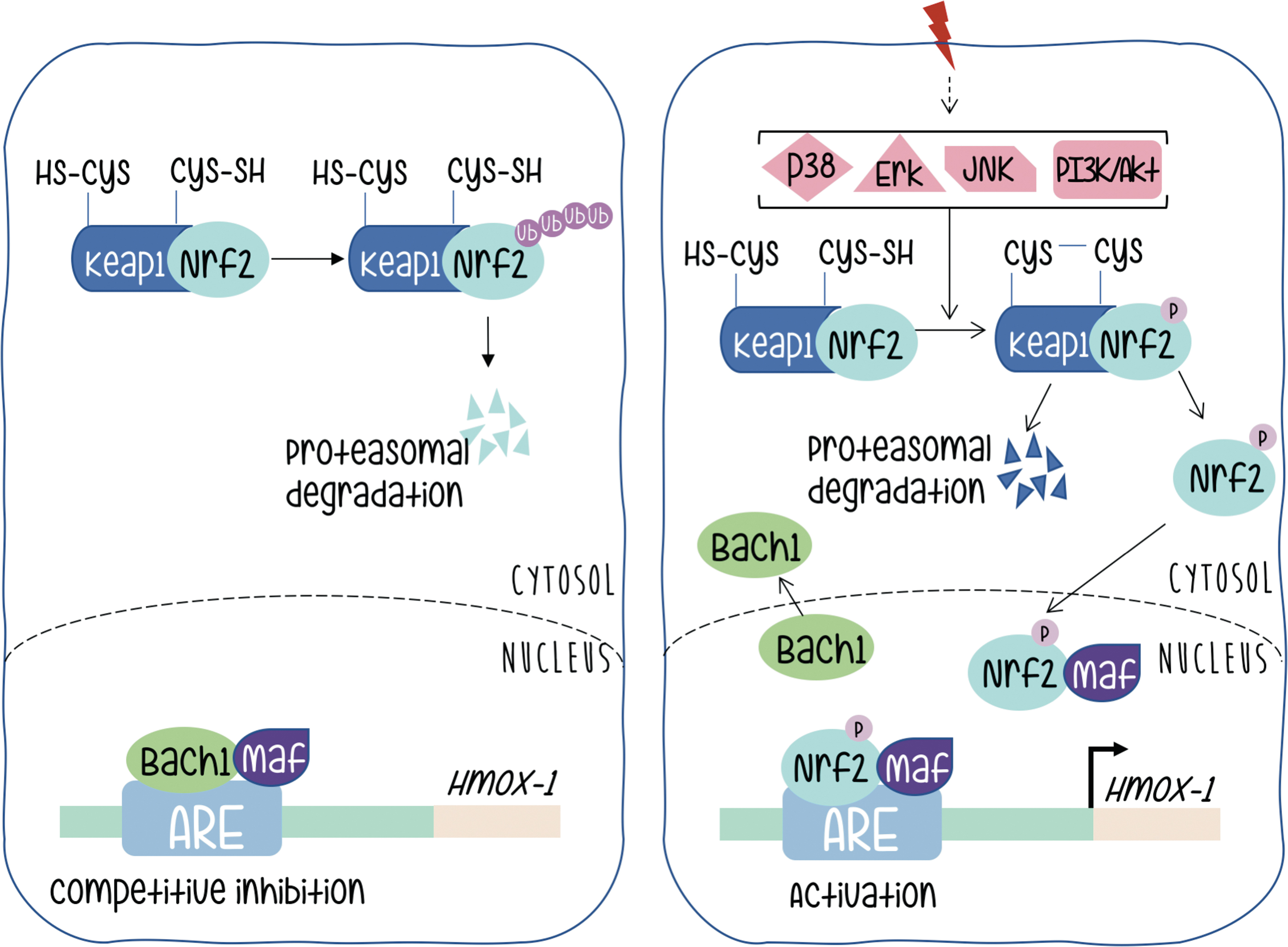
Activation of Nrf2 needs dissociation from its Keap partners. The mechanism by which Nrf2 is liberated from the Nrf2-Keap1 complex remains controversial, but probably involves phosphorylation and stabilization of Nrf2 protein, which otherwise has a half-life of just 30 min before being subjected to degradation. There are numerous studies suggesting that phosphorylation of Nrf2 may contribute to its regulation; some of them contradictory. This controversy is probably indicative of a strong cell context dependency. The amount of information on Nrf2 probably goes beyond the scope of this review, but we can briefly quote that Nrf2 contains serines, threonines, and tyrosines that can provide phosphorylation sites for various kinases. Protein kinase C can phosphorylate Nrf2 in serine 40, and disrupt the association between Nrf2 and Keap1 promoting the translocation of Nrf2 to the nucleus. It has also been reported that p38 can phosphorylate Nrf2, promote its association with Keap1, and thus prevent its nuclear translocation (10, 28, 49). It is clear that kinase activity might be necessary for Nrf2 activation in a variety of scenarios; however, if Nrf2 is the target of direct phosphorylation, the target residues, the identity of the kinases that take part in the process (even in an indirect manner) remains a subject of active interest.
Upon dissociation from Keap and translocation to the nucleus Nrf2 meets a new partner protein called Maf. This family of factors (as at least six members are described) contain the basic leucine zipper domain for DNA binding and dimerization. As extensively reported for Jun and Fos proteins and its specific binding to DNA motifs (named TRE) spread in promoters along mammalian genomes, Maf and Nrf2 form a heterodimer that binds to specific sequence motifs (called antioxidant response element [ARE] instead) in the HO-1 promoter (Fig. 3, right panel).
An additional level of regulation at the ARE sites in the HO-1 promoter is provided by Bach (62). This protein forms a heterodimer with Maf and remains bound to the ARE sites providing negative regulation (Fig. 3, left panel). Therefore, HO-1 activation requires both Nrf2 release from Keap and inactivation of Bach and release of Maf to be available to complex with Nrf2 upon its nuclear translocation. Silencing Bach1 by Bach1-siRNA significantly increased levels of HO-1 mRNA and protein. The heterodimer between Bach1–MafG (a representative member of the Maf family) recruits chromatin remodeling factors and DNA methyltransferases that contribute to gene silencing. Bach is downregulated by the heme group and microRNAs (miRNAs), both endogenous and expressed upon viral infection (12, 62). All these molecular components provide a rich environment for fine tuning of the regulation of HO-1 gene expression at its promoter.
Regulation of Gene Expression Beyond Promoter Activity
Current vision of gene expression regulatory processes is no longer confined to gene promoter activity, although this is certainly the most studied phenomena. Primary transcripts undergo extensive processing after being synthesized by RNA-polymerase II. These processes include capping, splicing, and polyadenylation, all of which are tightly regulated by proteins that bind to nucleic acids and at the same time are putative targets for signaling pathway regulation. To date, alternative mRNA processing, understood as the inclusion or exclusion of specific exons, regulation of poly(A) tail length, etc., is widely known. These processes allow transcripts with different characteristics to be generated from the same gene. Sometimes, the alternative processing results in different proteins being produced, while in other situations different processing alternatives may influence localization, transport, translation efficiency, and, very importantly, stability of the resulting mature mRNAs. Such differences are likely to be highly influential in cell fate, which lately results in consequences for tissue and organism development.
In humans, as well as mammals in general, most multiexonic genes are alternatively spliced and HO-1 is no exception. In addition to the widely reported HO-1 expressed as a 32 kDa protein, a novel alternative splice isoform of 14 kDa HO-1 has been documented (9). This novel isoform is generated through exclusion of exon 3 and is highly expressed in immortalized cells. Regarding expression, induction, and cellular sublocalization, there are slight differences between the two isoforms. While both are induced by UV and H2O2, there is a slight preference of UV irradiation to produce more of the 14 kDa HO-1 isoform. In addition, fluorescence microscopy analysis shows that UV irradiation leads to migration of the 32 kDa HO-1 from the cytoplasm to the nucleus while in contrast, the 14 kDa HO-1 was retained in the cytoplasm under this condition.
Experiments with overexpression of the 14 kDa HO-1 significantly reduced the cell number in the G1 phase and increased the number of cells in S and G2 phases compared with controls with empty vector or the 32 kDa HO-1 isoform. These results indicate that the 14 kDa form of HO-1 has a function in controlling cell proliferation. Telomere maintenance is essential for cancer cell immortalization, and shortened telomeres induce a cell-cycle arrest (9). The finding of this 14 kDa HO-1 isoform that increases relative telomere length both in vivo and in vitro can provide one mechanistic explanation for the positive role of HO-1 in tumorigenesis (for more information of the role of HO-1 in cancer, see Fig. 4).
A different splicing variant from that generated by exon 3 skipping involves the 5′ end of the HO-1 gene, particularly the report introduces the existence of a novel first exon 1a redefining the 5′ untranslated region (UTR). Expression of exon 1a can be induced in HepG2 hepatoma cells by hemin. This alternative in the 5′ UTR contributes to translational regulation as 5′ UTRs containing different alternatives of exon 1 differ in their translational efficiency. These findings provide another mechanistic feature for the regulation of HO-1 expression (32).
Differences in HO-1 expression are also reported at the protein regulation level through changes in cellular sublocalization and protein stability. HO-1 is anchored in the endoplasmic reticulum (ER) through a single transmembrane segment (TMS). ER localization is important for its full enzymatic function with the molecule being exposed on the cytoplasmic side (22). HO-1 forms oligomers attached to the ER and the TMS provides the interface for protein–protein interaction. Interference with interactions at the level of this particular domain results in protein destabilization and concomitant reduction in the enzymatic function of HO-1. Signaling pathways that involve 14-3-3 and STAT family members can stabilize HO-1 expression, preventing ubiquitination and proteasome-mediated degradation (73).
Through the process of alternative polyadenylation, a specific primary transcript has the potential to be cleaved and processed at alternative sites constituting different end points for the last exon (42). The resulting products end up with the addition of a poly(A) tail at different polyA sites [pA or poly(A) sites], and producing mature mRNAs with 3′ UTRs of different lengths. These alternative 3′ noncoding regions influence both translation efficiency and mRNA stability. The binding of specific proteins to well-defined AU-rich sequences (ARE) in these 3′ UTRs is critical. These proteins are known as AU-rich element RNA binding proteins (AUBPs). Just like transcription factors do to enhancer sequences in the DNA, they bind to the nucleic acid (RNA in this case) in a sequence-specific manner and, at the same time, are the targets of signal transduction pathways providing an additional opportunity for sensing the environment and respond regulating gene expression, although at a different level (60, 64, 66, 68, 70, 76).
Major representative AUBPs are members of the following families of proteins: HuR (human antigen R), AUF1 (AU-rich element RNA-binding protein 1), KSRP (K-homology splicing regulator protein), and TTP (tristetraprolin). This protein mRNA interaction can also be modulated by miRNAs depending on the gene under study.
Working with the mouse integrin-beta1 (Itgb-1) gene, we identified two distinct poly(A) consensus sequences located in the 3′ UTR, which suggested the existence of alternative cleavage and polyadenylation sites (50). The short isoform lacks AU-rich motifs and miRNA target sequences that are potentially implicated in the regulation of mRNA stability and translation efficiency. We further determined that the AU-binding protein HuR appears to selectively stabilize the longer isoform in the mammary gland. As for HO-1 database analysis performed in our laboratory confirms a structure at the 3′ flanking region of the gene that resembles that of Itgb-1 in the sense that shows two alternative polyadenylation sites and a AUBP binding motif (also known as ARE) that is present in the longer isoform of the HO-1 mRNAs but is absent in the shorter form rendering the alternative mRNAs for HO-1 different putative responses related to mRNA stability regulation (unpublished observation, study under progress) (Fig. 5).
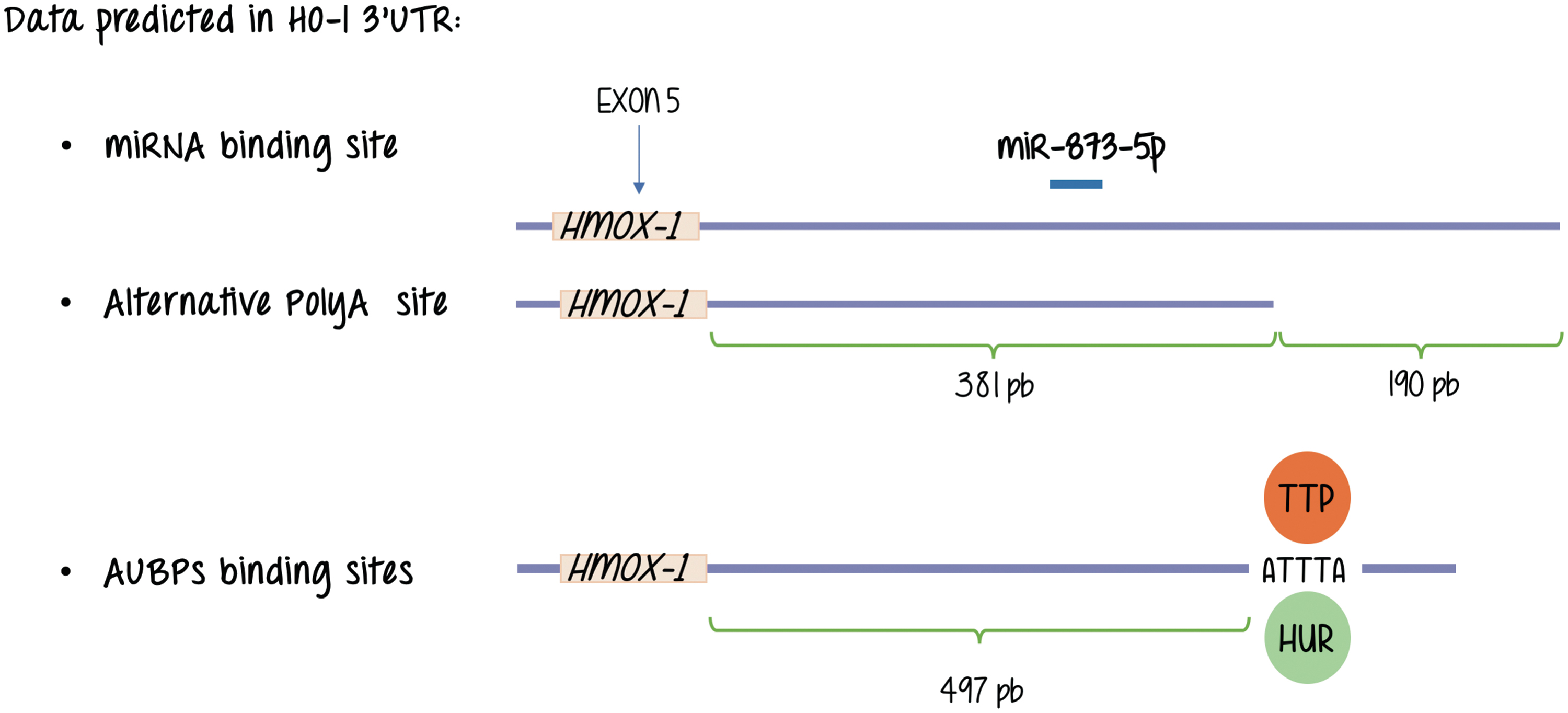
Reports showing changes in HO-1 mRNA stability depending on the physiological conditions of the cell show that nitric oxide (NO) exposure increases the stability of mRNAs encoding HO-1 in human and mouse fibroblasts (34). Using a microarray-based test intended to search for changes in mRNA stability in response to NO treatment in mouse NIH 3T3 fibroblasts treated with actinomycin D (to arrest transcription), the majority of mRNAs was found unaffected. However, around seven genes showed an increase in mRNA stability, including HO-1. Further analysis indicated the presence in the mouse sequences of at least one hit of a signature motif for the stabilizing RNA-binding protein (AUBP or RBP) HuR in accordance with our analysis. Aligned with that observation, a measurable fraction of HuR increased in the cytoplasm after NO treatment. However, among the seven transcripts, only HO-1 mRNA showed a robust increase in its level of association with HuR after treatment with NO. In support of the HuR involvement in HO-1 mRNA stability mechanisms, they observed that HO-1 mRNA and protein levels were significantly reduced upon silencing of HuR and, conversely, they were elevated upon HuR overexpression. Following a similar strategy, we found equivalent results for the regulation of mRNA stability of c-Fos, and this stabilizing effect of HuR proteins is now widely accepted for many mRNAs containing ARE regions (14). To our knowledge, this is the first report of HO-1 gene expression regulation by mRNA stabilization, the identification of HuR as an AUBP implicated in the NO response, and that HO-1 expression induction by NO is regulated by HuR.
We await the unraveling of an increasingly complex panorama in the knowledge of the molecular interactions implicated in this regard as our research related to these regulation instances progresses in the coming years.
As for miRNAs, we do not know any direct regulation of HO-1 expression by direct interactions with miRNAs. Figure 5 shows that database analysis performed in our laboratory showed binding of miR-873-5p, suggestive of a putative role of this miRNA in HO-1 expression, its study awaits further explorations. However, HO-1 expression and miRNAs are not unconnected issues as attenuation of Nrf2 and HO-1 expression through induction of miR-132 and miR-200c by ochratoxin A (OTA, a mycotoxin exhibiting nephrotoxic and potential carcinogenic activity) elevates reactive oxygen species levels and profibrotic TGF expression (74).
HO-1 Expression, Signaling Pathways, and Cell Biology
The cytoprotective actions of HO-1 and its products resulting from enzymatic activity have already been praised in this article. However, overexpression or unbalances in the expression or activity of HO-1 and related metabolic components can be harmful and lead to pathophysiological processes.
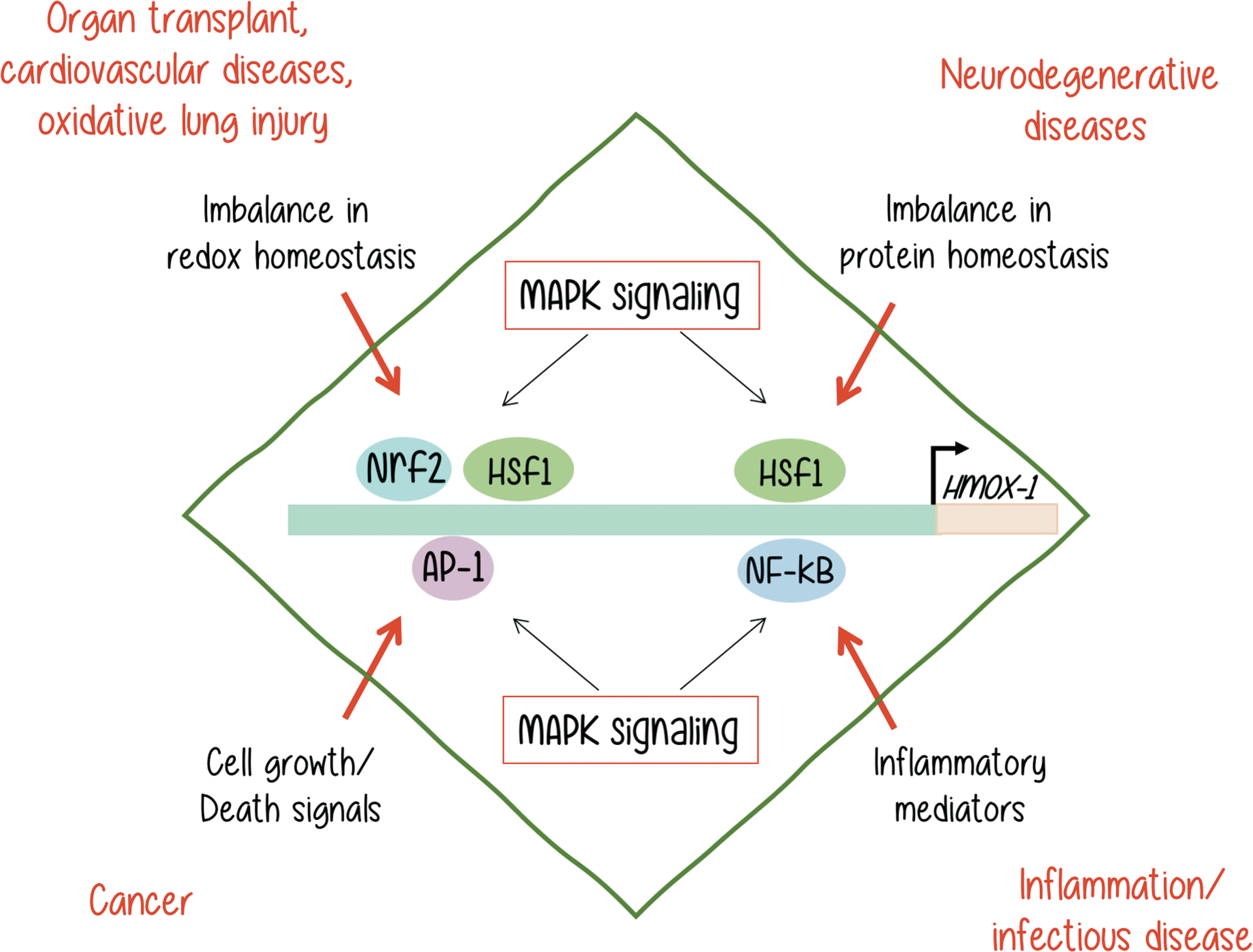
All in all, many signaling pathways regulate HO-1 expression with MAPKs being the most widely reported. MAPKs play an essential role in the transduction of environmental stimuli to the nucleus, and are capable of regulating the expression of genes involved in a variety of cellular processes, including cell proliferation, differentiation, programmed cell death, and, when unbalanced, different malignancies, being the most notorious, neoplastic transformation. MAPKs are classified into at least six subfamilies, the most extensively studied being Erk/MAPKs (Erk1 and Erk2), JNKs (JNK1, JNK2, and JNK3), and p38 kinases. While Erk1/2 and Erk5 (a lesser known MAPK of higher molecular weight) are considered to respond to growth signals, JNKs and p38s are activated by cellular stresses such as exposure to heat shock, protein synthesis inhibitors such as anisomycin, free radicals, ionizing radiation, and UV light. However, depending on the cell context under study, all stimuli can activate any MAPK with relative different intensities. Despite the wide knowledge accumulated on agonist-induced MAPK activation, the way stresses are sensed and where and how the signals are converted into stress-activated protein kinases activation with the consequent triggering of nuclear responses are still open questions, particularly when we refer to the control of expression of genes that are important for cellular homeostasis like HO-1 (14, 39, 44, 77).
As reviewed in an outstanding manner by Alam and Cook (2), different unbalances, which are shown in black letters in Figure 4, can lead to HO-1 overexpression and trigger the malignancies shown next in red letters on the same diagram. Although all of them involve MAPK activation, it is not yet clear if the MAPKs are directly or indirectly involved in each process. Neither we know for sure if MAPK activation may be sufficient to trigger the HO-1 expression that leads to a malignant outcome in every example. It is very clear that each process is dependent on a more complex signaling context.
If tumorigenesis is considered, expression of HO-1 is significantly elevated in tumors, compared with healthy tissues. These tumors include those originated in pancreatic cancer, melanoma, renal cell carcinoma, etc. Also, expression of HO-1 can be enhanced in tissues undergoing anticancer therapies (radio-, chemo-, and photodynamic therapy) (26). What the cell ignites as a cytoprotective mechanism turns out to be harmful instead as it results in decreased effectiveness of the treatment and a negative outcome for the organism. Moreover, in some cases, increased expression of HO-1 is found predominantly in macrophages, not only in tumor cells, as is the case for human gliomas. HO-1 expressed in inflammatory macrophages contributes to immune suppression. If we remember the fact that HO-1 contributes to VEGF secretion and concomitant vascularization, we find that HO-1 overexpression assembles or builds a scenario in which cytoprotective mechanisms, combined with immune suppression and angiogenesis, clearly favor tumor growth (26, 67).
Both over- and underproduction of HO-1 and HO activity can affect a broad spectrum of cells and biological systems. A recent review points out its biological role and clinical implications of dysregulation in obesity, hepatoprotection, and vascular disease (15, 16).
An unexpected turn in the study of the nature of HO-1 and its role in cellular biochemistry and functions is provided by its cellular localization and contribution to tumor biology. Generally, HO-1 localizes mainly in the cytoplasm where it is associated with ER, caveoli, or mitochondria (22). But HO-1 is also a molecular component of the nucleus. Nuclear localization added up to its potential role as an angiogenesis factor (which is dependent on cell context) and in immunosuppression contributes to poor prognosis as observed in several types of cancers (lung, head and neck). The exact mechanism of such localization is not known yet. Initially, it was suggested that in response to stress conditions, HO-1 cleaved and translocated to the nucleus mediating upregulation of genes linked to antioxidant response and cytoprotection. Nevertheless, HO-1 does not function as a transcription factor, and it is more likely to have an indirect effect on gene transcription, modulating transcription factor binding. Nuclear HO-1 promotes glycolysis in hyperoxia but fails to protect against heme toxicity. The role of nuclear HO-1 might then be to enhance glycolysis during hyperoxic stress, protecting against injury and displaying one more mechanism for the promotion of cell proliferation (26, 37). Indicating an exact role for HO-1 in tumor biology is not only complex but also diverse. HO-1 might have diametrically opposite effects regarding cancer as a pathology as antiproliferative effects of HO-1 have been observed for breast cancer (17) and prostate cancer (18, 56), cellular settings that showed an increase in proliferation of cancer cells upon pharmacological inhibition of HO-1 (26). Therefore, the role of HO-1 in cell proliferation, although predominantly positive, may be conditioned to cellular context.
Due to our commitment to the study of HO-1 in AIDS-related malignancies, we will dedicate special attention to this subject. The KS is the most frequent tumor in AIDS patients, characterized by proliferative lesions that contain spindle cells derived from the infection of endothelial cells by the KSHV (also denominated human herpesvirus-8) (6). One gene encoded by KSHV, open reading frame 74, encodes a G protein-coupled receptor (GPCR) named KSHV-GPCR or vGPCR, which is related to the mammalian IL-8 receptor CXCR2 (17). This viral hijacked receptor has an Asp1423Val mutation in a highly conserved Asp-Arg-Lys (DRY) sequence in homolog mammalian GPCRs, which enables its constitutive, ligand-independent activity. As a consequence, vGPCR is able to trigger constitutive downstream signal transduction and induce transformation in fibroblasts as well as angiogenesis in endothelial cells, and even develop human KS-resembling angioproliferative lesions (6, 45, 69). These findings and the elevated expression of HO-1 observed in KS lesions and KS-infected endothelial cells prompted us to investigate whether vGPCR could induce HO-1 expression, its role in vGPCR-dependent transformation, and the molecular components of signaling mediators.
Our summarized results indicate that the viral oncogene induced HO-1 mRNA and protein levels, and that HO-1 was highly expressed in mouse tumors derived from vGPCR-transfected cells. Our data indicate that targeted knockdown gene expression of HO-1 and chemical inhibition of HO-1 enzymatic activity impaired vGPCR-induced VEGF expression, survival, proliferation, and transformation both in cell culture and in a murine allograft tumor model. These findings uncovered the identity of HO-1 and putative signaling mediators that regulate HO-1 expression in the context of KSHV-vGPCR signaling as a potential therapeutic target in KS (39).
As shown in Figure 6, vGPCR contributes to KS development by switching on a complex network of signaling pathways, with the activation of HO-1 expression playing a central role. We have shown that expression of constitutively activated G12 and G13 subunits mimicked vGPCR-induced HO-1 expression and transformation, and that vGPCR activates HO-1 and transformation through both heterotrimeric and small GTP-binding proteins. Our data indicate that vGPCR activates the small GTPase RhoA, a known downstream effector of G12/G13, and that reduced expression or inhibition of RhoA impairs vGPCR-induced VEGF expression and secretion, cell survival and proliferation, and transformation both in cell culture and in a murine allograft tumor model. In addition, G13- and RhoA-induced tumorigenesis are dramatically impaired when HO-1 expression or activity is inhibited. These data show the G12/13/RhoA signal transduction pathway as mediator of vGPCR-induced tumor growth with a central role of HO-1 expression in this process (41).
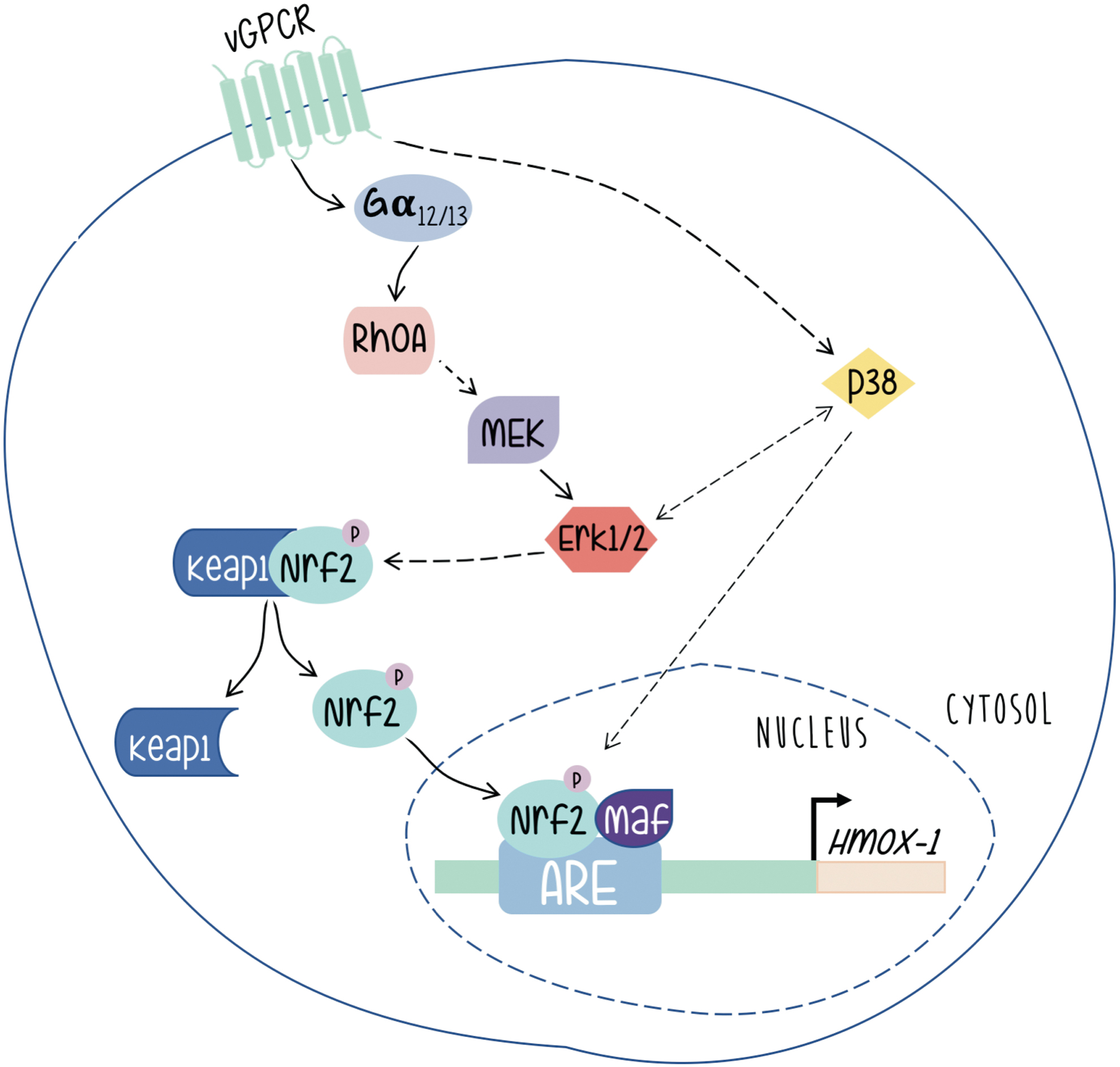
Works from different groups have shown that vGPCR can activate different signaling pathways in different models (6, 46, 72). We have shown that in a fibroblast model, vGPCR activates the ERK1/2 and p38 pathways. Interestingly, we have shown that vGPCR signaling stabilizes Nrf2 and increases the phosphorylation levels of Nrf2. Furthermore, vGPCR activates the ERK1/2 pathway, and has a positive effect on both Nrf2 transcriptional activation and nuclear translocation (59). A role of Erk1/2 in the upregulation of HO-1 expression is reported in other systems (47, 53). However, we have observed no effect on Nrf2 phosphorylation. Nevertheless, our results clearly show the involvement of ERK2 as an intermediary between the vGPCR-Ga12/13-RhoA axis and nuclear translocation of Nrf2 as well as its transcriptional activation and its impact on HO-1 expression.
General Considerations
In an attempt to deliver a concise synopsis of the events involved in HO-1 expression regulation as displayed in currently available literature, we have focused on the concepts that are most striking and consistently observed. We have introduced, to our knowledge, for the first time in a revision the concept of HO-1 gene expression regulation beyond gene promoter activity, a field that will surely grow over the next few years. We may have left out important details for the sake of clarity and to make a point in what we consider the basis of HO-1 gene regulation. However, a great deal of information, in addition to what is provided here, is available in the literature regarding HO-1 expression in particular systems or conditions. We invite the readers to use this material as a basis for understanding the subject and encourage them to consider data not included here.
We might however point out a few more concepts that are emergent in the present and might yield more contributions to the field of HO-1 gene expression in the next future.
It is noteworthy, and an unexpected paradox, that the sequence motifs for the enhancer elements present in the double-stranded DNA comprising the HO-1 promoter are named “ARE,” for Antioxidant Responsible Elements and, similarly, the sequence motifs that bind AUBPs in the 3′ UTRs of single-stranded mRNAs have also been named “ARE.” Despite the coincidence, we understand that it is clear for the reader that both systems may have analogies but work through absolutely different mechanisms.
An emerging concept is that the HO-1 gene is under complex regulatory control. A detailed understanding of these mechanisms, including delineation of the precise signaling pathways and their target transcription factors, will require more extensive work as, probably, a systems biology or multidisciplinary approach. However, gathering information on this regard may provide unique opportunities for a more refined modulation of a therapeutically important enzyme (63). It is important to point out that although intuitively associated and generally treated as such, increases in mRNA and protein levels do not always reflect increases in HO activity. It is well documented, by others and ourselves, the case that upon pharmacological inhibition of enzyme activity cells may respond by dramatically increasing expression levels but still keeping low activity levels in this particular condition (39, 60).
Targeted gene therapies have already been tested using approaches that involve Nrf2 or its regulatory elements. Their involvement in the antioxidant response also contributes to the design of these strategies. A strategy exploited in cancer suicide gene therapy by using a lentiviral vector expressing herpes simplex virus thymidine kinase (HSV-TK/GCV) under the regulation of ARE sequence binding Nrf2 is effective in reducing viability of human lung adenocarcinoma A549 cells (reviewed in Jazwa et al. 24).
An alternative strategy is being used in other pathologies, including cardiovascular diseases such as atherosclerosis and cardiac ischemia-reperfusion injury using ARE-driven oxidative stress-inducible lentiviral vectors for gene therapy (21).
In view of the information displayed in this review, HO-1 and related signaling components, particularly Nrf2, may be considered by now in the category of oncogenes (48). As already mentioned, the potentially beneficial functions of HO-1 may also play an important role in tumor development and progression. As HO-1 is even upregulated in tumors in comparison with healthy tissues, and its expression is further induced upon chemo- or radiotherapy, the otherwise beneficial effects of HO-1 result in decreased effectiveness of the treatment. As a consequence, blockade or inhibition of HO-1 or signaling intermediates involved in HO-1 induction or overexpression (especially Nrf2) are proposed as therapeutic targets for anticancer treatment in many types of tumors (55). Design of strategies is currently under progress in our laboratory and around the globe.
An important observation that arises from the fact that gene expression regulation occurs beyond promoter activation by means of alternative processes occurring during post-transcriptional regulation is provided as follows: in cancer cells, genetic alterations can activate proto-oncogenes leading to tumorigenesis. Mutations that alter the protein products of oncogenes are not essential as sometimes overexpression without alteration of the proto-oncogene is sufficient to transform cells. Comparing two types of cells (cancer cell lines to nontransformed cell lines), cancer cells often express substantial amounts of mRNA isoforms with shorter 3′ UTRs generated through alternative cleavage and polyadenylation. The shorter mRNA isoforms exhibit increased stability, produce more protein, and this is possibly due to the lack of AUBP and miRNA binding sites (as described above and shown in Fig. 5 for the AUBPs) with consequent loss of microRNA-mediated repression (43). These observations strengthen the need to further understand the mechanisms that regulate alternative polyadenylation in healthy cells and the unbalances that lead to disease.
Footnotes
Funding Information
This work was supported by the NIH grant CA221208, by grant PICT 2015 awarded by the National Agency of Scientific and Technological Promotion (ANPCyT), Argentina, a grants from University of Buenos Aires 20020150100200BA and a grant from Instituto Nacional del Cancer (Argentina) INC - AF III - 2017.